

Abstract
During the development of skeletal muscle, myoblasts withdraw from the cell cycle and differentiate into myotubes. The insulin-like growth factors IGF-I and IGF-II, through their cognate tyrosine kinase receptor (IGF-I receptor), are known to play a role in this process. After withdrawal of myoblasts from the cell cycle, IGF-I promotes muscle differentiation by inducing the expression or activity of myogenic regulatory factors (MyoD, myogenin) and effectors (p21). However, little is known about the intracellular mechanisms by which the IGF-I system regulates these factors during the process of myogenesis. Here we show that MKR mice, which express a dominant negative IGF-I receptor specifically in skeletal muscle, have marked muscle hypoplasia from birth to 3 weeks of age. This hypoplasia occurs concomitantly with a decrease in ERK immunoreactivity levels and decreases in MyoD and myogenin expression. BrdU immunocytochemistry showed a compensatory hyperplasia as MKR mice grew to adulthood. Interestingly, hyperplasia occurred concomitantly with an increase in p38, MyoD, myogenin, and p21 immunoreactivity levels, as well as a decrease in Twist levels. These findings suggest that regulation of these cellular elements by IGF-I may play a role in the development and differentiation of skeletal muscle in vivo.
Introduction
The development of skeletal muscle is a multistep process wherein pluripotent mesodermal cells give rise to myoblasts. Myoblasts permanently withdraw from the cell cycle and begin to express muscle-specific genes and proteins. These processes lead to the fusion of myoblasts into multinucleated differentiated myotubes. Prior to the onset of muscle differentiation, proliferating myoblasts express two muscle-specific basic helix-loop-helix (bHLH) transcription factors, MyoD and Myf5. Upon activation, MyoD and Myf5 induce myoblast withdrawal from the cell cycle and the expression of other myogenic bHLH regulatory transcription factors, including myogenin and MRF4 (1, 2).
The IGF system plays an essential role in the formation and maintenance of skeletal muscle. IGF-I and IGF-II and their cognate tyrosine kinase receptor (IGF-I receptor) have been shown to elicit major growth promoting effects (3).While IGF-II is essential for normal embryonic growth (4), IGF-I operates throughout prenatal and postnatal development (5). Mice lacking the IGF-I receptor exhibit marked muscle hypoplasia and die soon after birth because they failed to breathe (5). On the other hand, mice overexpressing IGF-I in muscle develop muscle hypertrophy (6–8). Unlike other growth factors, IGF-I stimulates both myoblast proliferation and differentiation (9, 10). In proliferating myoblasts, IGF-I increases the expression of the cell cycle progression factors (9). After withdrawal of myoblasts from the cell cycle, IGF-I promotes muscle differentiation by inducing the expression or activity of myogenic regulatory factors (11).
The proliferative versus the differentiating functions of IGF-I appear to be mediated by distinct intracellular signaling pathways (12). Studies suggest that the mitogen-activated protein (MAP) kinase pathway mediates cellular proliferation, whereas the phosphoinositide 3′-kinase (PI3K) pathway is activated during differentiation (12–14). However, recent evidence demonstrates a key role for the MAP kinase signaling pathways (ERK1/2 and p38) in muscle differentiation (15–18). At present, the mechanisms by which IGF-I–induced activation of these signaling pathways regulates muscle differentiation in vivo have not been determined. The muscle-specific bHLH transcription factors (MyoD, myogenin, MRF4, and Myf5) function by activating genes that are required for muscle determination and/or differentiation. One of the targets of MyoD is the gene encoding the cyclin-dependent protein kinase inhibitor p21 (also known as Waf1 and Cip1). In culture cells, p21 protein blocks progression through the cell cycle, and its expression is induced early in the differentiation process (19). In addition, p21 has been shown to be a survival factor through the IGF-I–induced PI3K pathway (20). Twist, another transcription factor from the bHLH protein family, acts as an inhibitor of muscle differentiation. This effect is thought to be mediated by blocking of DNA binding by MyoD (21, 22).
We used a transgenic mouse model that overexpresses a dominant negative IGF-I receptor specifically in skeletal muscle to better characterize the role of the IGF-I receptor in the development of skeletal muscle in vivo. We examined various IGF-I receptor–activated signaling pathways, and the expression of myogenic regulators and effectors in skeletal muscle from these animals. We show that mice with a functional inactivation of the IGF-I receptor in skeletal muscle exhibit reduced muscle mass and hypoplasia from birth to 3 weeks of age. However, as these mice grew to adulthood, a compensatory muscle hyperplasia developed, as shown with BrdU immunocytochemistry. Hyperplasia coincides with an increase in the amounts of the ERK1/2 (p44/42) and p38 MAP kinases.
Methods
Generation of MKR mice.
The generation of MKR mice has been previously described (23). Briefly, the muscle creatine kinase promoter, used to drive muscle-specific expression, was subcloned upstream of a mutant human IGF-I receptor cDNA. The mutation Lys1003 → Arg1003 abolishes ATP binding within the β subunit of the human IGF-I receptor and results in a kinase-inactive human IGF-I receptor that acts in a dominant negative fashion, as we have previously demonstrated both in vitro and in vivo (23, 24). The transgene for muscle creatine kinase promoter/human IGF-I receptor (MKR) was injected into the eggs of female FVB/N mice. All experiments were performed in homozygous mice, newborn to 8 weeks of age. Experiments in both male and female mice gave the same results. The data we present here include the results of experiments performed with male mice. Controls consisted of sex- and age-matched wild-type (WT) mice. Mice were kept under a 12-hour light/dark cycle and were fed standard diets. All studies were conducted in accordance with NIH guidelines, as approved by the Animal Care and Use Committee of NIDDK.
Growth and development.
The growth rate of mice was determined by measuring body weight from the time of birth to 8 weeks of age. Specific developmental parameters, including body length, bone length, and tissue wet weight were measured for the same time period (0–8 weeks of age). For body length measurement, anesthetized animals were stretched on top of a ruler. Femur length was measured from acetabulum to trochanter using x-ray radiography on dead animals. Tissue wet weights were measured on an electronic scale.
Muscle histology and morphometry.
Animals were anesthetized with Avertin (15 ml/kg body weight, injected intraperitoneally). Hindlimb muscles from newborn to 8-week-old WT and MKR mice were removed, fixed in a 4% paraformaldehyde solution, and embedded in paraffin. Serial 5-μm cross sections of tissue from each group were stained with hematoxylin and eosin. Morphometric analysis was performed on digital muscle cross sections taken with a Zeiss Axiovert S100 TV microscope with a video camera (Carl Zeiss Microimaging Inc., Thornwood, New York, USA) at magnifications of 2.5× and 20×. Analysis of the muscle cross section area, fiber diameter, number of fibers per area, and number of nuclei per myofiber was carried out on three consecutive sections from five animals in each group, using MacBAS version 2.52 software (Fuji Photo Film Co., Stamford, Connecticut, USA). Muscle protein concentration was determined from whole muscle homogenates using the BCA protein assay (Pierce Chemical Co., Rockford, Illinois, USA) and normalized to body weight.
Immunocytochemistry.
For intraperitoneal injections, BrdU (Sigma Chemical Co., St. Louis, Missouri, USA) was dissolved in 0.9% NaCl, and 50 mg/kg was administered once per day for 15 days to 3- and 6-week-old WT and MKR mice. Control animals received a saline injection. Animals were anesthetized and killed 24 hours after the last injection of BrdU. Animals were perfused transcardially with 4% paraformaldehyde in 0.1 M phosphate buffer (PB; pH 7.4). Hindlimb muscles were removed and postfixed for 24 hours at 4°C. Thirty-micrometer serial cross sections of muscles were cut with a vibratome (Leica Microsystems Nussloch GmbH, Nussloch, Germany) and immersed, free-floating, in 0.1 M PB. One series of sections was used for labeling with BrdU, and another series for hematoxylin and eosin staining.
For BrdU immunohistochemistry, sections were incubated in 0.1 M PB with 0.5% Triton X-100 and 0.1% BSA, and then blocked by incubating for 30 minutes in a solution of 97% methanol and 3% hydrogen peroxide. DNA was denatured with a 45-minute incubation in 2 N HCl at room temperature. Primary antibody used was mouse anti-BrdU (University of Iowa Developmental Studies Hybridoma Bank, Iowa City, Iowa, USA) at 1:2,500. The secondary antibody used was a biotinylated donkey anti-mouse IgG (Pierce Chemical Co.) at 1:1,000. This was followed by analysis with the peroxidase-based ABC system (Vector Laboratories Inc., Burlingame, California, USA) using 3,3′-diaminobenzidine tetrahydrochloride as the chromogen.
BrdU-positive cells were counted in a one-in-three series of sections (90 μm apart) with a 40× objective (Leica Microsystems Nussloch GmbH) throughout the proximal to distal part of the muscle. The same muscle areas and number of sections were studied for all the animals and all the experimental groups.
Treadmill running.
Animals were familiarized with the treadmill apparatus (Cibertec SA, Madrid, Spain) to minimize stress. Three- and six-week-old WT and MKR mice were divided into two groups: exercised and non-exercised (control). The exercised group ran for 1 hour/day at 18 m/min for 15 days. Control mice remained in the treadmill without running. In a second series of experiments, exercised and control mice received a daily intraperitoneal injection of BrdU for 15 days (see above). After running, animals were anesthetized and perfused as described above, and hindlimb muscles were processed for hematoxylin and eosin staining and morphometry, or for BrdU immunocytochemistry (see above).
Strength and fatigue testing.
The contractile properties of the gastrocnemius were determined in situ as described previously (25). Four- to five-week-old mice were anesthetized with sodium pentobarbital (100 mg/kg), and an incision was made through the skin from the ankle to the knee along the medial side of the right leg. The distal tendon of the gastrocnemius was isolated and threaded through a hole in the lever arm of a galvanometer. The tibial nerve was partially isolated approximately 2 mm from the gastrocnemius and secured to an electrode. The ankle was fixed in a clamp, and the skin was pulled up to form a reservoir for a pool of mineral oil. The entire apparatus was placed in a heated Plexiglas chamber kept at 37°C. Contractile properties were measured using a dual-mode servo-controlled moving iron galvanometer (model 305; Cambridge Technology Inc., Cambridge, Massachusetts, USA) with a step response time of 4 ms. The final measurement was the fatigue test, performed as described previously (26).
Western blot analysis.
After anesthesia, hindlimb muscles were removed and processed for immunoblotting as previously described (27), using antibodies against the following: phospho-p44/42 MAP kinase, phospho-p38 MAP kinase, Akt, phospho-Akt, and phospho-Jun N-terminal kinase (JNK) (all from New England Biolabs Inc., Beverly, Massachusetts, USA); ERK1/2, p38 MAP kinase, JNK, p21, MyoD, and myogenin (all from Santa Cruz Biotechnology Inc., Santa Cruz, California, USA); mouse β-actin (Sigma Chemical Co.); and rabbit Twist (provided by C.A. Glackin, Beckman Research Institute of the City of Hope, Duarte, California).
Northern blot analysis.
Northern blot analysis of samples containing 20 μg of total RNA were performed exactly as described previously (27). Blots were hybridized with cDNA probes (mouse MyoD and myogenin, provided by P. Rotwein, Oregon Health Sciences University, Portland, Oregon), or mouse Twist (27). The integrity and the quantification of different transcripts were assessed using an 18S RNA probe (Ambion Inc., Austin, Texas, USA).
Statistical analysis.
Results are expressed as mean ± SEM. Statistical analysis of the data was performed using a one-factor ANOVA and the Student t test.
Results
Impaired postnatal growth in MKR mice.
The generation of transgenic mice expressing a dominant negative IGF-I receptor specifically in skeletal muscle (MKR mice) has been previously described (23). Since the expression of muscle creatine kinase promoter is maximal immediately after birth (28), we studied the effect of functional inactivation of the IGF-I receptor in the postnatal development of skeletal muscle.
MKR mice showed growth retardation soon after birth, compared with WT animals (Figure 1a). We studied growth in MKR and WT mice by measuring the following parameters from birth to 8 weeks of age: total body weight, body length, bone length, and tissue wet weight. Compared with WT mice, MKR mice had 20% less total body weight from birth up to 5 weeks of age. From 5 to 8 weeks, the body weight was 10% lower in MKR mice than in WT mice (Figure 1b). Body length, measured as the nose-to-anus distance, was also reduced by 20% in MKR from birth to 5 weeks of age, as compared with WT mice. However, from 5 to 8 weeks of age, there was no difference in body length between the two groups (Figure 1c). Tissue wet weight was measured in organs expressing IGF-I receptors, namely, brain, lung, heart, liver, spleen, kidney, perigonadal white fat, and skeletal muscle. There were no differences between MKR and WT mice in wet weight of any of the tissues tested, except for skeletal muscle from birth to 8 weeks (data not shown). MKR mice showed a significant reduction in total muscle wet weight. The wet weight of hindlimb muscles (extensor digitorum longus [EDL], soleus, gastrocnemius, and quadriceps) were an average of 30% smaller in MKR mice from birth to 3 weeks of age, 20% smaller from 3 weeks to 5 weeks of age, and 10% smaller from 5 weeks to 8 weeks of age (Table 1). The femur length from acetabulum to trochanter was also approximately 30% less in MKR mice than in WT mice from birth to 3 weeks of age (the average femur length of WT mice at 3 weeks was 0.6 ± 0.03 cm, and that of MKR mice was 0.4 ± 0.03 cm; P < 0.01). In contrast, femur length was not different between the MKR and WT groups from the ages of 5 weeks to 8 weeks (data not shown).
Figure 1.

Growth characteristics of WT and MKR mice. (a) Two-, four-, and eight-week-old WT and MKR mice. (b) Body growth and (c) body length curves from birth to 8 weeks in WT and MKR mice. Open circles, WT; filled circles, MKR. Each point represents the mean ± SEM of at least 20 animals in each group. *P < 0.05; **P < 0.01.
Table 1.
Differences in hindlimb muscle weights from WT and MKR mice

Compensatory muscle hyperplasia in MKR mice.
To investigate whether the decrease in skeletal muscle mass in MKR mice arose from atrophy and/or hypoplasia, we quantified muscle size, fiber size, number of fibers per area, and number of nuclei per myofiber. The average cross-sectional area (CSA) of MKR soleus muscle (slow-twitch fiber type) was 20% smaller than that of WT mice from birth to 3 weeks of age, and 10% smaller than WT up to 8 weeks of age (Figure 2a and Figure 3a). The average number of myofibers per given area was similar in MKR and in WT animals from birth to 8 weeks of age (data not shown). The diameter of myofibers was similar between groups (data not shown). Moreover, at 8 weeks of age, the CSA of the EDL, a fast-twitch muscle, was smaller in MKR mice than in WT mice (WT, 1.8 ± 0.2 mm2; MKR, 1.56 ± 0.1 mm2; P < 0.01) (Figure 2a), whereas the number of myofibers per area was not different between the two groups (WT, 548 ± 12; MKR, 547 ± 15; P < 0.2) (Figure 2a). These parameters indicate that MKR muscles are hypoplastic. We also measured number of nuclei per myofiber. From 0 to 3 weeks of age, there was no difference in the number of nuclei per myofiber (Figure 2b). However, 5- and 8-week-old MKR mice had 15% and 20% more nuclei per myofiber, respectively, than did WT animals (Figure 2b). The protein content in soleus muscle as normalized to total body weight was also measured in WT and MKR mice. MKR mice had a 20% lower protein content in this muscle from birth to 3 weeks of age than WT animals had. In contrast, we found that protein content was 15% and 25% higher in the soleus muscle of MKR mice than in WT mice at 5 weeks and 8 weeks of age, respectively (Figure 2c). Protein content in EDL muscles showed a similar pattern to that in soleus muscles (data not shown).
Figure 2.

Characteristics of MKR versus WT muscle at 3, 5, and 8 weeks of age. (a) CSA of the soleus muscle. (b) Number of nuclei per myofiber. (c) Protein content of soleus muscle, expressed as mg/g body weight. White bars, WT mice, black bars, MKR mice. Data are expressed as mean ± SEM of five animals in each group. *P < 0.05; **P < 0.01.
Figure 3.
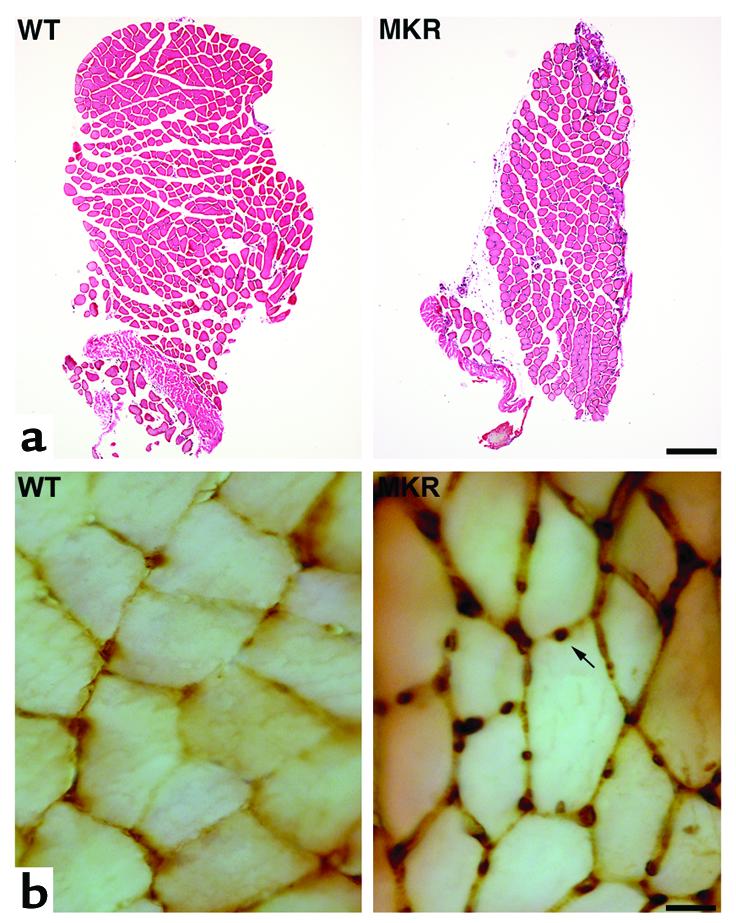
Cross sections of soleus muscle from 8-week-old WT and MKR mice. (a) CSA of MKR soleus muscle (right) compared with WT soleus muscle (left), stained with hematoxylin and eosin. Bar, 200 μm. (b) BrdU immunocytochemistry showed an enhanced number of nuclei per myofiber (arrow) in MKR (right) compared with WT muscles (left). Bar, 25 μm.
Newly formed cells were assessed by BrdU immunocytochemistry in soleus and EDL muscles of WT and MKR mice. MKR soleus muscle showed a higher number of BrdU+ nuclei at 5 weeks (WT, 1.0 ± 0.2; MKR: 2.4 ± 0.1; P < 0.01) and 8 weeks (WT, 1.0 ± 0.1; MKR, 3.2 ± 0.1; P < 0.01) of age (Figure 3b). Similar results were obtained in EDL muscle. Proliferation of myofibers in adult muscle is carried out by the satellite cells (29). Therefore, these results indicate that satellite cells are responsible for the hyperplasia that significantly contributed to the increase in muscle mass in adult MKR mice.
To induce muscle mass increase, we exercised 3- and 6-week-old WT and MKR mice for 15 days. Furthermore, to examine the effect of exercise training on satellite cell proliferation, we injected BrdU into running 3- and 6-week-old WT and MKR mice. Morphometric analysis revealed that unlike WT muscles, MKR muscles showed neither an increase in fiber diameter nor an increase in satellite cell proliferation (data not shown).
We considered the possibility that these structural changes in MKR mice might also be associated with functional changes in the contractile machinery of skeletal muscle. Hence, we measured muscle strength in MKR and WT mice. However, there was no difference in the specific tension of the gastrocnemius, indicating that overexpression of a kinase-inactive IGF-I receptor does not affect the functional contractile activity of this muscle (Table 2).
Table 2.
Contractile properties of the right gastrocnemius muscle in WT and MKR mice

Taken together, these data suggest that inactivation of IGF-I receptor specifically in skeletal muscle causes hypoplasia during the early postnatal stages. However, these mice appear to undergo a compensatory muscle hyperplasia upon reaching adulthood.
MAP kinase signaling pathways are upregulated in MKR mice.
To determine which signaling pathways are involved in the hypoplastic and hyperplastic responses in MKR mice, we evaluated several protein kinases implicated in the IGF-I signaling pathway, including ERK1/2, p38, JNK, and Akt. Both the phosphorylation state and the total amount of these proteins were analyzed in hindlimb muscles from MKR and WT mice aged 0–8 weeks. Representative blots are shown at 0–3 weeks, 5 weeks, and 8 weeks of age.
Levels of ERK1/2 immunoreactivity were 1.5-fold lower in MKR mice than in control mice from 0 to 3 weeks of age, and were increased twofold above WT mice at 5 weeks of age. By 8 weeks, there was no difference in ERK1/2 levels between groups (Figure 4a). The phosphorylation state of p44/42 was not different in MKR mice than in WT mice, as phospho-p44/p42 immunoreactivity closely paralleled the levels of total p44/42 immunoreactivity. That is, phospho-p44/p42 levels were 1.5-fold lower in the skeletal muscle of MKR mice than in WT mice from 0 to 3 weeks of age, increased twofold above WT mice at 5 weeks of age, and were not different between groups in adult animals. β-actin protein levels were similar in both groups of mice (Figure 4a).
Figure 4.

Comparison of ERK1/2, p44/42, p38, JNK, and Akt levels in WT versus MKR mice. Hindlimb muscles were removed from WT and MKR mice at the ages of 0–3 weeks, 5 weeks, and 8 weeks, and subjected to Western blot analysis. (a) Representative blots showing phospho-p44/42 MAP kinase (upper panel), ERK1/2 (middle panel), and β-actin (lower panel) immunoreactivity levels in MKR and WT mice. (b) Representative blots showing phospho-p38 (upper panel), p38 (middle panel), and β-actin (lower panel) immunoreactivity levels in MKR and WT mice. (c) Representative blots showing phospho-JNK (upper panel) and JNK (lower panel) immunoreactivity levels in MKR and WT mice. (d) Representative blots showing phospho-Akt (upper panel) and Akt (lower panel) immunoreactivity levels in MKR and WT mice. Each blot represents three independent experiments, using five animals in each group.
Recently, the stress-activated protein kinase p38 was reported to play a key role in muscle differentiation by activating MyoD (15–17). From 0 to 3 weeks of age, p38 levels were similar in WT and MKR mice. However, p38 immunoreactivity levels were significantly increased in MKR mice at the ages of 5 weeks and 8 weeks, by twofold and threefold, respectively, compared with WT mice (Figure 4b). Phospho-p38 immunoreactivity levels closely paralleled the increases in p38 protein levels in MKR mice, indicating that there was no net change in the phosphorylation state of p38. β-actin levels were similar in both groups of mice (Figure 4b).
We also analyzed the levels of expression and phosphorylation of JNK. Figure 4c shows that there were no differences between MKR and WT mice in either JNK or phospho-JNK immunoreactivity levels at any of the ages tested. We also measured Akt and phospho-Akt immunoreactivity levels in skeletal muscle of MKR and WT mice. Levels of phospho-Akt and Akt were similar in both groups from birth to 8 weeks of age (Figure 4d).
MyoD and myogenin are upregulated in the skeletal muscle of MKR mice.
We explored the question of whether the MKR phenotype was associated with upregulation of myogenic transcription factors in muscle. We evaluated the mRNA and protein levels of MyoD and myogenin, two myogenic transcription factors involved in early and late responses in differentiation, respectively, from birth to 8 weeks of age. As an example, we describe blots from three different ages. MyoD expression was twofold lower in 0- to 3-week-old MKR mice than in WT animals. In contrast, at the age of 5 weeks, MyoD levels were threefold higher in MKR mice than in WT mice. Similar levels of MyoD were expressed in MKR and WT mice at the age of 8 weeks (Figure 5a). MyoD protein levels showed an expression pattern similar to that of its mRNA levels (Figure 5b). Myogenin expression was 1.5-fold lower in MKR mice than in WT mice from 0 to 3 weeks of age. Myogenin levels progressively increased in MKR mice from the age of 5 weeks to levels that were threefold higher than those of WT controls by the age of 8 weeks (Figure 5c). A similar pattern was found for myogenin protein levels (Figure 5d). β-actin and 18S RNA levels were similar in both groups. Taken together, these data show that muscle inactivation of the IGF-I receptor induces delayed muscle differentiation.
Figure 5.

Delayed activation of MyoD and myogenin in MKR mice. Hindlimb muscles were removed from WT and MKR mice at the ages of 0–3 weeks, 5 weeks, and 8 weeks, and subjected to Northern or Western blot analysis, as indicated below. (a) Northern blot analysis of MyoD levels in MKR and WT mice (upper panel). 18S RNA levels were similar in all groups (lower panel). (b) Western blot analysis of MyoD (upper panel) and β-actin (lower panel) immunoreactivity levels in MKR and WT mice. (c) Northern blot analysis of myogenin levels in MKR and WT mice (upper panel). 18S RNA levels were similar in all groups (lower panel). (d) Western blot analysis of myogenin (upper panel) and β-actin (lower panel) immunoreactivity levels in MKR and WT mice. Data are expressed as the mean ± SEM of three independent experiments, using five animals in each group.
Differential responses of Twist and p21 proteins in MKR mice.
Twist inhibits muscle differentiation by blocking the DNA binding activity of MyoD (21, 22). To determine whether Twist is involved in the hypoplastic phenotype of the skeletal muscle in MKR mice, we measured Twist mRNA and protein levels in both groups of animals. These experiments showed that Twist mRNA and protein levels were threefold higher in 0- to 3-week-old MKR mice than in WT mice. No differences were found between the groups at other ages (Figure 6, a and b).
Figure 6.

Differential expression of p21 and Twist in MKR mice. Hindlimb muscles were removed from WT and MKR mice at the ages of 0–3 weeks, 5 weeks, and 8 weeks, and subjected to Northern or Western blot analysis, as indicated below. (a) Northern blot analysis of Twist levels in MKR and WT mice (upper panel). 18S RNA levels were similar in all the groups (lower panel). (b) Western blot analysis of Twist (upper panel) and β-actin (lower panel) immunoreactivity levels in MKR and WT mice. (c) Western blot analysis of p21 (upper panel) and β-actin (lower panel) immunoreactivity levels in MKR and WT mice. Data are expressed as the mean ± SEM of three independent experiments, using five animals in each group.
We also measured the levels of p21 in the skeletal muscle of MKR and WT mice from birth to 8 weeks of age. MyoD stimulates p21 expression in muscle, and activates its differentiation (19). Thus, we determined p21 levels in MKR and WT mice. Levels of p21 were 1.5-fold lower in 0- to 3-week-old MKR mice than in WT mice. However, p21 levels were twofold higher at the ages of 5 weeks and 8 weeks in MKR mice than in WT mice (Figure 6c). Taken together, all these data may explain the delayed and late differentiation of muscles of MKR mice.
Discussion
Here we show that functional inactivation of the IGF-I receptor specifically in skeletal muscle of mice resulted in a delay in the proliferation and differentiation processes in skeletal muscle cells. Although these mice showed hypoplasia and reduced muscle cell mass from birth to 3 weeks of age, a hyperplastic response was observed as mice grew to adulthood. Newly formed cells in MKR hindlimb muscles were demonstrated with BrdU immunocytochemistry. It is known that adult skeletal muscle cells are incapable of dividing, and increases in the number of nuclei in adult skeletal muscle are due to the proliferation and differentiation of latent undifferentiated muscle precursors called satellite cells (29). Therefore, the changes in muscle mass in MKR mice are due to the proliferation and differentiation of their satellite cells. Exercise-induced proliferation of satellite cells has been documented, and is thought to be mediated by IGF-I (30, 31). Consistent with the lack of IGF-I receptor activation, there was no increase of newly formed nuclei in the exercised MKR mice.
Previous studies demonstrated that mice with a targeted disruption of the IGF-I receptor exhibit marked skeletal muscle hypoplasia, and die soon after birth (5). In contrast, mice overexpressing IGF-I specifically in muscle develop increased muscle mass and hypertrophy (6–8). In accordance with our results, transfection with a kinase-inactive IGF-I receptor abolished IGF-I–induced proliferation and delayed differentiation in mouse myoblasts (32). Conversely, overexpression of human IGF-I receptors in rodent skeletal muscle cells enhanced IGF-I–induced proliferation and differentiation (10).
The signal transduction pathways and effectors of IGF-I receptor–regulated skeletal muscle differentiation in vivo have not been identified. Unlike other growth factors, IGF-I stimulates proliferation as well as differentiation of myoblasts (9, 10). It has been suggested that IGFs promote proliferation by activating the MAP kinase pathway and differentiation through the PI3K pathway (12–14). However, recent studies have reported that ERK1/2 MAP kinases are differentially regulated in mitogenesis and myogenesis, and that the stress-activated protein kinase p38 is critical for regulation of terminal differentiation (15–18). This discrepancy might be due to the distinct types of culture cells used in the various studies.
Our results indicate that in MKR mice, there is a multistep regulation of the myogenic program by the p38 and ERK MAP kinase pathways, wherein ERKs have a biphasic profile during myogenesis, and p38 is particularly activated for terminal differentiation. ERK1/2 levels were lower in 0- to 3-week-old MKR mice than in controls, in which hypoplasia was more prominent. While MKR mice are growing, ERK levels start to be activated and muscle cell mass to increase. At this point, p38 levels remain higher as MKR mice mature from 5 weeks to 8 weeks of age. This period coincides with the shift from the hypoplastic to the hyperplastic phenotype. Thus, lower ERK levels seem to be responsible for the decreased proliferation and hypoplasia responses in the skeletal muscle of MKR mice. Activation of p38 may be essential for terminal differentiation and compensatory hyperplasia in cooperation with ERKs in MKR mice. Thus, it is possible that coordinated regulation of the ERKs and p38 may contribute to this dual phenotype in MKR mice.
Like p38, JNK is a stress-activated protein kinase (18). There were no differences in JNK or phospho-JNK immunoreactivity levels in skeletal muscle between MKR and WT mice. This sharply contrasts with the differences in ERK and p38 levels between these two groups of animals. JNK has also been shown to be involved in the contractile activity in skeletal muscle (33). In accordance with these results with JNK, we found no differences in the strength of MKR skeletal muscles (an indicator of the status of the contractile apparatus) compared with WT.
Akt and phospho-Akt immunoreactivity levels were similar in MKR and WT mice. These results were not surprising, since basal insulin receptor substrate-1 phosphorylation was very similar in MKR mice and WT controls (23).
Taken together, these results indicate that the two MAP kinase signaling pathways (ERK and p38) must act in parallel, and both are required for the compensatory myogenic proliferation and differentiation in MKR mice. However, the PI3K pathway may also be important for muscle cell survival, since adult MKR mice, even showing a compensatory hyperplasia, still have reduced muscle mass.
Interestingly, we found that two muscle-specific transcription factors are also differentially expressed in MKR versus WT mice during the period in which the muscle phenotype changes from hypoplasia to hyperplasia. The mechanisms by which the mitogen-activated and stress-activated protein kinase signaling pathways may collaborate with myogenic regulatory factors to promote terminal differentiation in vivo have not been clearly defined. Knockout mouse models have been very useful in determining the function of myogenic regulatory factors in myogenesis. MyoD is expressed in myoblasts determined to have withdrawn from the cell cycle, and myogenin is involved in the late terminal differentiation process (1). These findings are in concordance with our results. That is, MyoD and myogenin levels are lower in MKR mice than in WT mice from birth to 3 weeks of age, when a hypoplastic phenotype is observed. At the age of 5 weeks, MyoD levels were increased in MKR mice compared with WT mice. At the age of 8 weeks, when a hyperplastic phenotype was observed in skeletal muscle, myogenin levels were higher in MKR mice than in WT mice.
In myocytes and other cell types in culture, p21 is induced early during differentiation via activation of MyoD, and this promotes withdrawal from the cell cycle (19, 20). Interestingly, the upregulation of ERK1/2 and p38 in skeletal muscle of MKR mice relative to WT mice closely parallels the differential expression of p21 and the transcription factors MyoD and myogenin in these mice. These differences in gene and protein expression also coincide with the transition from a hypoplastic to a hyperplastic phenotype in MKR mice.
We also found that the period of hypoplasia in MKR mice coincides with an upregulation of Twist protein and mRNA levels. Twist inhibits differentiation by blocking the DNA binding activity of MyoD (21, 22). We have previously shown that Twist can be induced by IGF-I through the p44/42 MAP kinase pathway (27). Similarly, in the MKR mice, Twist levels are upregulated in younger animals, in which the hypoplasia is more pronounced, and the levels of both p38 and ERK MAP kinases are downregulated relative to levels in WT mice. Taken together, these results suggest the possibility that hypoplasia in MKR mice may be due to a Twist-mediated inhibition of MyoD. Inhibition of MyoD, in turn, blocks the induction of p21 and thereby inhibits terminal differentiation. The ERK and p38 signaling pathways may mediate this process.
In conclusion, IGF-I receptors are important for the establishment and maintenance of the mature muscle phenotype in normal and regenerating muscle tissue. The present study establishes that the IGF-I receptor acts as an important regulator of muscle cell differentiation by regulating muscle-specific genes that control muscle cell mass. Therefore, IGF-I receptor represents a potential candidate target for gene therapy in muscle degeneration. In mature cells, IGF-I receptor signaling coordinates the necessary responses of muscle cells to changes in the functional requirements that occur during normal aging and after injury or atrophy. The concomitant regulation of muscle phenotype with the ERK and p38 signaling pathways together with myogenin, MyoD, Twist, and p21 suggests that these cellular elements may be coordinately involved in the process of muscle development. Clearly, further studies will be required to establish a causal role for these factors in the muscle phenotype exhibited by MKR mice. One or more of these factors in the IGF-I signaling pathway may also be useful targets for the development of novel therapeutic strategies to treat various muscular disorders. These studies represent an important early step toward delineating the molecular mechanisms that regulate muscle cell structure and function.
Acknowledgments
We thank S. Yakar and A.L. Castle for helpful discussions, J. Setser for technical assistance, P. Rotwein for the MyoD and myogenin probes, and C.A. Glackin for the anti-Twist antibody. A.M. Fernández was partially supported by a postdoctoral fellowship from the Ministerio de Educación y Cultura of Spain, and a Fogarty fellowship from NIH.
Footnotes
Ana M. Fernández’s present address is: Instituto Cajal, Consejo Superior de Investigaciones Científicas, Madrid, Spain.
References
- 1.Arnold HH, Braun T. Targeted inactivation of myogenic factor genes reveals their role during mouse myogenesis: a review. Int J Dev Biol. 1996;40:345–353. [PubMed] [Google Scholar]
- 2.Lassar AB, Skapek SX, Novitch B. Regulatory mechanisms that coordinate skeletal muscle differentiation and cell cycle withdrawal. Curr Opin Cell Biol. 1994;6:788–794. doi: 10.1016/0955-0674(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 3.LeRoith D, Werner H, Beitner-Johnson D, Roberts CT. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143–163. doi: 10.1210/edrv-16-2-143. [DOI] [PubMed] [Google Scholar]
- 4.DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin- like growth factor II gene disrupted by targeting. Nature. 1990;345:78–80. doi: 10.1038/345078a0. [DOI] [PubMed] [Google Scholar]
- 5.Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- 6.Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci USA. 1998;95:15603–15607. doi: 10.1073/pnas.95.26.15603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coleman ME, et al. Myogenic vector expression of insulin-like growth factor I stimulates muscle cell differentiation and myofiber hypertrophy in transgenic mice. J Biol Chem. 1995;270:12109–12116. doi: 10.1074/jbc.270.20.12109. [DOI] [PubMed] [Google Scholar]
- 8.Musaro A, et al. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet. 2001;27:195–200. doi: 10.1038/84839. [DOI] [PubMed] [Google Scholar]
- 9.Engert JC, Berglund EB, Rosenthal N. Proliferation precedes differentiation in IGF-I-stimulated myogenesis. J Cell Biol. 1996;135:431–440. doi: 10.1083/jcb.135.2.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quinn LS, Steinmetz B, Maas A, Ong L, Kaleko M. Type-1 insulin-like growth factor receptor overexpression produces dual effects on myoblast proliferation and differentiation. J Cell Physiol. 1994;159:387–398. doi: 10.1002/jcp.1041590302. [DOI] [PubMed] [Google Scholar]
- 11.Musaro A, Rosenthal N. Maturation of the myogenic program is induced by postmitotic expression of insulin-like growth factor I. Mol Cell Biol. 1999;19:3115–3124. doi: 10.1128/mcb.19.4.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J Biol Chem. 1997;272:6653–6662. doi: 10.1074/jbc.272.10.6653. [DOI] [PubMed] [Google Scholar]
- 13.Tamir Y, Bengal E. Phosphoinositide 3-kinase induces the transcriptional activity of MEF2 proteins during muscle differentiation. J Biol Chem. 2000;275:34424–34432. doi: 10.1074/jbc.M005815200. [DOI] [PubMed] [Google Scholar]
- 14.Jones NC, Fedorov YV, Rosenthal RS, Olwin BB. ERK1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J Cell Physiol. 2001;186:104–115. doi: 10.1002/1097-4652(200101)186:1<104::AID-JCP1015>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 15.Gredinger E, Gerber AN, Tamir Y, Tapscott SJ, Bengal E. Mitogen-activated protein kinase pathway is involved in the differentiation of muscle cells. J Biol Chem. 1998;273:10436–10444. doi: 10.1074/jbc.273.17.10436. [DOI] [PubMed] [Google Scholar]
- 16.Wu Z, et al. p38 and extracellular signal-regulated kinases regulate the myogenic program at multiple steps. Mol Cell Biol. 2000;20:3951–3964. doi: 10.1128/mcb.20.11.3951-3964.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zetser A, Gredinger E, Bengal E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J Biol Chem. 1999;274:5193–5200. doi: 10.1074/jbc.274.8.5193. [DOI] [PubMed] [Google Scholar]
- 18.Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. doi: 10.1016/s0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 19.Halevy O, et al. Correlation of terminal cell cycle arrest of skeletal muscle with induction of p21 by MyoD. Science. 1995;267:1018–1021. doi: 10.1126/science.7863327. [DOI] [PubMed] [Google Scholar]
- 20.Lawlor MA, Rotwein P. Insulin-like growth factor-mediated muscle cell survival: central roles for Akt and cyclin-dependent kinase inhibitor p21. Mol Cell Biol. 2000;20:8983–8995. doi: 10.1128/mcb.20.23.8983-8995.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hebrok M, Wertz K, Fuchtbauer EM. M-twist is an inhibitor of muscle differentiation. Dev Biol. 1994;165:537–544. doi: 10.1006/dbio.1994.1273. [DOI] [PubMed] [Google Scholar]
- 22.Spicer DB, Rhee J, Cheung WL, Lassar AB. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science. 1996;272:1476–1480. doi: 10.1126/science.272.5267.1476. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez AM, et al. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 2001;15:1926–1934. doi: 10.1101/gad.908001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato H, Faria TN, Stannard B, Roberts CT, Jr, LeRoith D. Role of tyrosine kinase activity in signal transduction by the insulin-like growth factor-I (IGF-I) receptor. Characterization of kinase-deficient IGF-I receptors and the action of an IGF-I-mimetic antibody (alpha IR-3) J Biol Chem. 1993;268:2655–2661. [PubMed] [Google Scholar]
- 25.Walters TJ, Sweeney HL, Farrar RP. Influence of electrical stimulation on a fast-twitch muscle in aging rats. J Appl Physiol. 1991;71:1921–1928. doi: 10.1152/jappl.1991.71.5.1921. [DOI] [PubMed] [Google Scholar]
- 26.Burke RE, Levine DN, Tsairis P, Zajac FE. Physiological types and histochemical profiles in motor units of the cat gastrocnemius. J Physiol. 1973;234:723–748. doi: 10.1113/jphysiol.1973.sp010369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dupont J, Fernandez AM, Glackin CA, Helman L, LeRoith D. Insulin-like growth factor 1 (IGF-1)-induced twist expression is involved in the anti-apoptotic effects of the IGF-1 receptor. J Biol Chem. 2001;276:26699–26707. doi: 10.1074/jbc.M102664200. [DOI] [PubMed] [Google Scholar]
- 28.Trask RV, Billadello JJ. Tissue-specific distribution and developmental regulation of M and B creatine kinase mRNAs. Biochim Biophys Acta. 1990; 1049:182–188. doi: 10.1016/0167-4781(90)90039-5. [DOI] [PubMed] [Google Scholar]
- 29.Seale P, Rudnicki MA. A new look at the origin, function, and “stem-cell” status of muscle satellite cells. Dev Biol. 2000;218:115–124. doi: 10.1006/dbio.1999.9565. [DOI] [PubMed] [Google Scholar]
- 30.Smith HK, Maxwell L, Rodgers CD, McKee NH, Plyley MJ. Exercise-enhanced satellite cell proliferation and new myonuclear accretion in rat skeletal muscle. J Appl Physiol. 2001;90:1407–1414. doi: 10.1152/jappl.2001.90.4.1407. [DOI] [PubMed] [Google Scholar]
- 31.Singleton JR, Feldman EL. Insulin-like growth factor-I in muscle metabolism and myotherapies. Neurobiol Dis. 2001;8:541–554. doi: 10.1006/nbdi.2001.0416. [DOI] [PubMed] [Google Scholar]
- 32.Cheng ZQ, et al. Functional inactivation of the IGF-I receptor delays differentiation of skeletal muscle cells. J Endocrinol. 2000;167:175–182. doi: 10.1677/joe.0.1670175. [DOI] [PubMed] [Google Scholar]
- 33.Aronson D, Dufresne SD, Goodyear LJ. Contractile activity stimulates the c-Jun NH2-terminal kinase pathway in rat skeletal muscle. J Biol Chem. 1997;272:25636–25640. doi: 10.1074/jbc.272.41.25636. [DOI] [PubMed] [Google Scholar]