

Abstract
We reported earlier that IL-1β, an NF-κB-regulated cytokine, was made by intestinal epithelial cells during detachment-induced apoptosis (anoikis) and that IL-1 was antiapoptotic for detached cells. Since surviving anoikis is a prerequisite for cancer progression and metastases, we are further exploring the link between anoikis and cytokines. Here we determined that multiple genes are expressed following detachment including a number of NF-κB-regulated products and therefore aimed to determine whetherNF- κB signalling plays any role in regulating apoptosis. Using Western blotting, we detected that IκBα becomes phosphorylated immediately following detachment and that levels of phospho-IκBα peaked within 20 min. Phosphorylation of IκBα was followed by Rel A (p65) nuclear translocation. Increased NF-κB activity following detachment was confirmed using the detection of NF-κB-promoted luciferase gene expression delivered by adenovirus infection. Infection of cells with adenovirus expressing a super-repressor IκBα protein and pharmacological inhibitors of NF-κB resulted in the failure to phosphorylate IκBα, a more rapid activation of caspases and earlier apoptosis. We also detected that IκB kinase α (IKKα) and not IKKβ became phosphorylated following detachment. Since IKKα is activated by NF-κB-inducing kinase (NIK), we overexpressed native NIK using an adenovirus vector that resulted in enhanced phospho-IκBα and nuclear p65 in detached cells compared to control detached cells but did not result in a significantly greater number of cells surviving to 24 h. We conclude that detachment directly activates NF-κB, which, in addition to launching an inflammatory cytokine wave, contributes to a delay in apoptosis in intestinal epithelial cells.
Keywords: anoikis, intestinal epithelial cell, apoptosis, NF-κB, detachment
Introduction
Patients with chronic inflammatory bowel diseases are at a greatly increased risk of developing colon cancer (Gillen et al., 1994), likely due to the combination of chronic insults to the epithelium and the ongoing exposure to factors that promote cell survival, as part of the reparative process. Cancer arises during these diseases as intestinal epithelial cells (IEC) become transformed and survive detached from the basement membrane. Detachment of normal adhesion-dependent cells leads to apoptosis, a process termed ‘anoikis’; overcoming anoikis, therefore, is paramount to cancer establishment and metastases (Sträter et al., 1996).
In order to study the mechanisms promoting epithelial survival during the stress of inflammation and, in particular, detachment, we used the anoikis-susceptible rat crypt-like line IEC-18. Roughly 40% of detached IEC-18 survive to 24 h if the cells are prevented from reattaching (Waterhouse et al., 2001). In addition, we showed that immediately following detachment, IEC-18 express IL-1β and the IL-1 receptor type II (Waterhouse and Stadnyk, 1999), and Miller and McGee (2002) showed that IL-6 was upregulated by detachment, suggesting that IEC are programmed to trigger inflammation. Others showed IL-8 expression due to detachment of bronchial epithelial cells (Shibata et al., 1996), implying that this may be a common feature of mucosal epithelial cells. We subsequently determined that IL-1 promotes survival of detached IEC-18 and thus the detached cells would seem to be expressing mediators that not only contribute to inflammation but also may affect apoptosis in vivo (Waterhouse et al., 2001). IL-1β, IL-6 and IL-8 expression are all regulated to some extent by the transcription factor NF-κB, but whether NF-κB becomes activated directly as a result of detachment has not been reported.
NF-κB is a transcriptional regulator of many genes associated with early immune, acute phase and inflammatory responses. It is a heterodimer composed of Rel A (p65) and NF-κB1 (p50) subunits in IEC (Jobin et al., 1997; Jobin et al., 1998a). The canonical activation pathway of NF-κB is regulated by an endogenous cytoplasmic inhibitor, IκB, which, in responding to certain stimuli, becomes phosphorylated at serine residues 32 and 36, by a multimolecular complex called IκB kinase (IKK), and then is selectively ubiquinated and degraded, leaving NF-κB to translocate into the nucleus (Baeuerle and Baltimore, 1996; Chen et al., 1996; Hochstrasser, 1996). In IEC, the activation of this pathway can be stimulated by cytokines, for example IL-1 and TNF-α (Bader and Nettesheim, 1996), or by infection (Savkovic et al., 1997; Elewaut et al., 1999). In various other cell types, an alternative pathway of activation exists, where NF-κB-inducing kinase (NIK) activates IKKα, which in turn mediates the decay of p100, ultimately allowing p52/RelB nuclear translocation (Beinke and Ley, 2004). Finally, a third, NIKdependent route to IκB phosphorylation and p65 nuclear translocation was described for some receptors (Ramakrishnan et al., 2004).
There are mixed results implicating NF-κB in various models of apoptosis, with both pro- and antiapoptotic effects observed. Activating NF-κB in detached IEC with exogenous stimuli, for example using trefoil factor (Chen et al., 2000), or IL-1 can delay anoikis but whether NF-κB is activated directly by detachment has not been reported. Considering that IEC detachment is associated with reputed NF-κB-mediated cytokine gene expression, we hypothesized that NF-κB activation would be intrinsic to detachment. In the present study, we report that NF-κB signalling indeed occurs following detachment and that activation of this pathway delays apoptosis. Moreover, the pattern of activation is NIKdependent nuclear translocation of p65, resembling the new emerging pathway of NF-κB activation.
Results
Detaching IEC-18 results in caspase activation and apoptosis
As reported by others, the detachment-induced apoptotic ‘death signal’, the progressive activation of caspases (Alnemri et al., 1996; Salvesen and Dixit, 1997), was detected by Western blot using rabbit anti-active caspase-3 antisera (Figure 1a) or by using the substrate Z-Glu-Lys(biotinyl)-Asp-CH2-DMB (zEK(bio)Daomk) for detection of multiple activated caspases (Figure 1b). Increased caspase activation was positively associated with increased cell apoptosis judged by morphology within 4 h post detachment (Figure 1c) and negatively associated with the number of 3,3′ dihexyloxacarbocyanine iodide (DiOC6)-positive (viable) cells following an overnight incubation (Figure 1d). The DiOC6 measure indicates compromised mitochondria permeability (Zamzami et al., 1996) and has been used to follow apoptosis during anoikis (Douma et al., 2004). These multiple means of measuring apoptosis establish that we can detect significant apoptotic changes within 4 h of detachment.
Figure 1.
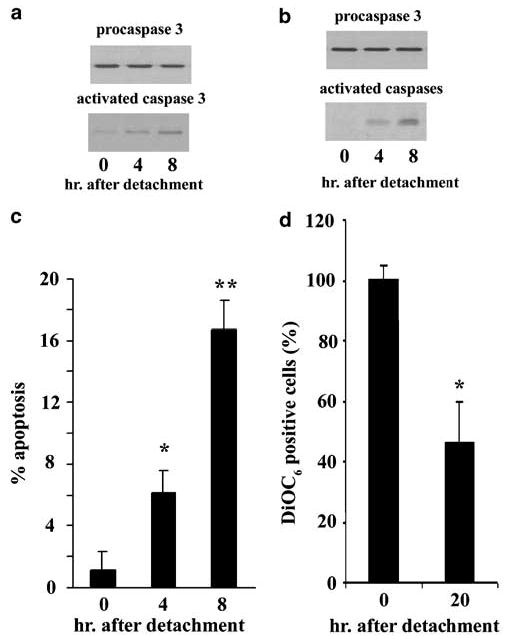
IEC-18 intestinal epithelial cells undergo apoptosis following detachment (anoikis). The activation of caspases correlates with increasing time as detached cells. IEC-18 were detached by trypsinization and reincubated in rotating polypropylene tubes for the times indicated and then lysed. Lysates (60 μg) were assayed by Western blotting for activated caspase 3 (a) or multiple active caspases detected with zEK(bio)D-aomk (b). In both cases, the blots were stripped and then probed with antibody to procaspase 3, demonstrating equal protein loading in each lane. The antibody used to detect activated caspase 3 does not detect procaspase 3. (c) Apoptosis is evident from cell morphology. Cells recovered at the times indicated were settled onto slides, stained and then assessed for apoptosis microscopically. A total of 500 cells were counted per slide and the mean and standard deviation were calculated from three slides, each prepared from a separate tube of cells. All experiments were conducted at least three times, each with similar results. (d) Apoptotic cells fail to stain with DiOC6. Declining DiOC6 staining indicates compromised integrity of the mitochondria. The figure includes the mean and standard error of the mean of five experiments. ANOVA was conducted with post hoc testing when there are two experimental groups compared to the control or time 0 treatment; otherwise, the Student’s t-test was used to compare the experimental group with the control group of cells. *P < 0.05; **P < 0.01 when compared to 0 h
Gene expression by detached IEC-18
To gain further insights into the IEC-18 response to the stress of detachment, we conducted gene microarray analysis using mRNA prepared from cells harvested in a similar time frame as the caspase assays, up to 8 h. The array results revealed that a considerable number of genes were expressed including well-known NF-κBmediated products (Supplementary Table 1). In the annotated list are cytokines/chemokines (e.g. TNF-β, fractalkine, RANTES, IL-15), inflammatory molecules (VCAM-1, iNOS, CD44), transcription factors (NF-κB, IRF-7, STAT1, 2), proteases (MMP-2) and apoptotic molecules (BimEL), most of which have been confirmed as made by epithelial cells under other stimuli. We tested seven of the NF-κB-regulated genes using reverse transcription–polymerase chain reaction (RT–PCR) detection and indeed all seven were increased during anoikis although the time-course kinetics and perhaps the magnitude of changes may not exactly match our predictions from the array results (Figure 2). We also repeated our earlier finding that IL-1β and IL-1RII are made (Waterhouse and Stadnyk, 1999) as well as the observation by Miller and McGee (2002) that IL-6 is made (Figure 2).
Figure 2.
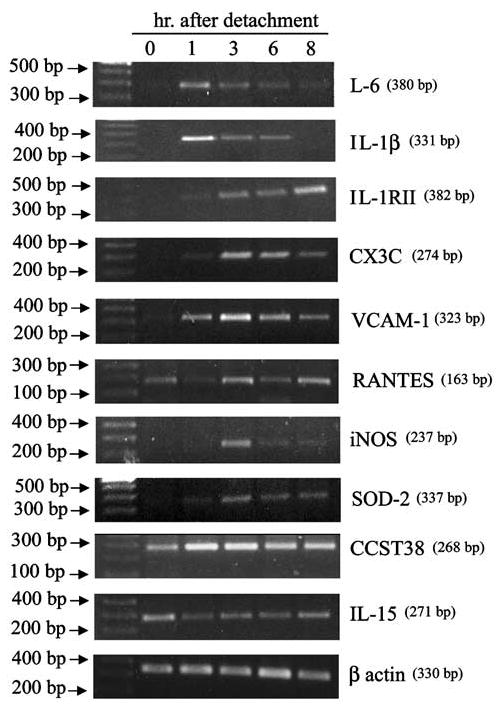
RT–PCR detection of mRNA for NF-κB-mediated genes following detachment. Cells were detached and established on poly-HEMA-coated plates for the length of time indicated on the gel and then harvested and total RNA isolated. The primer sequences used for detection of each molecule predicted to increase by microarray are available in Supplementary Table 2. The experiment was conducted twice using cells on poly-HEMA and once using cells in roller culture, and all the molecules increased in all the experiments
Detachment induces phosphorylation of IκBα and nuclear translocation of p65
Directly assaying for distal events in NF-κB activation, we observed that detachment induced the immediate phosphorylation of IκBα (Figure 3a). Levels of phospho- IκBα reached a peak by 10 min, at which time a decline in protein levels was also evident (Figure 3a). After incubation overnight as detached cells and with equal amounts of protein loaded into each lane of the gel, phospho-IκBα returned to levels seen in attached cells (Figure 3b). The decline in IκBα was temporary as it returned to attached cell levels by 1 h, presumably due to new protein synthesis. The detection of nuclear p65 protein was immediate but the peak concentration was reached between 30 min and 1 h (Figure 3b), similar to the course of IκBα phosphorylation. Levels then slowly declined, until it was no longer detectable by about 20 h (Figure 3b). Nuclear NF-κB activity was also detectable using an adenovirus construct expressing luciferase downstream of three NF-κB binding sites, shown in Figure 3c as increased luciferase at 4 h relative to immediately detached cells. Thus, NF-κB activation is rapid and transient during anoikis.
Figure 3.
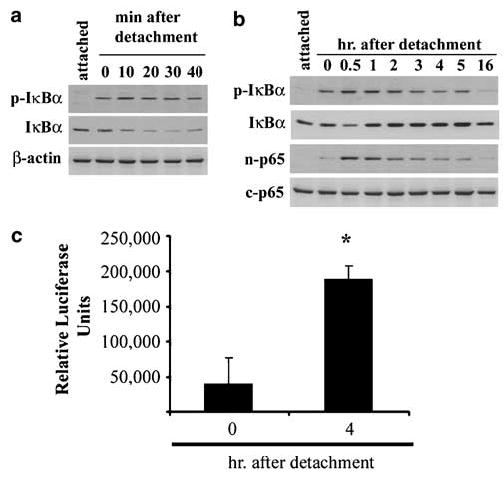
Detachment induces the activation of NF-κB. (a) Phosphorylated IκBα becomes detectable as a result of detachment and total protein levels decline over the first 30 min. p-IκB = phosphorylated-IκBα. (b) The protracted time course reveals that protein levels of IκBα recover by about 1 h following detachment. Secondly, increasing amounts of p65 protein accumulate in the nucleus coincidental with increased phosphorylated IκBα. n-p65, nuclear p65; c-p65, cytoplasmic p65. (c) IEC-18 were infected with adenovirus expressing NF-κB-promoted luciferase prior to detachment, then detached and harvested immediately or after 4 h, and luciferase activity was measured. *P < 0.01
Inhibition of NF-κB activation results in increased caspase activation and apoptosis
The data above support a model whereby the decay of nuclear NF-κB parallels cell death. We then sought to confirm directly whether specific inhibition of the canonical pathway would affect the timing of apoptosis in cells after detachment. Adenoviral infection of IEC- 18 with IκBAA, the mutated, super-repressor form of IκBα, prevented nuclear accumulation of p65 relative to detached green fluorescent protein (GFP)-expressing or uninfected cells at the same time point of 1 h (Figure 4a). Infection with the super-repressor also accelerated apoptosis measured by DiOC6 staining (Figure 4b) and enhanced activation of caspase 3 (Figure 4c). The functional inhibition of NF-κB activity was confirmed using coinfections with the IκBAA- and luciferase-expressing viruses, detected as a lack of increased luciferase in detached cells (Figure 4d). Therefore, the activation of NF-κB appears essential for the cells to survive the period immediately following detachment. As an alternative approach to inhibiting NF-κB, we treated cells with gliotoxin (0.5 μg/ml) or curcumin (50 μm), potent pharmacological inhibitors of NF-κB signalling (Pahl et al., 1996; Ward et al., 1999), during trypsinization and after detachment, and then measured nuclear p65 and caspase activation. Figure 5a shows that both chemicals attenuated IκBα phosphorylation and p65 nuclear accumulation with a concomitant enhancement of caspase activation. Detached cells undergo a dramatic widespread tyrosine dephosphorylation (Maher, 1993, and data not shown) and in the course of our investigations into the role of phosphatases in anoikis, we have determined that the tyrosine phosphatase inhibitor phenylarsine oxide (PAO) but not pervanadate inhibits detachment-induced NF-κB signalling. Figure 5a shows that cells incubated in 0.5 μm PAO during trypsinization and while detached failed to phosphorylate IκBα or translocate p65 to the nucleus but showed high levels of caspase activity. The acceleration of apoptosis by the pharmacological agents was also evident from the morphology of the cells harvested after 8 h of detachment (Figure 5b). We conclude from these results that intrinsic activation of distal components of the canonical pathway (p65) serves to directly delay apoptosis of the detached cells.
Figure 4.
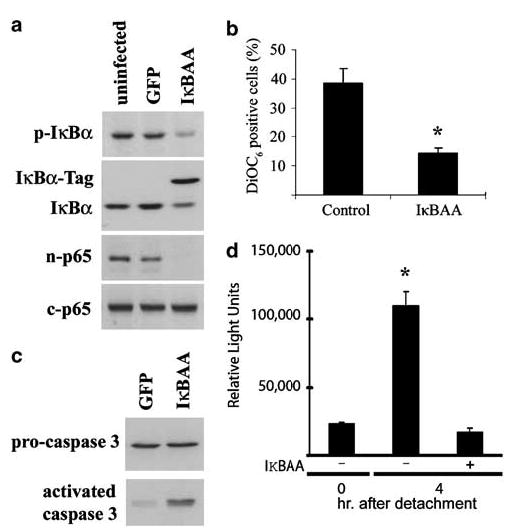
Infection of IEC-18 with adenovirus expressing a mutated form of IκBα (IκBAA) interrupts NF-κB activation in detached cells. (a) Infection with IκBAA but not virus expressing GFP blocks the nuclear accumulation of p65 in detached cells harvested after 1 h. (b) A greater percentage of IκBAA-infected IEC-18 are apoptotic, measured as DiOC6 staining at 16 h post detachment. (c) Greater caspase 3 is detected in IκBAA-expressing cells than control GFP-expressing virus-infected cells harvested at 4 h post detachment. (d) Infection with IκBAA prevents the detachment-induced expression of an NF-κB-driven luciferase reporter gene. *P < 0.05
Figure 5.
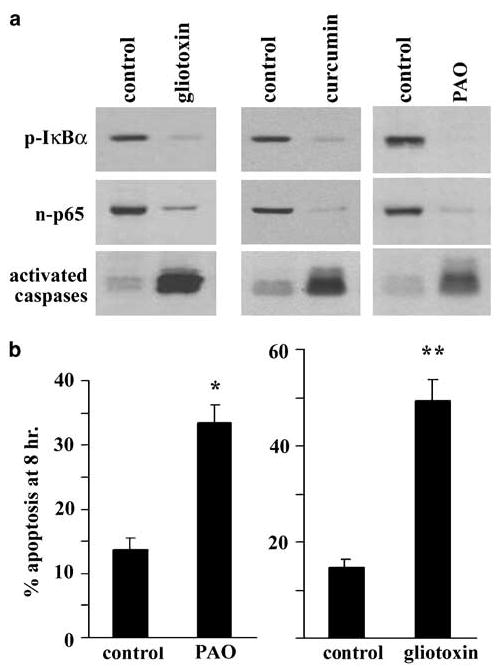
(a) Pharmacological inhibitors of NF-κB prevent phosphorylation of IκBα and the nuclear accumulation of p65 (assayed at 1 h) as well as accelerate the activation of caspases, detected with the pan activated caspase marker zEK (bio) D-aomk using 4 h lysates. Cells were incubated in each inhibitor during trypsinization and throughout the period of detachment until harvest. The blots were stripped and probed for actin, which showed similar levels of protein in each lane (not shown). (b) The proapoptotic effect of gliotoxin and PAO quantitated using the morphology of detached cells after 8 h. Gliotoxin was used at 0.5 μg/ml, curcumin at 50 μm and PAO at 0.5 μm. *P < 0.05, **P < 0.01
Considering the early NF-κB activation following detachment, we explored whether other signalling cascades, reputedly upstream of IκBα phosphorylation in other systems, may impact on the detachment-induced activation of NF-κB. We treated cells during trypsinization and throughout the period of detachment with various pharmacological inhibitors of protein kinases including the tyrosine kinase inhibitors tyrphostin AG126 (75 μm), genestein (75 μm) and herbimycin A (5 μm); inhibitors of phosphatidylinositol-3 kinase, LY294002 (1 μm) and wortmannin (0.1 μm); and the MAP kinase inhibitors SB203580 (5 μm) and PD98059 (50 μm), but none prevented the phosphorylation of IκBα or nuclear p65 accumulation measured 1 h after detachment (data not shown).
Detachment results in IKKα phosphorylation
The canonical pathway to Rel A activation involves the heterotrimeric complex IKK consisting of IKKα, IKKβ and IKKγ. IKKβ reputedly phosphorylates IκBα, releasing p65 (Beinke and Ley, 2004). Using Western blotting, we detected rapid IKKα but not IKKβ phosphorylation in the detached IEC-18 (Figure 6a). We then used our pharmacological approach to probe whether phosphorylated IKKα was functional in p65 nuclear accumulation. Cells incubated in gliotoxin showed intact IKKα phosphorylation following detachment, while cells incubated in PAO showed lower levels than detached control cell levels (Figure 6b). This indicated that gliotoxin likely acts directly to inhibit IKKα activity. Incubation in PAO, on the other hand, resulted in increased catabolism of the phospho-IKKα protein, which registered as a steady decline in total IKKα levels, presumably as more of the steady-state protein became phosphorylated (Figure 6c). The sum of these observations implicates IKKα in the p65 nuclear translocation.
Figure 6.
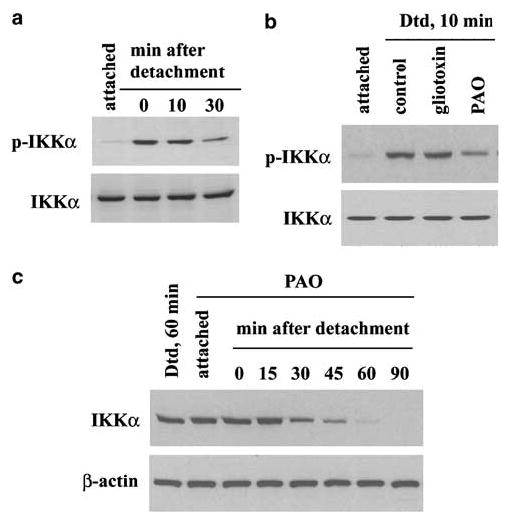
IKKα becomes phosphorylated as a function of detachment. (a) IKK rapidly becomes phosphorylated following detachment, without a discernable change in total IKK protein. p- IKK: phosphorylated IKK. (b) Incubation of cells in PAO (0.5 μm) but not gliotoxin (0.5 μg/ml) prior to detachment results in less phosphorylated IKKα after detachment. (c) Preincubation of cells in PAO results in a rapid and specific decline of total IKKα protein in detached cells. The attached control cells were harvested after 60 min incubation in PAO. Dtd: detached
Finally, considering the antiapoptotic effect of intrinsic NF-κB activation early after detachment and the antiapoptotic effect of the NF-κB-activating cytokine IL-1 (Waterhouse et al., 2001), we explored whether enhancing NF-κB activation, specifically, was sufficient for cells to overcome anoikis. We chose to infect cells prior to detachment with adenovirus expressing native NIK since this molecule signals through IKKα in the alternative pathway yet reportedly can activate the canonical NF-κB pathway resulting in nuclear p65 (see Ramakrishnan et al., 2004). We then measured parameters of detachment-induced apoptosis. Adherent IEC-18 ectopically overexpressing NIK did not show higher constitutive NF-κB activation as measured by coinfection with NF-κB-mediated luciferase-expressing virus (data not shown). On the other hand, cells infected for up to 2 days with virus expressing NIK and then detached possessed greater amounts of phosphorylated IκBα at 1 h (Figure 7a) and 24 h post detachment (not shown), increased levels of nuclear p65 (Figure 7b) and lower levels of activated caspase 3 at 24 h (Figures 7c), confirming that NIK signalling is linked to the activation of p65 by detachment. However, despite an apparent increase in DiOC6 staining, the increase in numbers of cells surviving at 24 h post detachment did not reach statistical significance compared to cells infected with virus expressing GFP (Figure 7d). The decline in surviving numbers of cells expressing the IκB super-repressor did reach statistical significance, also shown in Figure 4. Therefore, while inhibiting NF-κB accelerated apoptosis, heightening the activation of NFκB alone, at least to the magnitude achieved by overexpression of NIK, failed to prolong significantly the survival of detached cells.
Figure 7.
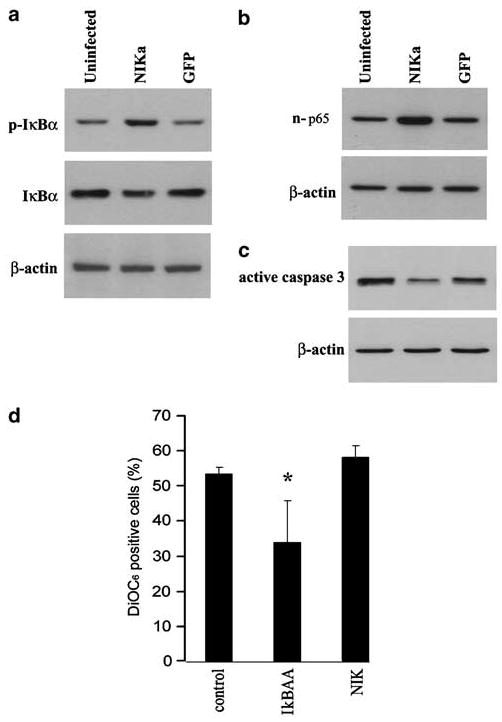
Enhanced NF-κB activation fails to increase the numbers of IEC-18 cells surviving detached. (a) Infection of cells with adenovirus overexpressing native NIK protein for 24 h prior to detachment results in greater levels of phospho-IκBα than detached cells harvested at 1 h. NIK-overexpressing cells show greater levels of nuclear NF-κB (b) and less activation of caspase 3 (c) than uninfected cells or cells infected with virus expressing GFP and then detached for 24 h. (d) NIK-overexpressing detached cells do not survive in greater numbers than detached uninfected cells, measured as DiOC6 staining after 24 h. The number of cells surviving expressing IκBAA, included for comparison, was significantly reduced, *P < 0.05
Discussion
Epithelial cells progress from the dividing stage deep in the crypt to rise to the villus tip in the small intestine or the surface of the crypt in the colon, where they undergo apoptosis. Inflammation can change the dynamics of this relationship including influencing whether the cells continue into apoptosis (Shanmugathasan and Jothy, 2000). Indeed, carcinomas arise from the chronically inflamed bowel with greater frequency than they do from the uninflamed bowel (Gillen et al., 1994; Karlén et al., 1999), indicating mechanisms occur that may circumvent the normal apoptotic routine. We have therefore been studying the relationship between inflammatory mediator production and apoptosis of IEC, using the IEC-18 line as a model of undifferentiated crypt cells. We have shown that IL-1β is transiently produced by detached IEC-18 (Waterhouse and Stadnyk, 1999) and others have reported that IL-6 is expressed (Miller and McGee, 2002). Now, our gene array results expand the list of detachment-induced genes to include other molecules important in inflammation as well as transcription factors and regulators of apoptosis, many of which are transcriptionally activated by NF-κB. Here we confirm that detachment directly activates NF-κB and that this activation impacts on the early course of anoikis. NF-κB rapidly became activated in the period when the cells were detaching, as was evident from the phosphorylated IκBα at the earliest time we harvested cell lysates, time 0. This was not due to trypsinization per se, since the activation also occurred during scraping of the cells (data not shown). We then show, using genetic and pharmacological approaches, that interrupting NF-κB activation accelerated apoptosis of the detached cells, directly implicating this transcription factor in the regulation of cell fate. Together, these data support the conclusion that NF-κB plays an essential role in IEC survival early under the stress of detachment.
NF-κB activation has been described in multiple systems but whether it protects cells or promotes apoptosis appears to depend on the cell type and method used to induce apoptosis (Chen et al., 2000; Gill and Windebank, 2000; Kuhnel et al., 2000; Yang et al., 2001; reviewed in Weaver et al., 2002). This varied response is undoubtedly related to the fact that NF-κB promotes the expression of both antiapoptotic and proapoptotic molecules. NF-κB activation also reportedly occurs during cell reattachment and this has been exploited as a target to prevent the survival of metastatic cells (Scaife et al., 2002).
The canonical pathway leading to nuclear p65 utilizes IKKβ (Beinke and Ley, 2004), yet we detected early phosphorylation of IKKα, which would be consistent with activation of the alternative pathway. However, NF-κB signalling consisting of proximal alternative pathway events (NIK activation of IKKα) engaging the distal events of the canonical pathway (phosphorylation of IκBα and nuclear translocation of p65) seems to be an emerging hybrid of these two pathways. This pattern of activation was recently highlighted by Ramakrishnan et al. (2004), who proposed that a NIK-dependent arm of the canonical pathway is triggered by certain receptors. Jijon et al. (2004) reported that NIK-dependent p65-mediated gene expression was dependent on p38 MAPK phosphorylation of p65. We also show that overexpression of NIK in the IEC-18 cells resulted in greater nuclear p65 in detached cells, and while p38 is activated by detachment (Vachon et al., 2001; Rosen et al., 2002, and results not shown), NF-κB activation was not prevented by the p38 inhibitor SB203580. Furthermore, the increase did not result in a significantly greater number of cells surviving to 24 h post detachment. This outcome contrasts with our previous results and the results of others showing that stimulating NF-κB activity in detached cells is antiapoptotic (Chen et al., 2000; Waterhouse et al., 2001). This may be due to the magnitude of activation or the exogenous NF-κB triggers possibly activating other signalling pathways that complement the effect of NF-κB acting through the canonical pathway.
Knowing that NF-κB is activated by detachment is important because it is purportedly the axis between chronic inflammation and tumorigenesis (Clevers, 2004; van der Woude et al., 2004), although this is widely appreciated to be through indirect roles such as enhancing iNOS or COX-2 expression. Activated NF-κB can be found at the invasive margin of colorectal tumors in situ (Evertsson and Sun, 2002) and inhibition of NF-κB in transformed cells is one strategy taken to promote apoptosis, for example in breast carcinomas (Biswas et al., 2003). Yet, we believe our finding of intrinsic detachment-induced NF-κB implicates this factor directly in epithelial apoptosis. Early intrinsic activation may serve in a number of cellular functions as part of the normal physiology of the IEC. One role may be during mitosis, when cells round up and dissociate focal adhesions from the substrate, presumably becoming temporarily detached. During mitosis, focal adhesion kinase (FAK) becomes tyrosine dephosphorylated but serine phosphorylated (Ma et al., 2001), resulting in the decoupling of FAK from downstream signalling molecules. FAK normally provides survival signals through the upregulation of Bcl-2 homologs, and fibroblasts transfected with a constitutively active FAK become anoikis resistant (Frisch et al., 1996). FAK regulation may crosstalk with NF-κB activation to support survival through mitosis, and in other cell systems, FAK indeed leads to AKT activation and AKT in turn activates NF-κB. However, our data using LY294002 and wortmannin would suggest that this is not the case in detachment-induced NF-κB activation.
A second function for intrinsic activation by detachment is to launch the inflammatory response. The large number of NF-κB-mediated genes that are important in the inflammatory/immune response (Figure 2 and Supplementary Table 1) is a testament to this possible role for a cell that resides at the boundary with the environment. Presumably, detachment-induced activation of NF-κB may not be a response of all epithelia but only of those directly interfacing with the external environment such as the lung, gut and genitourinary tract. This programming of the inflammatory response happens to be compatible with a role for ceramide in activating NF-κB. Ceramide liberation by neutral sphingomyelinases induced RelA/p50 activation and IL-8 expression in a human colon carcinoma line (Colell et al., 2002). Whether detachment leads to increased ceramide is presently under investigation. The shortterm survival of IEC would ensure the induction of inflammatory mediators, thus alerting the host of damage or stressors to the epithelium.
Beyond these hypotheses, the mechanism leading to activation of NF-κB by detachment remains unknown. The immediacy of the activation suggests that endogenous signalling molecules are responsible and not soluble molecules acting on surface receptors. Our finding of NIK-dependent activation of p65 may help to focus the search for more proximal activators. Ultimately, mapping this pathway will identify potential controlling points over the fate of IEC that may be used to promote restitution or possibly reverse anoikis resistance in the precancerous stages of colorectal cancer.
Materials and methods
Materials
The IEC-18 cell line and mycoplasma PCR-based detection kit were purchased from American Type Culture Collection (Rockville, MD, USA). Cell culture polystyrene flasks and multiwell plates were from Becton-Dickenson (Lincoln, NJ, USA). DMEM, HEPES, l-glutamine, newborn cow serum (NBCS), penicillin, streptomycin and bovine insulin were purchased from Life Technologies (Burlington, ON, Canada). Nitrocellulose membrane and ECL Western blot detecting reagents were purchased from Amersham Pharmacia Biotech Inc. (Baie d’Urfe, QC, Canada). Rabbit anti-phosphorylated IKK, anti-activated caspase 3 and anti-IκBα antisera, and mouse anti-phosphorylated IκBα monoclonal antibodies (mAb) were from Cell Signaling Technology (Beverly, MA, USA). The anti-phosphorylated IKK antisera detects both IKKα and IKKβ. As only IKKα becomes phosphorylated as a result of detachment, we confirmed the detection of phospho- IKKβ using IL-1-treated cells. The biotinylated caspase inhibitor zEK(bio)D-aomk was from BioChem ImmunoSystems (Montreal, QC, Canada). Rabbit anti-caspase 3, IKKα and RelA (p65) antisera were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The protein assay kit (Bradford method) and other reagents for electrophoresis were from BioRad Laboratories Ltd (Mississauga, ON, Canada). HRP-conjugated goat anti-mouse IgG, anti-rabbit IgG and rabbit anti-β-actin sera and all the remaining reagents were obtained from Sigma-Aldrich Canada Ltd (Oakville, ON, Canada).
Cell culture
The IEC-18 cell line was maintained in DMEM supplemented with 10 mm HEPES, 2 mm l-glutamine, 5% heat-inactivated NBCS, 50 U/ml penicillin and 50 μg/ml streptomycin (hereafter referred to as complete DMEM) at 37°C supplemented with 5% CO2. The passage and periodical Mycoplasma testing were carried out as described earlier (Stadnyk et al., 1995; Waterhouse and Stadnyk, 1999).
Experiments were performed 1 week post passage on confluent cells in six- or 12-well polystyrene plates. For experimental treatments, media were removed from wells and the cells washed once with PBS. Adherent control cells were collected in lysis buffer (see below) at this point. Cell detachment was achieved by treatment with 0.25% trypsin/1 mm EDTA (in PBS) for 10 min at 37°C followed by gentle pipetting, until cells detached. Other methodologies for detaching the cells, that is, using 5 mm EDTA alone, or cell scrapers, had also been tested, with similar results (data not shown). The detached cells were collected, washed once with PBS and resuspended in complete DMEM (considered as time ‘0’). Cells from each well of the culture plate were incubated in a polypropylene tube (5 × 105/ml) rotating at 37°C, for the times indicated. In some experiments, media were supplemented with various chemical inhibitors during trypsinization and during the following incubation. All experiments were conducted at least three times.
Microarray analyses
Detached IEC were plated at a density of 5 × 105 cells/ml on poly-HEMA-coated 35 mm plates in duplicate for 0, 2, 4 or 8 h, when total RNA was extracted using RNeasy (Qiagen). For each of the eight resulting samples, cRNA probes were generated by reverse transcription followed by in vitro transcription incorporating biotin labelling as part of the standard Affymetrix protocol. The probes were then hybridized to the Affymetrix Rat Genome U34 A chips, which interrogate 8740 mRNA transcripts and EST clusters from the UniGene database (Build 34). After hybridization and staining, the chips were scanned by laser. The final data set consisted of a total of eight scan files, each obtained using the Affymetrix GeneChip® software. Each qualifier in these files is associated with an intensity that is a measure of the corresponding transcript abundance. The output files were further processed into a format that adds an estimate of the standard deviation of the noise for each intensity (Theilhaber et al., 2001). The eight postprocessed scan files, arranged in order of the anoikis time course, were concatenated into a single file, with duplicates forming adjacent columns. Replicates were combined by computing the median of the replicate intensities for each qualifier. The final step in the data assembly consisted of obtaining expression ratios for each qualifier for 2, 4 and 8 h compared to 0 h using the PFOLD algorithm, which utilizes both intensity and noise data. The number of profiles in the IEC anoikis time course was reduced by retaining only those with the most significant variation. The data here represent the top 30 upregulated genes following 2, 4 and 8 h of detachment compared to time 0, all of which show greater than threefold increases in concentration.
RNA extraction and relative RT–PCR
Total cellular RNA was extracted from cells harvested at the indicated times using Trizol reagent (Life Technologies) following the manufacturer’s instructions. RT and PCR for the rat molecules were performed as described previously (Waterhouse and Stadnyk, 1999). Briefly, 1 μg of total cellular RNA from each sample was reverse transcribed using Maloney murine leukemia virus reverse transcriptase (Invitrogen) with 0.01 mm of each dNTP and 1 μg random hexamers (both from Pharmacia). The RT product was diluted 1 : 10 and 4 μl used for measurement of β-actin; or else an equal volume was used as template for all other molecule determinations. The PCR mix contained (in final concentrations) 50 mm KCl, 20 mm Tris-HCl, pH 8.4, 2.5 mm MgCl2, 0.1 μg/ml bovine serum albumin, 0.2 mm dNTPs and 2.5 pmol of each primer. Following agarose gel separation, the amplicon was photographed using ethidium bromide staining under UV light. The primer sequences developed for the detection of these rat molecules are listed in Supplementary Table 2.
Physical assessment of apoptosis
After the timed incubation, aliquots of 100 000 detached cells were removed from the culture and washed once with PBS. These cells were then resuspended in 0.1 ml of PBS and placed onto a polylysine-coated glass slide. Cells were left to adhere for 20 min at room temperature and then fixed with 2% paraformaldehyde and stained with H&E. Cells with karyopyknosis or karyolysis were considered as apoptotic cells. A total of 500 cells were counted from each treatment group and the mean of three or four experiments and standard deviation are shown.
Western blotting
Cell lysates, prepared by dissolving the cells with RIPA buffer or as described in Results, were assayed for protein concentrations using Bio-Rad Bradford protein assay reagents and then analysed by Western blotting, as described in detail elsewhere (Bonner et al., 2001).
Assay for caspase and IκBα activation
Caspase activation was assayed using a biotin-conjugated caspase-specific inhibitor zEK(bio)D-aomk according to the method of Yamashita et al. (1999), with some modifications. Cells were washed once with ice-cold PBS and then lysed with the caspase assay buffer (CAB: 1% Triton X-100, 20 mm HEPES, pH 7.5, 5 mm MgCl2, 5 mm EGTA, 5 mm EDTA, 5 mm DTT, 2 mm PMSF, 5 μg/ml leupeptin, 5 μg/ml pepstatin, 10 μg/ml aprotinin) (Niwa et al., 1999). Lysates were clarified by centrifugation at 12 000 g for 10 min and assayed for protein concentrations. Lysates (100 μg/sample) were incubated with 2 μm zEK (bio) D-aomk in CAB for 10 min at 37°C, followed by termination of the reactions by adding 5 volumes of cold acetone (−20°C). The proteins were then pelleted by centrifugation at 12 000 g for 10 min, dissolved in SDS–PAGE sample buffer, separated on 15% gels by SDS–PAGE and transferred to nitrocellulose membranes. The activated caspases that bound covalently with zEK (bio) D-aomk were detected by HRP-conjugated streptavidin followed by ECL Western blot detection reagents.
For assessment of IκBα activation, cells were lysed with M2 buffer (20 mm Tris, pH 7.0, 0.5% NP-40, 250 mm NaCl, 3 mm EGTA, 3 mm EDTA, 2 mm DTT, 5 mm PMSF, 5 μg/ml leupeptin, 5 μg/ml pepstatin, 10 μg/ml aprotinin, 0.2 mm Na3VO4, 2.5 mm NaF, 10 μm PAO) (Lin et al., 2000), and then equal amounts of protein (35 μg/sample) analysed by Western blotting using mouse anti-phosphorylated IκBα or a rabbit anti-IκBα protein Ab as described above.
Assay for p65 nuclear accumulation
These experimental procedures were essentially as conducted by Ward et al. (1999), with some modifications. Equal numbers of cells (106/sample) were washed once with ice-cold PBS/DFP and then treated with Buffer A (10 mm Tris, pH 7.8, 10 mm KCl, 1.5 mm EDTA, 0.5 mm DTT, 1 mm PMSF, 0.1 mm Na3VO4, 5μg/ml leupeptin, 5 μg/ml pepstatin, 10 μg/ml aprotinin, 2.5 mm NaF, 10 μm PAO) for 10 min on ice. Then, 0.1 volume of 10% NP-40 was added and the cells vortex mixed for 4 s, followed by centrifugation at 12 000 g for 2 min. The supernatants were aspirated and the pellets washed once with Buffer A. The nuclei were collected by centrifugation as above and the nuclear proteins extracted with Buffer B (20 mm Tris, pH 7.8, 150 mm NaCl, 50 mm KCl, 1.5 mm EDTA, 5 mm DTT and protease and phosphatase inhibitors as in Buffer A) for 1 h at 4°C. The residues of the nuclei were removed by centrifugation at 12 000 g for 10 min at 4°C and the nuclear extracts were analysed by Western blot using a rabbit anti-p65 Ab as described above.
Adenovirus constructs, infection and luciferase assay
Replication-defective adenovirus lacking segments of the E1 and E3 regions were constructed to encode a CMV-driven, mutated IκBα resistant to degradation (Ad5IκBAA), as described previously (Jobin et al., 1998b). Ad5 expressing GFP was used as an infection control, while virus expressing NIK (Ad5NIK) was used to enhance the levels of activated NF-κB (Russo et al., 2004). NF-κB-luciferase virus (Ad5κb- Luc) was used to assess NF-κB-mediated gene expression, and was described previously (Russo et al., 2004). Viruses were replicated by infection of E1-transformed HEK 293 cells and purified from cell lysate as described elsewhere (Bett et al., 1994). Titers were estimated by OD260 absorbance and viral stocks were stored at −20°C in storage buffer (5 mm Tris pH 8.0, 50 mm NaCl, 500 μm MgCl2, 25% glycerol).
In preparation for infection, IEC-18 were grown to near confluence and infected overnight with recombinant adenovirus at a multiplicity of infection (MOI) of ~45PFU per IEC in DMEM supplemented with 2% NBCS and penicillin, streptomycin, l-glutamine and HEPES. Cells were rinsed in PBS and were either trypsinized or incubated with fresh media. Infected cell viability was monitored by DiOC6 staining, detected by flow cytometry. Adenovirus-infected adherent cells showed a maximum of 4% apoptosis or death by any measure.
In experiments measuring luciferase activity, IEC were infected as above with an MOI of 20 overnight. Cells were then washed and as described, or were subsequently infected with Ad5IκBAA to inhibit NF-κB-mediated gene expression. Equal cell numbers were collected in luciferase assay lysis buffer and stored at −20°C for further analysis (Enhanced Luciferase Assay Kit, BD Biosciences, Franklin Lakes, NJ, USA). Samples were analysed as per the manufacturer’s instructions.
Flow cytometry
Adherent or detached cells, at a density of 5 × 105 cells/ml, were incubated with 40 nm DiOC6 (Molecular Probes, Eugene, OR, USA) in complete media for 10 min at 37°C. Aliquots of 0.5–1.0 × 106 cells were prepared for analysis using a Becton Dickenson FACScan and 1 × 104 events were analysed for each sample. Gates were set presuming high viability of freshly detached untreated cells using forward and side light scatter properties as well as DiOC6 labelling in the FL-1 channel. Gated freshly detached but otherwise untreated cells served as 100% viable controls for experimental comparison. Furthermore, adherent adenovirus-infected cells served as viability controls for detached infected cells. Flow cytometry data was analysed using Winlist 5.0 (Verity House, Topsham, ME, USA).
Acknowledgments
This research was supported by a grant from the Natural Sciences and Engineering Research Council of Canada (NSERC). SR Yan is supported by a Research Associateship from the IWK Health Centre and RR Joseph is supported by a scholarship from NSERC. We thank Hana James and Karen Conrod for their technical help.
Footnotes
Supplementary Information accompanies the paper on Oncogene website (http://www.nature.com/onc)
References
- Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- Bader T, Nettesheim P. J Immunol. 1996;157:3089–3096. [PubMed] [Google Scholar]
- Baeuerle PA, Baltimore D. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Beinke S, Ley SC. Biochem J. 2004;382:393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett A, Haddara W, Prevec L, Graham FL. Proc Natl Acad Sci USA. 1994;91:8802–8806. doi: 10.1073/pnas.91.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas DK, Martin KJ, McAlister C, Cruz AP, Graner E, Dai S-C, Pardee AB. Cancer Res. 2003;63:290–295. [PubMed] [Google Scholar]
- Bonner S, Yan SR, Byers DM, Bortolussi R. Infect Immun. 2001;69:3143–3149. doi: 10.1128/IAI.69.5.3143-3149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-H, Lu Y, De Plaen IG, Wang L-Y, Tan X-D. Biochem Biophys Res Commun. 2000;274:576–582. doi: 10.1006/bbrc.2000.3176. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Parent L, Maniatis T. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Clevers H. Cell. 2004;118:671–674. doi: 10.1016/j.cell.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Colell A, Coll O, Mari M, Fernández-Checa JC, Garcia-Ruiz C. FEBS Lett. 2002;526:15–20. doi: 10.1016/s0014-5793(02)03106-x. [DOI] [PubMed] [Google Scholar]
- Douma S, van Laar T, Zevenhoven J, Meuwissen R, van Garderen E, Peeper DS. Nature. 2004;430:1034–1040. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- Elewaut D, DiDonato JA, Kim JM, Truong F, Eckmann L, Kagnoff MF. J Immunol. 1999;163:1457–1466. [PubMed] [Google Scholar]
- Evertsson S, Sun X-F. Int J Mol Med. 2002;10:547–550. [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui P-Y. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill JS, Windebank AJ. Neurobiol Dis. 2000;7:448–461. doi: 10.1006/nbdi.2000.0312. [DOI] [PubMed] [Google Scholar]
- Gillen CD, Walmsley RS, Prior P, Andrews HA, Allan RN. Gut. 1994;35:1590–1592. doi: 10.1136/gut.35.11.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M. Cell. 1996;84:813–815. doi: 10.1016/s0092-8674(00)81058-2. [DOI] [PubMed] [Google Scholar]
- Jijon H, Allard B, Jobin C. Cell Signal. 2004;16:1023–1032. doi: 10.1016/j.cellsig.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Jobin C, Haskill S, Mayer L, Panja A, Sartor RB. J Immunol. 1997;158:226–234. [PubMed] [Google Scholar]
- Jobin C, Hellerbrand C, Licato LL, Brenner DA, Sartor RB. Gut. 1998a;42:779–787. doi: 10.1136/gut.42.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobin C, Panja A, Hellerbrand C, Iimuro Y, Didonato J, Brenner DA, Sartor RB. J Immunol. 1998b;160:410–418. [PubMed] [Google Scholar]
- Karlén P, Löfberg R, Broström O, Leijonmarck CE, Hellers G, Persson PG. Am J Gastroenterol. 1999;94:1047–1052. doi: 10.1111/j.1572-0241.1999.01012.x. [DOI] [PubMed] [Google Scholar]
- Kuhnel F, Zender L, Paul Y, Tietze MK, Trautwein C, Manns M, Kubicka S. J Biol Chem. 2000;12:6421–6427. doi: 10.1074/jbc.275.9.6421. [DOI] [PubMed] [Google Scholar]
- Lin Y, Devin A, Cook A, Keane MM, Kelliher M, Lipkowitz S, Liu ZG. Mol Cell Biol. 2000;20:6638–6645. doi: 10.1128/mcb.20.18.6638-6645.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Richardson A, Schaefer EM, Parsons JT. Mol Biol Cell. 2001;12:1–12. doi: 10.1091/mbc.12.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher PA. Proc Natl Acad Sci USA. 1993;90:11177–11181. doi: 10.1073/pnas.90.23.11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TA, McGee DW. Immunology. 2002;105:101–110. doi: 10.1046/j.0019-2805.2001.01352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa M, Hara A, Kanamori Y, Matsuno H, Kozawa O, Yoshimi N, Mori H, Uematsu T. Eur J Pharmacol. 1999;371:59–67. doi: 10.1016/s0014-2999(99)00145-4. [DOI] [PubMed] [Google Scholar]
- Pahl HL, Krauss B, Schulze-Osthoff K, Decker T, Traenckner EB, Vogt M, Myers C, Parks T, Warring P, Muhlbacher A, Czernilofsky AP. J Exp Med. 1996;183:1829–1840. doi: 10.1084/jem.183.4.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan P, Wang W, Wallach D. Immunity. 2004;21:477–489. doi: 10.1016/j.immuni.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Rosen K, Shi W, Calabretta B, Filmus J. J Biol Chem. 2002;277:46123–46130. doi: 10.1074/jbc.M207883200. [DOI] [PubMed] [Google Scholar]
- Russo MP, Schwabe RF, Sartor RB, Jobin C. Cell Signal. 2004;16:741–750. doi: 10.1016/j.cellsig.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Dixit VM. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- Savkovic SD, Koutsouris A, Hecht G. Am J Physiol Cell Physiol. 1997;273:C1160–C1167. doi: 10.1152/ajpcell.1997.273.4.C1160. [DOI] [PubMed] [Google Scholar]
- Scaife CL, Kuang J, Wills JC, Trowbridge DB, Gray P, Manning BM, Eichwald EJ, Daynes RA, Kuwada SK. Cancer Res. 2002;62:6870–6878. [PubMed] [Google Scholar]
- Shanmugathasan M, Jothy S. Pathol Int. 2000;50:273–279. doi: 10.1046/j.1440-1827.2000.01047.x. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Nakamura H, Kato S, Tomoike H. J Immunol. 1996;156:772–777. [PubMed] [Google Scholar]
- Stadnyk AW, Sisson GR, Waterhouse CCM. Exp Cell Res. 1995;220:298–303. doi: 10.1006/excr.1995.1319. [DOI] [PubMed] [Google Scholar]
- Sträter J, Wedding U, Barth TFE, Koretz K, Elsing C, Möller P. Gastroenterology. 1996;110:1776–1784. doi: 10.1053/gast.1996.v110.pm8964403. [DOI] [PubMed] [Google Scholar]
- Theilhaber J, Bushnell S, Jackson A, Fuchs R. J Comput Biol. 2001;8:585–614. doi: 10.1089/106652701753307502. [DOI] [PubMed] [Google Scholar]
- Vachon PH, Harnois C, Grenier A, Dufour G, Bouchard V, Han J, Landry J, Beaulieu J-F, Vézina A, Dydensborg AB, Gauthier R, Côté A, Drolet J-F, Lareau F. Gastroenterology. 2001;123:1980–1991. doi: 10.1053/gast.2002.37072. [DOI] [PubMed] [Google Scholar]
- van der Woude CJ, Kleibeuker JH, Jansen PLM, Moshage H. Apoptosis. 2004;9:123–130. doi: 10.1023/B:APPT.0000018794.26438.22. [DOI] [PubMed] [Google Scholar]
- Ward C, Chilvers ER, Lawson MF, Pryde JG, Fujihara S, Farrow SN, Haslett C, Rossi AG. J Biol Chem. 1999:4309–4318. doi: 10.1074/jbc.274.7.4309. [DOI] [PubMed] [Google Scholar]
- Waterhouse CCM, Joseph RR, Stadnyk AW. Exp Cell Res. 2001;269:109–116. doi: 10.1006/excr.2001.5303. [DOI] [PubMed] [Google Scholar]
- Waterhouse CCM, Stadnyk AW. Cell Immunol. 1999;193:1–8. doi: 10.1006/cimm.1999.1468. [DOI] [PubMed] [Google Scholar]
- Weaver VM, Lelièvre S, Lakins JN, Chrenek MA, Jones JCR, Giancotti F, Werb Z, Bissell MJ. Cancer Cell. 2002;2:205–216. doi: 10.1016/s1535-6108(02)00125-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita K, Takahashi A, Kobayashi S, Hirata H, Mesner PW, Kaufmann SH, Yonehara S, Yamamoto K, Uchiyama T, Sasada M. Blood. 1999;93:674–685. [PubMed] [Google Scholar]
- Yang CH, Murti A, Pfeffer SR, Kim JG, Donner DB, Pfeffer LM. J Biol Chem. 2001;276:13756– 13761. doi: 10.1074/jbc.M011006200. [DOI] [PubMed] [Google Scholar]
- Zamzami N, Susin SA, Marchetti P, Hirsch T, Gómez- Monterrey I, Castedo M, Kroemer G. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]