

Each day respiration moves 15,000 liters of air containing hundreds of microorganisms over the 150-m2 respiratory epithelial surface. Most inhaled microbes are trapped on the mucus layer coating the nasal epithelium and upper respiratory tract and are transported by ciliary motion to the pharynx, where they are swallowed. Organisms that reach the alveolar compartment are deposited on the alveolar lining layer (ALL), a thin aqueous film containing pulmonary surfactant that lines the gas-exchanging surface of the pulmonary epithelium. After “touchdown,” complex offensive and defensive strategies are initiated by both the invader and the host. Sensing of the physiologic body temperature and the pH and ionic strength of the ALL by the organism triggers a program of gene expression designed to optimize survival under adverse conditions. These include upregulation of microbial genes required for proliferation and host evasion and downregulation of genes that regulate less necessary functions.
The lung response to this threat is coordinated by the pulmonary epithelium and alveolar macrophages, which release cytokines and chemokines to recruit additional inflammatory cells to the airspace. However, a single organism capable of dividing every 20 minutes will give rise to a population of millions within hours, so control of proliferation must begin well before reinforcements arrive. For the naive host, the primary antimicrobial defenses in the ALL are the resident alveolar macrophages and protein components of the innate immune system, including the cell wall–degrading enzyme lysozyme, the iron-chelating protein lactoferrin, and specific membrane-permeabilizing members of the defensin, cathelicidin, and pentraxin families. Macrophages are activated by innate immune receptors such as CD14, which detect LPS, peptidoglycan, and other molecules displayed on the surface of microorganisms. Phagocytosis is triggered by pattern recognition receptors on the surface of the phagocyte, including the mannose receptor and the macrophage scavenger receptor, which identify signature carbohydrate structures on microbial surfaces. If the microbe has been previously encountered, recognition may also proceed through opsonization by specific antibody and Fc receptor–dependent uptake. These events result in extracellular microbial incapacitation and internalization into activated phagolysosomes, which kill the organism through exposure to lethal reactive oxygen species and lytic enzymes.
Lung-specific requirements for host defense
Similar mechanisms of microbial clearance are encountered at the other major environmental interfaces, including the gastrointestinal tract and the skin, but the alveolar boundary is clearly the most vulnerable interface. For several reasons, host defense in the lung is exceptionally, if not uniquely, challenging. First, the surface area to be defended is enormous, about 60 times greater than the skin (2.5 m2). Although the gut is roughly equivalent in size (200 m2), it is connected in series to the environment, which allows for deployment of sequential defenses, such as salivary amylase, gastric acid, and bile. In contrast, the alveoli are exposed to the environment in parallel, and each unit must be self-sufficient in the defense against inhaled threats. Second, in the alveoli, the objective is sterility rather than maintenance of a normal flora. Third, the pool of resident inflammatory leukocytes available in the airspace for immediate control of infection is relatively small. Fourth, physical barriers or harsh chemical environments (such as are found in the skin, with its cornified epithelial layers, and in the gut, with its regular infusions of antiseptic detergents in the form of bile) cannot be tolerated in the delicate alveolar space. Fifth, the risk of dissemination is greater in the lung than at any other environmental boundary, since only two cell layers (the alveolar epithelium and the capillary endothelium) and small amounts of interstitial tissue separate the invader from the bloodstream. Finally, even moderate degrees of inflammation and exudation threaten the primary function of the organ.
From the perspective of the host, an optimal defensive strategy would include preemptive control of microbial proliferation and immediate clearance, preferably within a few cell divisions. The innate immune cellular and molecular constituents of the alveolar lining fluid must do more than rapidly kill and clear pathogens; they must also execute a finely tuned inflammatory response, one that is sufficient to contain infection without inducing harmful degrees of alveolar exudation and cellular infiltration.
Surfactant proteins and pulmonary host defense
The elaborate mechanisms that have developed to meet these stringent lung-specific requirements are not completely understood, but components of the surfactant lining layer have recently received increasing attention as primary immunomodulators in the alveolar space. By weight, surfactant is composed of 90% phospholipids and 10% protein, including the hydrophilic surfactant proteins A (SP-A) and D (SP-D), and the hydrophobic surfactant proteins B (SP-B) and C (SP-C) (1). After secretion by alveolar type II cells and nonciliated bronchiolar cells into the ALL, the components of surfactant form phospholipid enriched membranes at the air-liquid interface, which spread readily and resist compression during cyclical respiratory expansion and contraction of the alveolus. These properties of surfactant result in enhanced lung compliance during inspiration, which reduces the work of breathing, and very low alveolar surface tension at end expiration, which helps to maintain airspace patency.
For the first three decades after Pattle discovered the surface active properties of surfactant (1), most surfactant research focused on the biophysical properties of the film. It did not escape the attention of pulmonary scientists that the anatomical location of surfactant placed it at the front lines of defense for inhaled pathogens, however, and early reports demonstrated that surfactant phospholipids had modest immunoregulatory properties. The discovery and molecular characterization of the surfactant proteins in the 1980s revealed extensive sequence similarities between SP-A and SP-D and the then newly identified acute-phase reactant of serum called mannose-binding protein (MBP) (2). The overall structural organization was also similar to MBP, in that all three proteins contained N-terminal collagen-like regions involved in trimerization, and ligand-binding C-type lectin domains (CLDs) at the C-terminus. MBP and SP-A assemble into 18-subunit oligomers composed of six disulfide-linked trimers that are aligned in a “flower bouquet” pattern, and SP-D forms a cruciform structure composed of four trimers radiating from a disulfide-linked hub. Based on their structural similarities, SP-A, SP-D, and MBP have been grouped into the human contingent of the “collectin” (collagen-lectin) family, which also includes bovine serum proteins conglutinin and CL-43 (3).
MBP has served as a useful model for understanding the structure and function of SP-A and SP-D. Low serum levels of MBP due to mutations in the collagen-like region result in an infantile illness characterized by recurrent infections, persistent diarrhea, and failure to thrive, suggesting a systemic host defense role (4). Functional studies show that MBP is an antibody-independent opsonin that binds a broad range of Gram-negative and Gram-positive organisms and enhances macrophage-dependent microbial clearance. Recognition of diverse microbial species by MBP is mediated by the CLD, which selectively binds to carbohydrates such as mannose and fucose, which are prevalent on glycolipids and glycoproteins that decorate the surface of microbes, but not to sialic acid and other carbohydrates that commonly occur as terminal sugars on mammalian cell surface molecules (5). Although binding by individual CLDs is relatively weak, the oligomeric organization of MBP greatly enhances binding affinity to the repeating glycoconjugates on the surface of microbes (6). This property provides MBP with a second mechanism to discriminate between self and nonself, since the more widely spaced glycoconjugates on mammalian cells are poor ligands for MBP. Complex formation between the CLD of MBP and microbial ligands activates two MBP-associated serine proteases, MASP-1 and MASP-2, which trigger the complement cascade and decorate the surface of the organism with iC3b (7). The microbe-associated complement and MBP interact with the complement and collectin receptors on the surface of monocytes, respectively, and promote phagocytosis and killing.
Studies to determine if SP-A and SP-D are the functional counterparts of MBP in the lung have been challenging, in part because they are found in the lung fluid, a compartment that is clearly less accessible than the serum, where MBP resides. Moreover, no human diseases of altered immunity associated with an inherited deficiency of SP-A or SP-D have been reported. A primary role for SP-A and SP-D as broad-spectrum pulmonary opsonins is certainly intuitively appealing, since air-breathing creatures are naive to many inhaled organisms, and the uninflamed alveolar space does not contain significant quantities of the systemic “all-purpose” opsonins MBP or complement. A multitude of in vitro studies have shown that the pulmonary collectins bind and aggregate bacterial, fungal, viral, and mycobacterial organisms, directly activate macrophages, and enhance the in vitro phagocytosis and killing of a variety of pulmonary pathogens (for review see ref. 8). The temporal pattern and spatial distribution of pulmonary collectin expression are also consistent with roles in host defense. Both proteins are synthesized in the alveolar type II cells and nonciliated bronchiolar cells of the distal pulmonary epithelium. SP-D mRNA and protein have also been detected in some extrapulmonary mucosal surfaces, including the pancreatic ducts, the intrahepatic biliary ducts and gall bladder of the gastrointestinal tract, the sweat glands and epithelial cells of the skin, and the parotid and lachrymal glands. The mRNA and protein levels of SP-A and SP-D increase dramatically just prior to birth and in response to lung injury, infection, or endotoxin challenge.
Some reported collectin functions are less consistent with host defense roles, however. For instance, it is claimed that for some organisms, collectins suppress phagocytosis; other studies suggest that SP-A and SP-D have opposite effects on host cell function (9). In addition, the functional comparisons between the lung collectins and MBP are clearly limited, since there have been no reports of complement activation by SP-A or SP-D. Finally, in vitro studies of SP-A and SP-D have suggested a bewildering array of diverse, nonimmunologic roles for the proteins. For example, SP-A has been reported to regulate surfactant homeostasis by controlling the secretion and uptake of surfactant by type II cells (10, 11), to protect the surface activity properties of surfactant from functional inhibitors (12), to inhibit phospholipase A2 (13), and to be required for the formation of surfactant aggregates such as tubular myelin, a highly structured form of surfactant lipids and proteins that serves as the major reservoir of surfactant material in the alveoli (Figure 1) (14).
Figure 1.

Tubular myelin in host defense. Surfactant phospholipids that are secreted into the alveolar space form lattice-like arrays of intersecting membranes called tubular myelin (a). Tubular myelin formation is dependent on SP-A, which is visible as electron-dense structures at the corners of the lattice (30). (b)The images suggest a model in which the C-type lectin domains of SP-A are buried at the membrane intersections and the N-terminal domains extend toward the centers of individual squares. The association of SP-A with tubular myelin places the collectin in the first line of defense against alveolar pathogens, in a configuration that does not disrupt surface activity.
Assessment of the relative physiologic importance of these laboratory activities of the pulmonary collectins has recently become feasible through the development of genetically-engineered experimental animal models. Evidence derived from these in vivo experiments does not support a primary role for collectins in surfactant surface activity but strongly supports their critical contribution to innate pulmonary defense and orchestration of inflammatory responses of the lung following infection (see Table 1).
Table 1.
Roles of lung collectins in pulmonary homeostasis

Animal models of pulmonary collectin function
Spa gene-targeted mice.
Animal models of collectin deficiency and excess have been generated, respectively, by targeted insertion of a gene disrupting neomycin cassettes using homologous recombination and by lung-specific overexpression of surfactant proteins under the control of the human surfactant protein C promoter (15–19). Resting and exercising SP-A–deficient mice (Spa–/–) and resting mice that overexpress rat SP-A all possess normal pools of surfactant, suggesting that SP-A does not regulate intraalveolar surfactant component levels in vivo. The surfactant aggregates isolated from Spa–/– mice consist of unusual extended sheets and vesicles lacking tubular myelin figures. Nonetheless, under physiologic conditions, surfactant from the Spa–/– mice adsorbs rapidly to form stable films with properties nearly identical to those of surfactant from control mice.The surface activities of surfactant isolated from the SP-A–overexpressing mice and Spa–/– mice are more and less resistant to serum protein inhibitors, respectively. However, SP-A deficiency does not compromise lung function in oxygen-exposed animals, even though the levels of proteinaceous surfactant inhibitors are higher in the alveolar wash taken from Spa–/– lungs than in that taken from wild-type controls.
The most compelling phenotypic characteristic in the Spa–/– mice is a pulmonary host defense defect. These mice exhibit delayed microbial clearance after intratracheal inoculation with group B Streptococcus (GBS) (20), Haemophilus influenza (21), respiratory syncytial virus (RSV) (22), and Pseudomonas aeruginosa (23). For each organism, the absence of SP-A results in an enhanced inflammatory response with increased or earlier neutrophil infiltration, and increased lavage cytokine levels including TNF-α and IL-6. GBS-treated Spa–/– mice, but not control littermates, show increased colony-forming units in the spleen, suggesting that SP-A plays a role in the prevention of bacterial dissemination. The bacterial clearance defect in the Spa–/– mice has been traced to the alveolar macrophage, which ingests fewer microbes and releases less superoxide and hydrogen peroxide than do macrophages from the wild-type mice. Bacterial clearance and macrophage function can be restored by coadministration of SP-A with the microbial inoculum in mice infected with GBS and RSV. Deficiency of SP-A is also associated with increased susceptibility to chronic infection in immunosuppressed Spa–/– mice exposed to the fungus Pneumocystis carinii (24).
Spd gene-targeted mice.
Unlike the Spa–/– mice, which have normal lung structure, surfactant function, and surfactant metabolism, the Spd–/– mouse manifests a complex and surprising lung phenotype. These mice develop a progressive alveolar proteinosis and distal airspace dilation, associated with increased levels of tissue and macrophage-associated metalloproteinases, macro-phage-derived oxidants, and phospholipids (25) — phenotypes that can be corrected by introducing the rat SP-D transgene into respiratory epithelial cells of Spd–/– mice. Overexpression of the rat SP-D in wild-type mice does not affect lung structure or surfactant pool sizes. Spd–/– mice exhibit decreased alveolar macrophage uptake of H. influenza and GBS (21), as well as of influenza A virus (26), without a significant difference in overall microbial clearance. As with the Spa–/– mice, deficiency of SP-D is associated with increased inflammation, including an increase in the inflammatory cell infiltrate and proinflammatory cytokine levels after intratracheal bacterial or viral inoculation (26).
Mechanisms of pulmonary collectin function
In vivo studies with Spa and Spd gene-targeted mice support roles for the pulmonary collectins as opsonins that enhance bacterial clearance and killing and as immunomodulators that regulate the cellular recruitment and activation arms of the inflammatory response (Figure 2). The mechanisms involved in these newly identified SP-A and SP-D functions are complex and only partially understood. Accumulating data indicate that the pulmonary collectins affect every phase of the inflammatory response to pulmonary pathogens, exerting direct effects on the growth and viability of microbes, presentation of the microbial antigens to inflammatory cells, and modulation of leukocyte phagocytic, proteolytic, chemotactic, and oxidant responses. Many of the upregulated genes responsible for oxidant and proinflammatory cytokine production are targets of the transcription factor NF-κB. Interestingly, recent studies demonstrated that NF-κB is spontaneously activated via oxidant-sensitive pathways in alveolar macrophages from the Spd–/– mice (27).
Figure 2.
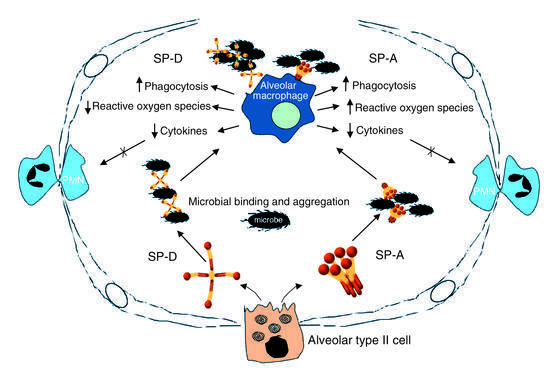
Pulmonary collectins orchestrate host defense, inflammation, and oxidant production. Schematic of alveolar defense roles for SP-A and SP-D suggested by in vitro studies and genetically engineered animal models. SP-A and SP-D are secreted by alveolar type II cells into the alveolar lumen. The presentation of microorganisms to inflammatory cells is determined in part by SP-A and SP-D, which aggregate and opsonize diverse viral, fungal, and bacterial species. SP-A and SP-D preserve gas exchange by limiting inflammatory cell infiltration and alveolar exudation, both by increasing the rate of clearance of microorganisms from the lung and by directly modulating inflammatory cytokine and oxidant responses.
For most organisms that have been studied, binding of pulmonary collectins to the microbial surface is mediated by the CLD. A notable exception is the herpes simplex virus, which appears to bind to SP-A via the N-linked oligosaccharides on the SP-A molecule (28). The microbial surface CLD ligands reported for SP-A include bacterial LPS, the capsular polysaccharides of certain Gram-negative organisms including Klebsiella, and the major surface glycoprotein from P. carinii; microbial ligands for SP-D include LPS, mycobacterial lipoarabinomannan, and influenza virus neuraminidase- and hemagglutinin-associated oligosaccharides. The effect of collectin binding on the physiology and viability of microorganisms has not been well studied, but recent evidence suggests that SP-D may have a direct effect on metabolism and hyphal growth of Candida albicans (29). Binding of SP-A and SP-D to viruses such as RSV and influenza virus, respectively, induce aggregation, enhance macrophage clearance, and reduce infectivity. Microbial aggregation is a common but not invariable consequence of collectin binding, which may enhance clearance by gathering organisms and presenting them as aggregates that are more readily internalized into phagocytes. Finally, the affinity of SP-A for phospholipid interfaces may facilitate interactions with inhaled microbes. SP-A is located in the corners of the tubular myelin lattice, with the CLDs buried at membrane intersections and the N-terminal regions of the molecule extending toward the center (Figure 1). While multiple in vivo studies suggest that SP-A does not directly contribute to the surface properties of pulmonary surfactant, mutations of SP-A disrupt tubular myelin formation and cooperative SP-A/surfactant interactions that maintain optimal surface activity (30). These data suggest that a primary antimicrobial function for tubular myelin may be to concentrate SP-A at the interface in a configuration that does not interfere with the surface active properties of surfactant.
Interaction of pulmonary collectins with LPS
The pulmonary collectins have also been reported to modulate the response to pulmonary infection that is triggered by LPS (31–33). The optimal pulmonary response to LPS during bacterial infection would include upregulation of the LPS signal that alerts the host to legitimate pathogens, and downregulation of LPS responses that promote inappropriate and excessive inflammation and compromise gas exchange. SP-A and SP-D bind to LPS by interaction of their CLDs with the lipid A moiety and core oligosaccharide domains of LPS, respectively. In vitro exposure of macrophages to LPS upregulates expression of the SP-A receptor (34), and intratracheal instillation of LPS increases the mRNA and protein levels of SP-A and SP-D (35). Alveolar macrophages release reactive oxygen species and proinflammatory cytokines in response to challenge with LPS.
These responses are variably dampened and enhanced by SP-A and SP-D in different laboratories and under differing conditions. SP-A–deficient mice have been reported to produce more nitric oxide and proinflammatory cytokines than do wild-type animals following treatment with intratracheal LPS (23, 36). This response suggests an important role for SP-A as an immunomodulator with a dampening function, opposite in direction to the effect of the serum LPS signal-amplifying protein LPS-binding protein (LBP). Like LBP, however, recent data suggest that SP-A may regulate LPS responses through a direct interaction with the cell surface LPS receptor CD14 (37).
Interaction of pulmonary collectins with inflammatory cells
Binding and internalization.
Both collectin-deficient mouse models are defective in phagocytosis. Kinetic studies demonstrating binding of SP-A and SP-D to the surface of macrophages and type II cells indicate the presence of one or more cell surface receptors for each protein, but there is no consensus regarding the molecular identity of the receptors responsible for microbial clearance. The best-characterized host defense receptor for SP-A is a 210-kDa cell surface protein (38) that can mediate the uptake and killing of Mycobacterium bovis. Both SP-A and SP-D bind to the putative collectin opsonin receptor gp-340 (also known as salivary agglutinin), a macrophage scavenger receptor family member with both soluble and alveolar macrophage membrane–bound components that has recently been shown to bind and aggregate some bacteria (39).
Oxidant responses.
Alveolar macrophage production of lethal reactive oxygen species and nitric oxide is dysregulated in animal models of pulmonary collectin deficiency. The mechanism by which the collectins modulate oxidant production is an active area of investigation. Recent data suggest that nitric oxide production induced by SP-A or LPS treatment of isolated alveolar macrophages is increased when cells are pretreated with IFN-γ, and decreased if the cells are not primed (40). Hence, inappropriate deployment of reactive nitrogen species (and perhaps other oxidants) by alveolar macrophages in the intact animal is prevented by a requirement for activation in multiple steps. The first step in this pathway may be upregulation of inducible nitric oxide synthase production through an IFN-γ–mediated increase in NF-κB. The role of the pulmonary epithelium in the generation of oxidant responses to infectious challenge requires further study (41).
Cytokine production and inflammatory cell recruitment
Cytokine production in the lungs of the infected Spa–/– and Spd–/– mice is increased compared with wild-type littermates. SP-A has been reported to stimulate TNF-α release from monocytes via NF-κB–dependent pathways (42), while, in other laboratories, it has been reported to decrease cytokine release from LPS-stimulated isolated alveolar macrophages. Recent evidence that the underlying activation state of the macrophage determines SP-A macrophage responses in vitro may explain some of these experimental differences (40). Although the macrophage has been the focus of studies involving collectin modulation of cytokine production, some of the elevated levels of cytokines in the collectin-deficient mouse models likely derive from the pulmonary epithelium. Alveolar type II cells have receptors for collectins, internalize microorganisms, produce reactive oxygen species and metalloproteinases, and communicate with intravascular and intra-alveolar leukocytes through production of a variety of cytokines. Data from several laboratories suggest that signaling between pulmonary epithelial cells and inflammatory cells in the alveolar space contributes to the response to lumenal microbial threats.
Summary
The data presented here suggest that SP-A and SP-D perform distinct, as well as complementary, host defense functions in the lung. Although both proteins are agglutinins, opsonins, and inflammatory immunomodulators, they interact uniquely with various pathogens and inflammatory cells. For instance, SP-A and SP-D appear to have opposite effects on the phagocytosis of Mycobacterium tuberculosis and on regulation of oxidant responses from alveolar macrophages. The pulmonary collectins partition into different microdomains within the airway lining; SP-D appears to prefer the aqueous phase of the ALL but may be weakly associated with the surface of epithelial cells, whereas SP-A is enriched in tubular myelin and at surfactant membrane interfaces. While the initial view held that tubular myelin would be required for surfactant regulation and function, it now appears more likely that it serves a primary host defense role as a solid-phase, high-density SP-A array that optimizes the surface properties of surfactant and the interception of inhaled microbes at the air-lung interface.
Implications for medical therapeutics
Few areas in science have experienced as great a shift in focus as the study of the pulmonary collectins. Genetically engineered animal models have overturned concepts of the importance of SP-A in the biophysical activity of surfactant, exchanged the roles of SP-A and SP-D in surfactant homeostasis, introduced a new requirement for SP-D for the maintenance of lung structure, and, most notably, redirected attention to the immunomodulatory functions of the pulmonary collectins. The finding that collectin replacement in the SP-A– and SP-D–deficient animals corrected defects in dysregulated cellular functions and microbial clearance suggests a possible role for these proteins in human therapies. Patients suffering with diseases associated with lung injury and infection, many of which are known to be associated with reductions in the levels of SP-A and SP-D, may benefit from the anti-inflammatory and antimicrobial properties of the collectins. As with all advances in medicine, the surest route to meaningful new treatment strategies is through a more complete understanding of the basic mechanisms of SP-A and SP-D action in the lung.
Acknowledgments
This work was supported by NIH grants to F.X. McCormack (HL-61612) and J.A. Whitsett (HL-61646), and by a Merit Grant to McCormack from the Department of Veterans Affairs, Washington, D.C., USA.
References
- 1.Pattle RE. Properties, function and origin of the alveolar lining layer. Nature. 1955;175:1125–1126. doi: 10.1038/1751125b0. [DOI] [PubMed] [Google Scholar]
- 2.Drickamer K, Dordal MS, Reynolds L. Mannose-binding proteins isolated from rat liver contain carbohydrate-recognition domains linked to collagenous tails. Complete primary structures and homology with pulmonary surfactant apoprotein. J Biol Chem. 1986;261:6878–6887. [PubMed] [Google Scholar]
- 3.Sastry K, Ezekowitz RA. Collectins: pattern recognition molecules involved in the first line of host defense. Curr Opin Immunol. 1993;5:59–63. doi: 10.1016/0952-7915(93)90082-4. [DOI] [PubMed] [Google Scholar]
- 4.Super M, Thiel S, Lu J, Levinsky RJ, Turner MW. Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet. 1989;2:1236–1239. doi: 10.1016/s0140-6736(89)91849-7. [DOI] [PubMed] [Google Scholar]
- 5.Kuhlman M, Joiner K, Ezekowitz RA. The human mannose binding protein functions as an opsonin. J Exp Med. 1989;169:1733–1745. doi: 10.1084/jem.169.5.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weis WI, Drickamer K. Trimeric structure of a C-type mannose-binding protein. Structure. 1994;2:1227–1240. doi: 10.1016/S0969-2126(94)00124-3. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda K, Sannoh T, Kawasaki N, Kawasaki T, Yamashina I. Serum lectin with known structure activates complement through the classical pathway. J Biol Chem. 1987;262:7451–7454. [PubMed] [Google Scholar]
- 8.Wright JR. Immunomodulatory functions of surfactant. Physiol Rev. 1997;77:931–962. doi: 10.1152/physrev.1997.77.4.931. [DOI] [PubMed] [Google Scholar]
- 9.Walenkamp AM, Verheul AF, Scharringa J, Hoepelman IM. Pulmonary surfactant protein A binds to Cryptococcus neoformanswithout promoting phagocytosis. Eur J Clin Invest. 1999;29:83–92. doi: 10.1046/j.1365-2362.1999.00429.x. [DOI] [PubMed] [Google Scholar]
- 10.Dobbs LG, et al. Pulmonary surfactant and its components inhibit secretion of phosphatidylcholine from cultured rat alveolar type II cells. Proc Natl Acad Sci USA. 1987;84:1010–1014. doi: 10.1073/pnas.84.4.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rice WR, Ross GF, Singleton FM, Dingle S, Whitsett JA. Surfactant-associated protein inhibits phospholipid secretion from type II cells. J Appl Physiol. 1987;63:692–698. doi: 10.1152/jappl.1987.63.2.692. [DOI] [PubMed] [Google Scholar]
- 12.Cockshutt AM, Weitz J, Possmayer F. Pulmonary surfactant protein A enhances the surface activity of lipid extract surfactant and reverses inhibition by blood proteins in vitro. Biochemistry. 1990;29:8424–8429. doi: 10.1021/bi00488a032. [DOI] [PubMed] [Google Scholar]
- 13.Fisher AB, Dodia C, Chander A. Inhibition of calcium independent phospholipase A2 by surfactant protein A. Am J Physiol. 1994;267:L335–L341. doi: 10.1152/ajplung.1994.267.3.L335. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki Y, Fujita Y, Kogishi K. Reconstitution of tubular myelin from synthetic lipids and proteins associated with pig pulmonary surfactant. Am Rev Respir Dis. 1989;140:75–81. doi: 10.1164/ajrccm/140.1.75. [DOI] [PubMed] [Google Scholar]
- 15.Korfhagen TR, et al. Altered surfactant function and structure in SP-A gene targeted mice. Proc Natl Acad Sci USA. 1996;93:9594–9599. doi: 10.1073/pnas.93.18.9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korfhagen TR, et al. Surfactant protein-D regulates surfactant phospholipid homeostasis in vivo. J Biol Chem. 1998;273:28438–28443. doi: 10.1074/jbc.273.43.28438. [DOI] [PubMed] [Google Scholar]
- 17.Fisher JH, et al. Pulmonary-specific expression of SP-D corrects pulmonary lipid accumulation in SP-D gene-targeted mice. Am J Physiol Lung Cell Mol Physiol. 2000;278:L365–L373. doi: 10.1152/ajplung.2000.278.2.L365. [DOI] [PubMed] [Google Scholar]
- 18.Elhalwagi BM, et al. Normal surfactant pool sizes and inhibition-resistant surfactant from mice that overexpress SP-A. Am J Respir Cell Mol Biol. 1999;21:380–387. doi: 10.1165/ajrcmb.21.3.3676. [DOI] [PubMed] [Google Scholar]
- 19.Botas C, et al. Altered surfactant homeostasis and alveolar type II cell morphology in mice lacking surfactant protein D. Proc Natl Acad Sci USA. 1998;95:11869–11874. doi: 10.1073/pnas.95.20.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LeVine AM, et al. Surfactant protein A-deficient mice are susceptible to group B streptococcal infection. J Immunol. 1997;158:4336–4340. [PubMed] [Google Scholar]
- 21.LeVine AM, et al. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol. 2000;165:3934–3940. doi: 10.4049/jimmunol.165.7.3934. [DOI] [PubMed] [Google Scholar]
- 22.LeVine AM, et al. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J Clin Invest. 1999;103:1015–1021. doi: 10.1172/JCI5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LeVine AM, et al. Surfactant protein-A-deficient mice are susceptible to Pseudomonas aeruginosainfection. Am J Respir Cell Mol Biol. 1998;19:700–708. doi: 10.1165/ajrcmb.19.4.3254. [DOI] [PubMed] [Google Scholar]
- 24.Linke MJ, et al. Immunosuppressed surfactant protein A-deficient mice have increased susceptibility to Pneumocystis cariniiinfection. J Infect Dis. 2001;183:943–952. doi: 10.1086/319252. [DOI] [PubMed] [Google Scholar]
- 25.Wert SE, et al. Increased metalloproteinase activity, oxidant production, and emphysema in surfactant protein D gene-inactivated mice. Proc Natl Acad Sci USA. 2000;97:5972–5977. doi: 10.1073/pnas.100448997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LeVine AM, et al. Surfactant protein D enhances clearance of influenza A virus from the lung in vivo. J Immunol. 2001;167:5868–5873. doi: 10.4049/jimmunol.167.10.5868. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida M, Korfhagen T, Whitsett J. SP-D regulates NF-κB and matrix metalloproteinase production in alveolar macrophages via oxidant sensitive pathways. J Immunol. 2001;166:7514–7519. doi: 10.4049/jimmunol.166.12.7514. [DOI] [PubMed] [Google Scholar]
- 28.van Iwaarden JF, et al. Binding of surfactant protein A to herpes simplex virus type 1-infected cells is mediated by the carbohydrate moiety of SP-A. J Biol Chem. 1992;267:25039–25043. [PubMed] [Google Scholar]
- 29.van Rozendaal BA, et al. Role of pulmonary surfactant protein D in innate defense against Candida albicans. J Infect Dis. 2000;182:917–922. doi: 10.1086/315799. [DOI] [PubMed] [Google Scholar]
- 30.Ikegami M, et al. The collagen-like region of surfactant protein A (SP-A) is required for correction of surfactant structural and functional defects in the SP-A null mouse. J Biol Chem. 2001;276:38542–38548. doi: 10.1074/jbc.M102054200. [DOI] [PubMed] [Google Scholar]
- 31.Kalina M, Blau H, Riklis S, Kravtsov V. Interaction of surfactant protein A with bacterial lipopolysaccharide. Am J Physiol. 1995;268:L144–L151. doi: 10.1152/ajplung.1995.268.1.L144. [DOI] [PubMed] [Google Scholar]
- 32.van Iwaarden JF, et al. Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem J. 1994;303:407–411. doi: 10.1042/bj3030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuan S, Rust K, Crouch E. Interactions of surfactant protein D with bacterial lipopolysaccharides. J Clin Invest. 1992;90:97–106. doi: 10.1172/JCI115861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chroneos Z, Shepherd VL. Differential regulation of the mannose and SP-A receptors on macrophages. Am J Physiol. 1995;269:L721–L726. doi: 10.1152/ajplung.1995.269.6.L721. [DOI] [PubMed] [Google Scholar]
- 35.McIntosh JC, Swyers AH, Fisher JH, Wright JR. Surfactant proteins A and D increase in response to intratracheal lipopolysaccharide. Am J Respir Cell Mol Biol. 1996;15:509–519. doi: 10.1165/ajrcmb.15.4.8879185. [DOI] [PubMed] [Google Scholar]
- 36.Borron P, et al. Surfactant-associated protein A inhibits LPS-induced cytokine and nitric oxide production in vivo. Am J Physiol Lung Cell Mol Physiol. 2000;278:L840–L847. doi: 10.1152/ajplung.2000.278.4.L840. [DOI] [PubMed] [Google Scholar]
- 37.Sano H, et al. Surfactant proteins A and D bind CD14 by different mechanisms. J Biol Chem. 2000;275:22442–22451. doi: 10.1074/jbc.M001107200. [DOI] [PubMed] [Google Scholar]
- 38.Chroneos ZC, et al. Purification of a cell-surface receptor for surfactant protein A. J Biol Chem. 1996;271:16375–16383. doi: 10.1074/jbc.271.27.16375. [DOI] [PubMed] [Google Scholar]
- 39.Prakobphol A, et al. Salivary agglutinin, which binds Streptococcus mutans and Helicobacter pylori, is the lung scavenger receptor cysteine-rich protein gp-340. J Biol Chem. 2000;275:39860–39866. doi: 10.1074/jbc.M006928200. [DOI] [PubMed] [Google Scholar]
- 40.Stamme C, Walsh E, Wright JR. Surfactant protein A differentially regulates IFN-gamma- and LPS-induced nitrite production by rat alveolar macrophages. Am J Respir Cell Mol Biol. 2000;23:772–779. doi: 10.1165/ajrcmb.23.6.4083. [DOI] [PubMed] [Google Scholar]
- 41.Kagnoff MF, Eckmann L. Epithelial cells as sensors for microbial infection. J Clin Invest. 1997;100:6–10. doi: 10.1172/JCI119522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koptides M, Umstead TM, Floros J, Phelps DS. Surfactant protein A activates NF-kappa B in the THP-1 monocytic cell line. Am J Physiol. 1997;273:L382–L388. doi: 10.1152/ajplung.1997.273.2.L382. [DOI] [PubMed] [Google Scholar]