

Abstract
Heterozygosity for C1 inhibitor (C1INH) deficiency results in hereditary angioedema. Disruption of the C1INH gene by gene trapping enabled the generation of homozygous- and heterozygous-deficient mice. Mating of heterozygous-deficient mice resulted in the expected 1:2:1 ratio of wild-type, heterozygous, and homozygous-deficient offspring. C1INH-deficient mice showed no obvious phenotypic abnormality. However, following injection with Evans blue dye, both homozygous and heterozygous C1INH-deficient mice revealed increased vascular permeability in comparison with wild-type littermates. This increased vascular permeability was reversed by treatment with intravenous human C1INH, with a Kunitz domain plasma kallikrein inhibitor (DX88), and with a bradykinin type 2 receptor (Bk2R) antagonist (Hoe140). In addition, treatment of the C1INH-deficient mice with an angiotensin-converting enzyme inhibitor (captopril) increased the vascular permeability. Mice with deficiency of both C1INH and Bk2R demonstrated diminished vascular permeability in comparison with C1INH-deficient, Bk2R-sufficient mice. These data support the hypothesis that angioedema is mediated by bradykinin via Bk2R.
Introduction
Hereditary angioedema (HAE) develops in individuals who are heterozygous for deficiency or dysfunction of C1 inhibitor (C1INH). This disease is characterized by recurrent episodes of angioedema involving the skin or the oropharyngeal, laryngeal, or gastrointestinal mucosa. Prior to the use of attenuated androgens such as danazol and stanazolol as chronic therapy for HAE, as many as one-third of patients died from asphyxiation secondary to laryngeal edema.
C1INH belongs to the serpin family of serine protease inhibitors. It is the only inhibitor of C1r and C1s, the classical complement pathway proteases (1). It also regulates kinin generation via inactivation of factors XIIa and plasma kallikrein, and intrinsic coagulation via inactivation of factor XIa (2–6). In HAE, the low levels of active C1INH in plasma lead to unregulated activation of the complement and contact cascades and the development of angioedema with its associated complications. Complement system activation results in decreased levels of C4 and C2, while contact system activation results in cleavage of high molecular weight kininogen.
In the past several years a great deal has been learned about the structure, genetics, mechanism of action, and inhibitory spectrum of C1INH. However, the pathophysiology of the increased vascular permeability of HAE has remained controversial for over 30 years. It is believed that angioedema results from uncontrolled activation of either the classical complement pathway with generation of a vasoactive peptide (C2 kinin) released from C2, and/or from contact system activation with release of bradykinin from high-molecular-weight kininogen (7–11). Although the majority of the available data support a role for bradykinin, the critical question is whether bradykinin alone could account for the symptoms of patients with HAE, or other mediators are also involved.
To investigate the role of C1INH in vivo, and to determine whether bradykinin is involved in the induction of vascular permeability in edema formation, we obtained mice in which the C1INH gene was targeted for disruption in murine embryonic stem cells using gene trapping (12). Neither homozygous nor heterozygous C1INH-deficient mice had an obvious phenotype. However, when compared with wild-type littermates, these animals clearly had increased vascular permeability that could be reversed by treatment with human C1INH, with an inhibitor of contact system activation (DX88), or with a bradykinin type 2 receptor (Bk2R) antagonist (Hoe140). Inhibition of bradykinin inactivation with captopril enhanced vascular permeability in C1INH-deficient mice, but mice doubly deficient in both C1INH and the Bk2R did not demonstrate increased vascular permeability.
Methods
C1INH gene targeting.
Mice in which the C1INH gene was targeted by gene trapping were obtained from a library of randomly targeted embryonic stem cell lines from Lexicon Genetics Inc. (The Woodlands, Texas, USA). This method uses random insertional mutagenesis with a fragment of DNA coding for a reporter or selectable marker gene as mutagen (13, 14). Embryonic stem cells were infected with recombinant retrovirus produced from a Moloney murine leukemia virus–based packaging cell line. Puromycin-selected clones were lysed to obtain RNA to be used in the rapid amplification of cDNA ends (3′ RACE) (12). Direct sequencing of 3′ RACE products gave a high quality sequence 80–700 nucleotides long. The sequence tag from each clone was compared to sequences in the GenBank database for sequence similarities. This indicated that the C1INH gene was targeted 210 bp upstream of exon 7. The gene trap vector inserted into the C1INH gene is shown schematically in Figure 1. Targeted 129/SvEvBrd embryonic stem cells were injected into C57BL/6 albino blastocysts. The chimeras (129/SvEvBrd) were then crossed with C57BL/6 albinos to produce the heterozygotes.
Figure 1.

Targeting the C1INH gene in mice by random insertional mutagenesis (12). The white boxes indicate C1INH gene exons (Ex). The retroviral vector with the galactosidase/neomycin phosphotransferase fusion gene (geo), the puromycin N-acetyl transferase gene (puro), and other components are inserted within intron 6, 210 bp in the 5′ direction from exon 7, and are indicated with shaded boxes. 1f, 2f, 1r, and 2r represent PCR primers. LTR, long term repeat; SA, splice acceptor sequence; IRES, internal ribosome entry site; pA, polyadenylation sequence; PGK, phosphoglycerate kinase promoter; SD, splice donor sequence.
Bk2R knockout mice.
Mice in which Bk2R is not expressed due to targeted disruption of the Bk2R gene (15) were obtained from The Jackson Laboratory (Bar Harbor, Maine, USA). Bk2R genotyping was performed by PCR using two primer pairs, one of which amplified a 361-bp fragment within the coding sequence of the Bk2R gene (forward primer, TGTCCTCAGCGTGTTCTTCC; reverse primer, GGTCCTGAACACCAACATGG). The other pair (forward primer, CTTGGGTGGAGAGGCTATTC; reverse primer, GGTCCTGAACACCAACATGG) amplified a 280-bp fragment within the inserted portion of the targeting vector. In the targeted gene, the entire Bk2R coding sequence was replaced. Therefore, amplification of DNA from wild-type and homozygous Bk2R-deficient mice resulted in single bands of 361 bp and 280 bp, respectively, while amplification of DNA from heterozygous mice yielded both bands.
PCR amplification of the mouse C1INH gene.
PCR amplification was performed with Taq polymerase and other reagents in the Platinum PCR SuperMix (Invitrogen Corp., Carlsbad, California, USA). Each cycle of denaturation (93°C for 30 seconds), annealing (59°C for 30 seconds), and extension (65°C for 2 minutes) was repeated 40 times. Two forward and two reverse primers were used to genotype the animals. One forward primer (1f) (CTCTGTTATTACATAGCTAGTCAAGG) amplified from a point near the 5′ end of intron 6, while the other (2f) (CTTGCAAAATGGCGTTACTTAAGC) used a sequence within the long terminal repeat. The two reverse primers amplified from sequences near the 3′ end of intron 6 (CAGATACCCCTTATTAGCTCTTCCT, primer 1r) and within exon 7 (TACCACGATCACAAAGCTCAGGTTG, primer 2r). Using these primers, amplification of genomic DNA from wild-type mice produced bands of 300 bp (1f → 2r) and 210 bp (1f → 1r) (Figure 1 and Figure 2a). Genomic DNA from the homozygous-deficient mice produced bands of 200 bp (2f → 2r) and 150 bp (2f → 1r) (Figure 1 and Figure 2a).
Figure 2.

Genotype/phenotype analysis. (a) The genotype of the C1INH-targeted mice was determined by PCR amplification using the two primer pairs described in Methods. Amplification in the C1INH+/+ mice resulted in bands of 300 bp and 210 bp; in the C1INH+/– mice, bands of 300 bp, 210 bp, 200 bp, and 150 bp resulted; in the C1INH–/– mice, bands of 200 bp and 150 bp resulted. (b) Detection of C1INH by Western blot analysis of mouse serum using antiserum to mouse C1INH. NEG, negative control; M, molecular weight marker.
Western blot analysis.
Mouse plasma (20 μl, 1:800 dilution) was added to an equal volume of SDS sample buffer containing 200 mM DTT and boiled for 3 minutes. Samples were resolved by SDS PAGE (7.5% polyacrylamide concentration) and transferred by electrophoresis onto a nitrocellulose membrane (NOVEX LC2001; Invitrogen Corp.). After blocking, membranes were incubated with primary rabbit anti-mouse C1INH IgG (16), followed by secondary goat anti-rabbit IgG–horseradish peroxidase. A chemiluminescent Western blotting detection kit (Pierce Chemical Co., Rockford, Illinois, USA) was used according to the manufacturer’s protocol to visualize the labeled protein.
Vascular permeability assay.
Evans blue dye (30 mg/kg in 100 μl PBS; Sigma Chemical Co., St. Louis, Missouri, USA) was injected into the tail vein of 7- to 8-week-old mice. In some experiments, after 1 minute, mustard oil (Sigma Chemical Co.) diluted to 5% in mineral oil was applied to the dorsal and ventral surfaces of the ear with a cotton swab; the application process was repeated 15 minutes later. Photographs were taken 30 minutes after injection of Evans blue dye. After the mice were euthanized by CO2 inhalation, ears and feet were removed, blotted dry, and weighed. The Evans blue dye was extracted from the ears and feet with 1 ml of formamide overnight at 55°C and measured spectrophotometrically at 600 nm (17). In one group of experiments, intestinal vascular permeability was evaluated by extraction of Evans blue dye from equal weights of small intestinal tissue. Data were expressed as mean ± SEM. Comparisons of the amounts of dye extravasation were evaluated by the unpaired Student t test. P values smaller than 0.05 were considered significant. In some experiments, immediately prior to the infusion of Evans blue dye, mice were treated with one of the following agents: human C1INH (100 μg; Advanced Research Technologies, San Diego, California, USA), the engineered Kunitz domain plasma kallikrein inhibitor DX88 (18) (10 μg; kindly provided by Shirish Hirani, Dyax Corp., Cambridge, Massachusetts, USA), the Bk2R antagonist Hoe140 (30 μg; Sigma Chemical Co.), the Bk1R antagonist des-Arg9,[Leu8]-bradykinin (30 μg; Sigma Chemical Co.), and the angiotensin-converting enzyme (ACE) inhibitor captopril (20 μg; Sigma Chemical Co.). All experiments were performed in compliance with relevant laws and institutional guidelines and were approved by the Center for Blood Research Animal Care and Use Committee.
Results
Phenotype of the C1INH-deficient mice.
The C1INH-deficient mice were genotyped by PCR amplification using the two indicated primer pairs (Figure 1 and Figure 2a). Both the heterozygous and homozygous C1INH-deficient mice appeared normal at birth, and subsequently developed and bred normally. Both the C1INH+/– and the C1INH–/– mice (males and females) were fertile. The growth of both heterozygous and homozygous-deficient mice appeared normal, and no more newborn deaths were observed among the C1INH+/– and C1INH–/– mice than among their wild-type littermates. The mean weight of six C1INH+/+ male mice was 24.2 g, while that of seven C1INH+/– male mice was 26.1 g and that of seven C1INH–/– male mice was 26.4 g. The mean weights of both the C1INH+/– and C1INH–/– male mice were significantly different from the mean weight of the C1INH+/+ male mice (P < 0.002 and P < 0.0002, respectively). However, the female mice showed no significant difference in weight (20.3 g, 19.8 g, and 19.9 g for C1INH+/+, C1INH+/–, and C1INH–/–, respectively; P = 0.68 for both C1INH+/– and C1INH–/– compared with C1INH+/+.
Western blot analysis demonstrated that the C1INH+/– mice expressed lower levels of C1INH protein in plasma than did C1INH+/+ littermates, while C1INH protein was not detected in plasma from C1INH–/– mice (Figure 2b). As is the case in humans with HAE, the level of C1INH in the C1INH+/– mice appeared to be less than 50% of normal. In addition, Western blot analysis of mouse serum following incubation with human C1s showed no complex formation in serum from null mice when probed with antibody to C1s or C1INH, while complexes were readily apparent in serum from wild-type and heterozygous-deficient mice (data not shown). Because of limited availability of reagents, ELISA for determination of serum C4 levels was performed for only three C1INH–/– and four C1INH+/– mice. The serum C4 levels of the C1INH–/– mice were 38%, 41%, and 59% of the levels measured in wild-type littermate controls. The serum C4 levels of the C1INH+/– mice were more variable (49%, 76%, 120%, and 49% that of wild-type controls). The mean serum C4 level of the C1INH–/– mice differed significantly from that of the wild-type mice (P < 0.01). Consistent with these low C4 levels, total hemolytic complement activity in serum from the null mice was also decreased in comparison with the wild-type mice (data not shown). These data provide indirect evidence suggesting insufficient regulation of complement system activation, as is the case with the human disease.
Mating of heterozygous-deficient mice (a total of 144 offspring) resulted in 38 C1INH+/+ mice (26%), 75 C1INH+/– mice (52%), and 31 C1INH–/– mice (22%), which corresponded to the expected Mendelian ratio of 1:2:1. The average number of pups per litter was seven. There was no alteration in the ratio of males to females (71 females, 73 males).
Increased vascular permeability in C1INH-deficient mice.
Evans blue dye was injected intravenously into the tail vein, and extravasation of dye from the vasculature was measured. Over a period of 15–30 minutes, the skin of the normal C1INH+/+ mice developed a slight blue coloration that was particularly apparent around the nose and eyes, and on the feet (Figure 3, a and c). Under baseline conditions, the C1INH+/– (Figure 3, b and c) and C1INH–/– mice (Figure 3c) had markedly increased vascular permeability compared with the wild-type mice. The blue coloration of the eyes, nose, and feet was observed within minutes after the Evans blue dye injection and was much more intense in the heterozygous and homozygous-deficient mice than in the wild-type mice.
Figure 3.

Analysis of vascular permeability. Extravasation of Evans blue dye at 15–30 minutes was much more extensive in C1INH-deficient mice (b) than in wild-type mice (a), particularly after the application of mustard oil to the left ears of the mice. (c) The difference in the amount of extravasation was clearly demonstrated by the rear footpads of mice of each genotype. Administration of human C1INH resulted in reduced vascular permeability, as did the combination of Bk2R deficiency together with C1INH deficiency.
The increased vascular permeability was less apparent on the ears than on the feet and noses of all the mice. In fact, there was no difference in baseline vascular permeability in the ears of C1INH–/– mice in comparison with the wild-type. However, the difference between wild-type and deficient mice was enhanced by the topical application of mustard oil to the ear after injection of Evans blue dye (Figure 3, a and b). Mustard oil is a local irritant that induces plasma leakage and inflammation. However, even with mustard oil treatment, the blue coloration was less intense in the wild-type mice (Figure 3a) than in the C1INH+/– mice (Figure 3b) and the C1INH–/– mice (not shown).
The amount of extravasated dye was quantitated by spectrophotometric analysis. Evans blue dye was extracted from the ears and rear paws of the mice with formamide and was read at an absorbance of 600 nm. This revealed that the amount of extravasated Evans blue dye in the rear paws was significantly greater in both the C1INH+/– and C1INH–/– mice than in the C1INH+/+ mice (Figure 4) (P = 0.0009 and P = 0.002, respectively). Similarly, 30 minutes after application of mustard oil to the ears, the amount of Evan’s blue dye increased two- to fivefold in the C1INH+/– and in the C1INH–/– mice compared with the wild-type mice (Figure 5), even though the baseline values in the two were very similar. The increases in both the C1INH+/– and C1INH–/– mice were significant in comparison with both the mustard oil–treated C1INH+/+ mice (P = 0.007 and P = 0.0003) and with the untreated controls of the same genotype (P = 0.003 for the C1INH+/– mice and P = 0.0002 for the C1INH–/– mice). Intestinal vascular permeability was analyzed in a limited number of mice (Figure 6). Permeability in this tissue was also increased in the C1INH–/– mice in comparison with C1INH+/+ mice, although the difference between the two was of borderline statistical significance (P = 0.05), probably as a result of the small number of mice studied. These data clearly indicate that the C1INH-deficient mice not only have increased vascular permeability, but also have an increased permeability response to an external irritant.
Figure 4.

Spectrophotometric analysis of vascular permeability in footpads. Quantitation of Evans blue dye extracted from the paws of mice of the indicated genotypes, either treated or not treated with intravenous C1INH (C1I), DX88 (DX), or Hoe140 (Hoe).
Figure 5.

Spectrophotometric analysis of the vascular permeability response to application of mustard oil to the ears of mice of the indicated phenotypes and treatments.
Figure 6.

Spectrophotometric analysis of vascular permeability in the small intestine. Quantitation of extravasated Evans blue dye in C1INH–/– mice treated with an ACE inhibitor (captopril), a Bk1R antagonist (des-Arg9,[Leu8]-bradykinin; Bk1RA), and a Bk2R antagonist (Hoe140).
Intravenous injection of human C1INH reverses the increased vascular permeability in C1INH-deficient mice.
In order to provide evidence that the increased vascular permeability observed in the C1INH+/– and C1INH–/– mice was a direct result of C1INH deficiency rather than an unforeseen associated defect, human C1INH (100 μg in 100 μl PBS) was injected into the tail veins of mice, followed by injection of Evans blue dye within 10 minutes. Mice treated with human C1INH became slightly blue and appeared essentially identical to the wild-type mice, as demonstrated by the footpads shown in Figure 3c. This observation was confirmed by spectrophotometric analysis of Evans blue dye extravasation in the foot (Figure 4). In both the C1INH+/– and C1INH–/– mice, the differences in Evan’s blue dye between the untreated and C1INH-treated groups were statistically significant (P = 0.003 and P = 0.002, respectively). The mice infused with C1INH were also treated with mustard oil on their ears, and their permeability responses were quantitated spectrophotometrically. The heterozygous and homozygous-deficient mice reconstituted with human C1INH showed a diminished response to mustard oil in comparison with the C1INH+/– and C1INH–/– mice that had not been treated with C1INH (Figure 5) (P = 0.02 for the C1INH+/– C1INH-treated versus untreated mice, and P = 0.006 for the C1INH–/– C1INH-treated versus untreated mice).
The increased vascular permeability in C1INH-deficient mice is mediated by Bk2R.
To test the hypothesis that increased vascular permeability in C1INH+/– and C1INH–/– mice resulted from bradykinin mediated via Bk2R, C1INH-deficient mice were treated with the plasma kallikrein inhibitor DX88, with the Bk2R antagonist Hoe140, and with the ACE inhibitor captopril (Figure 4 and Figure 6). Plasma kallikrein inhibition, which would prevent contact system–mediated bradykinin generation, reversed the increased vascular permeability in C1INH–/– mice (Figure 4) (P = 0.004 for C1INH–/– mice treated with DX88 versus untreated C1INH–/– mice). Similarly, interference with the ability of Bk2R to interact with its ligand also reduced vascular permeability (P = 0.008 and P = 0.0007 for treated versus untreated C1INH–/– mice, vascular permeability in the feet and intestines, respectively) (Figure 4 and Figure 6). On the other hand, treatment with the Bk1R antagonist, des-Arg9,[Leu8]-bradykinin, had no effect on vascular permeability (Figure 6). Bk1R is expressed primarily by tissues during inflammation, and therefore would not be expected to mediate the increased vascular permeability. Treatment with captopril resulted in a dramatic increase in vascular permeability (P = 0.002 treated versus untreated).
In addition, C1INH knockout mice were crossed with Bk2R knockout mice (Figure 7). Initially, C1INH-deficient mice were crossed with Bk2R null mice. C1INH+/–, Bk2R+/– mice that resulted from these crosses were mated. These crosses resulted in all the predicted genotypes. The C1INH+/–, Bk2R+/– and C1INH–/–, Bk2R–/– mice all appeared normal. Vascular permeability in the C1INH+/–, Bk2R–/– and C1INH–/–, Bk2R–/– mice was strikingly less than that in the C1INH+/–, Bk2R+/+ or C1INH–/–, Bk2R+/+ mice and was essentially the same as that observed in the C1INH+/+ mice (Bk2R+/+, Bk2R+/–, or Bk2R–/–), as shown both by visual inspection of the footpads (Figure 3c) and by spectrophotometric analysis of extravasated Evans blue dye in both the footpads and the mustard oil–treated ears (Figure 4 and Figure 5). The difference in dye extravasation in C1INH–/–, Bk2R–/– mice compared with C1INH–/–, Bk2R+/+ mice was significant in both the footpads (P = 0.002) and the mustard oil–treated ears (P = 0.0004).
Figure 7.
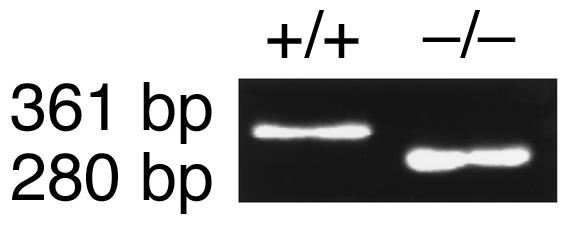
Genotype analysis of C1INH–/–, Bk2R–/– mice. The genotypes of the double knockout mice were determined by PCR amplification using the primer pairs indicated in Methods. Amplification in the C1INH+/+, Bk2R+/+ mice resulted in a C1INH+/+ genotype as shown in Figure 2a, while the Bk2R+/+ genotype showed a single band of 361 bp. Amplification in the C1INH–/–, Bk2R–/– mice resulted in a C1INH–/– genotype as shown in Figure 2a, while the Bk2R–/– genotype showed a single band of 280 bp.
Discussion
The C1INH-deficient mouse model described here shares a number of characteristics with HAE. In particular, the studies have shown that C1INH deficiency in the mouse, as in the human, results in increased vascular permeability. In addition, many of the mice had decreased C4 levels in association with the decreased C1INH levels. Furthermore, the observed increased vascular permeability could be reversed with intravenous infusion of C1INH. On the other hand, no episodes of angioedema involving the skin have been observed in the deficient mice. However, preliminary observations suggest that these mice may, in fact, develop acute attacks of angioedema. Five mice (two C1INH+/– and three C1INH–/–) have been observed with abdominal distension secondary to edema of the wall of the small intestine. It is not clear if such episodes are self-limited, because the mice were euthanized. Such episodes have not been observed in any wild-type littermates. However, it is not possible to reach definitive conclusions based on these observations because insufficient numbers of mice have been observed for prolonged periods. Currently, groups of C1INH+/+, C1INH+/–, and C1INH–/– mice are being observed throughout their life spans for the development of such episodes.
Another distinction between this animal model and HAE is that no human with homozygous deficiency of C1INH has ever been described. Therefore we had hypothesized that C1INH–/– mice would not be viable. However, as described here, these mice survive and are phenotypically essentially identical to the C1INH+/– mice. There are at least two potential explanations for this similarity. First, it is possible that below a threshold C1INH level, activation of the proteolytic pathways is completely unregulated, and that further decreases in the level of C1INH have no additional effect. Alternatively, it is possible that other protease inhibitors in the mouse contribute to regulation of the complement and/or contact system proteases to a greater extent than in the human. Preparation of the reagents required to perform the studies designed to test these hypotheses is in progress.
A primary goal of these studies was to test the hypothesis that bradykinin is the primary mediator of angioedema. Several earlier studies implicated the classical complement system in the generation of angioedema. Mixtures of C1s, C4, C2, and plasmin produced a kinin-like activity, referred to as C2 kinin, that was similar to the kinin activity isolated from plasma of patients with HAE (19). In addition, intradermal injection of C1s induced angioedema with no itching, no pain, and no signs of inflammation (20). This response was decreased or absent in both humans and guinea pigs deficient in C2. Intravenous infusion of C2 in C2-deficient guinea pigs restored the response to C1s (7).
However, other studies have suggested that C2 may not be the source of the kinin-like activity generated in HAE plasma. Fields et al. showed that the kinin was resistant to tryptic digestion but was destroyed by carboxypeptidase B, which is more consistent with the hypothesis that the source of the kinin activity is bradykinin rather than the C2 kinin (21). Another study was unable to generate kinin activity from C2 by cleavage with purified C1s (22). Blister fluid obtained from suction blisters induced on the skin of patients with HAE contained large amounts of active kallikrein, while fluid similarly obtained from normal individuals did not (23). Subsequently, Shoemaker et al. demonstrated that the contact system, but not the complement system, was required for generation of the kinin activity from angioedema plasma following incubation at 37°C (9). Furthermore, bradykinin was the only kinin that could be isolated from such plasma, and no kinin activity could be generated from normal plasma incubated under the same conditions. Other evidence suggesting that bradykinin might be a primary mediator of angioedema is the observation that members of a family with a mutant C1INH (Ala443 → Val) that inhibits the contact system proteases, but is a poor inhibitor of the complement proteases, have never developed angioedema (10). Finally, more recently, Nussberger et al. have shown that bradykinin can be detected in the plasma of HAE patients during attacks of angioedema (11). Taken together, these previous reports suggest that mediators of angioedema are generated through activation of the contact system, but the possibility that mediators resulting from complement system activation may play a role has not been disproved.
The studies reported here provide additional strong evidence that bradykinin contributes substantially to the vascular permeability in the C1INH-deficient mice via Bk2R. Previous studies have indicated that Bk2R mediates bradykinin-induced vascular permeability (24, 25). In this report, the C1INH-deficient mice were protected from enhanced vascular permeability by an inhibitor of plasma kallikrein, by a Bk2R antagonist, and by Bk2R-deficiency. This deficiency corrected both the generalized increased vascular permeability and the exaggerated vascular permeability response to the mustard oil skin irritant. Furthermore, treatment of C1INH–/– mice with an ACE inhibitor increased vascular permeability. To our knowledge, no ligand for Bk2R other than bradykinin has been described. These data, therefore, strongly suggest that the symptoms of angioedema are mediated by bradykinin via Bk2R. The receptor that mediates the C2 kinin–induced enhancement of vascular permeability has not been identified. Therefore it remains theoretically possible that the C2 kinin could play a role in the vascular permeability response. In either case, the results described here suggest that bradykinin receptor antagonists may be effective in the treatment of HAE.
Acknowledgments
This work was supported by NIH grants HD-22082 and HD-33727. The authors thank Patrick Andre, Minghe Ma, and Brendan Mulligan for outstanding technical support.
Footnotes
See the related Commentary beginning on page 1007.
References
- 1.Sim RB, Reboul A, Arlaud GJ, Villiers CL, Colomb MG. Interaction of 125I-labeled complement components C1r and C1s with protease inhibitors in plasma. FEBS Lett. 1979;97:111–115. doi: 10.1016/0014-5793(79)80063-0. [DOI] [PubMed] [Google Scholar]
- 2.Ratnoff O, Pensky J, Ogston D, Naff G. The inhibition of plasmin, plasma kallikrein, plasma permeability factor, and the C’1r subcomponent of complement by serum C’1 esterase inhibitor. J Exp Med. 1969;129:315–331. doi: 10.1084/jem.129.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forbes C, Pensky J, Ratnoff O. Inhibition of activated Hageman factor and activated plasma thromboplastin antecedent by purified C1 inactivator. J Lab Clin Med. 1970;76:809–815. [PubMed] [Google Scholar]
- 4.Gigli I, Mason JW, Colman RW, Austen KF. Interaction of plasma kallikrein with the C1 inhibitor. J Immunol. 1970;104:574–581. [PubMed] [Google Scholar]
- 5.Schreiber AD, Kaplan AP, Austen KF. Inhibition by C1INH of Hageman factor fragment activation of coagulation, fibrinolysis, and kinin generation. J Clin Invest. 1973;52:1402–1409. doi: 10.1172/JCI107313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wuillemin WA, et al. Inactivation of factor XIa in human plasma assessed by measuring factor XIa-protease inhibitor complexes: major role for C1-inhibitor. Blood. 1995;85:1517–1526. [PubMed] [Google Scholar]
- 7.Strang CJ, Auerbach KS, Rosen FS. C1s-induced vascular permeability in C2-deficient guinea pigs. J Immunol. 1986;137:631–635. [PubMed] [Google Scholar]
- 8.Strang C, et al. Angioedema induced by a peptide derived from complement component C2. J Exp Med. 1988;168:1685–1698. doi: 10.1084/jem.168.5.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shoemaker LR, Schurman SJ, Donaldson VH, Davis AE., III Hereditary angioneurotic edema: characterization of plasma kinin and vascular permeability-enhancing activities. Clin Exp Immunol. 1994;95:22–28. doi: 10.1111/j.1365-2249.1994.tb06009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zahedi R, Bissler JJ, Davis AE, III, Andreadis C, Wisnieske JJ. Unique C1 inhibitor dysfunction in a kindred without angioedema. II. Identification of an Ala443→Val substitution and functional analysis of the recombinant mutant protein. J Clin Invest. 1995;95:1299–1305. doi: 10.1172/JCI117780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nussberger J, et al. Plasma bradykinin in angio-oedema. Lancet. 1998;351:1693–1697. doi: 10.1016/S0140-6736(97)09137-X. [DOI] [PubMed] [Google Scholar]
- 12.Zambrowicz BP, et al. Disruption and sequence identification of 2,000 genes in mouse embryonic stem cells. Nature. 1998;392:608–611. doi: 10.1038/33423. [DOI] [PubMed] [Google Scholar]
- 13.Friedrich G, Soriano P. Insertional mutagenesis by retroviruses and promoter traps in embryonic stem cells. Methods Enzymol. 1993;225:681–701. doi: 10.1016/0076-6879(93)25044-3. [DOI] [PubMed] [Google Scholar]
- 14.Gossler, A., and Zachgo, J. 1993. Gene and enhancer trap screens in ES cell chimeras. In Gene targeting: a practical approach. A.L. Joyner, editor. Oxford University Press. New York, New York, USA. 181–227.
- 15.Borkowski JA, et al. Targeted disruption of a B2 bradykinin receptor gene in mice eliminates bradykinin actions in smooth muscle and neurons. J Biol Chem. 1995;270:13706–13710. doi: 10.1074/jbc.270.23.13706. [DOI] [PubMed] [Google Scholar]
- 16.Lener M, Vinci G, Duponchel C, Meo T, Tosi M. Molecular cloning, gene structure and expression profile of mouse C1 inhibitor. Eur J Biochem. 1998;254:117–122. doi: 10.1046/j.1432-1327.1998.2540117.x. [DOI] [PubMed] [Google Scholar]
- 17.Thurston G, et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286:2511–2514. doi: 10.1126/science.286.5449.2511. [DOI] [PubMed] [Google Scholar]
- 18.Markland W, Ley AC, Ladner RC. Iterative optimization of high-affinity protease inhibitors using phage display. 2. Plasma kallikrein and thrombin. Biochemistry. 1996;35:8058–8067. doi: 10.1021/bi952629y. [DOI] [PubMed] [Google Scholar]
- 19.Donaldson V, Rosen F, Bing D. Role of the second component of complement (C2) and plasmin in kinin release in hereditary angioneurotic edema (H.A.N.E.) plasma. Trans Assoc Am Physicians. 1977;90:174–183. [PubMed] [Google Scholar]
- 20.Klemperer MR, Donaldson VH, Rosen FS. Effect of C’1 esterase on vascular permeability in man: studies in normal and complement-deficient individuals and in patients with hereditary angioneurotic edema. J Clin Invest. 1968;47:604–611. doi: 10.1172/JCI105756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fields T, Ghebrehiwet B, Kaplan A. Kinin formation in hereditary angioedema plasma: evidence against kinin derivation from C2 and in support of “spontaneous” formation of bradykinin. J Allergy Clin Immunol. 1983;72:54–60. doi: 10.1016/0091-6749(83)90052-0. [DOI] [PubMed] [Google Scholar]
- 22.Smith M, Kerr M. Cleavage of the second component of complement by plasma proteases: implications in hereditary C1-inhibitor deficiency. Immunology. 1985;56:561–570. [PMC free article] [PubMed] [Google Scholar]
- 23.Curd JG, Prograis LJ, Cochrane CG. Detection of active kallikrein in induced blister fluids of hereditary angioedema patients. J Exp Med. 1980;152:742–747. doi: 10.1084/jem.152.3.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schachter M, et al. New synthetic antagonists of bradykinin. Br J Pharmacol. 1987;92:851–855. doi: 10.1111/j.1476-5381.1987.tb11390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Whalley ET, Nwator IA, Steward JM, Vavrek RJ. Analysis of the receptors mediating the vascular actions of bradykinin. Naunyn Schmiedeberg’s Arch Pharamcol. 1987;336:430–433. doi: 10.1007/BF00164878. [DOI] [PubMed] [Google Scholar]