

Abstract
Dominant mutations in sarcomere protein genes cause hypertrophic cardiomyopathy, an inherited human disorder with increased ventricular wall thickness, myocyte hypertrophy, and disarray. To understand the early consequences of mutant sarcomere proteins, we have studied mice (designated αMHC403/+) bearing an Arg403Gln missense mutation in the α cardiac myosin heavy chain. We demonstrate that Ca2+ is reduced in the sarcoplasmic reticulum of αMHC403/+ mice, and levels of the sarcoplasmic reticulum Ca2+-binding protein calsequestrin are diminished in advance of changes in cardiac histology or morphology. Further evidence for dysregulation of sarcoplasmic reticulum Ca2+ in these animals is seen in their decreased expression of the ryanodine receptor Ca2+-release channel and its associated membrane proteins and in an increase in ryanodine receptor phosphorylation. Early administration of the L-type Ca2+ channel inhibitor diltiazem restores normal levels of these sarcoplasmic reticular proteins and prevents the development of pathology in αMHC403/+ mice. We conclude that disruption of sarcoplasmic reticulum Ca2+ homeostasis is an important early event in the pathogenesis of this disorder and suggest that the use of Ca2+ channel blockers in advance of established clinical disease could prevent hypertrophic cardiomyopathy caused by sarcomere protein gene mutations.
Introduction
Hypertrophic cardiomyopathy is a prevalent autosomal dominant human disorder caused by mutations in contractile protein genes (for reviews, see refs. 1, 2). Over 200 DNA defects have been identified in ten disease genes that encode constituents of the cardiac muscle sarcomere. While much is known about these primary causes of hypertrophic cardiomyopathy, little is understood about the signaling events initiated by the mutant sarcomere that result in the hypertrophic response. Notably, there is a considerable latency period in the clinical expression of hypertrophic cardiomyopathy; ventricular hypertrophy evolves insidiously in genetically affected individuals, often beginning around pubescence. However, once established, the hypertrophic phenotype remains throughout life and causes a range of symptoms including chest pain, palpitations, and, in some, sudden death (3, 4). Sudden death can be the presenting manifestation of disease, and unrecognized hypertrophic cardiomyopathy remains the most frequent cause of sudden cardiac death in athletes (5, 6). Although a variety of pharmacologic agents are used to treat symptoms and arrhythmias in hypertrophic cardiomyopathy, therapies have not been identified that prevent expression of disease in individuals with sarcomere protein gene mutations. Study of animal models of hypertrophic cardiomyopathy (7–12) has indicated potential signaling pathways that may contribute to components of the cardiac pathology caused by sarcomere gene mutations. However, the identification of key early molecular events in disease pathogenesis has remained elusive. To define the early molecular steps in the pathogenesis of hypertrophic cardiomyopathy, αMHC403/+ mice were studied. αMHC403/+ mice carry an Arg403Gln missense mutation in one allele of the cardiac myosin heavy chain and model a severe human disease mutation (13). Like affected humans, αMHC403/+ mice gradually develop myocardial hypertrophy, myocyte disarray, and fibrosis (14). Biochemical and biophysical studies indicate that αMHC403/+ mice have abnormal Ca2+ homeostasis: myofibrillar preparations from mutant mice require higher Ca2+ concentrations to achieve the same contractile force as controls (15), and αMHC403/+ mice treated with agents that affect myocyte Ca2+ levels exhibit dramatically accelerated hypertrophic pathology and decreased survival (16). Precise regulation of Ca2+ is essential for myocyte function (17). Cyclic fluxes of Ca2+ between three compartments — cytoplasm, sarcoplasmic reticulum (SR), and sarcomere — account for excitation-contraction coupling. Depolarization triggers entry of small amounts of Ca2+ through the L-type Ca2+ channels located on the cell membrane, which in turn prompts SR Ca2+ release by cardiac ryanodine receptors (RyR’s), a process termed calcium-induced Ca2+ release. The resulting rapid rise in cytosolic levels fosters Ca2+–troponin-C interactions and triggers sarcomere contraction. Activation of the ATP-dependent calcium pump (SERCA) recycles cytosolic Ca2+ into the SR to restore sarcomere relaxation. To understand the mechanism by which calcium dysregulation occurs in αMHC403/+ myocytes, we studied Ca2+-binding proteins in specific myocyte compartments and monitored the hypertrophic response to the Arg403Gln missense mutation. We report that changes in Ca2+-binding protein levels occur in advance of disease, and we demonstrate that restoration of these protein levels by the L-type Ca2+ channel inhibitor diltiazem prevents clinical expression of hypertrophic cardiomyopathy in αMHC403/+ mice.
Methods
Mice.
αMHC403/+ mice were generated as described (13) and were bred and maintained on the 129/SvEv genetic background. Selected mice were treated with diltiazem added to their drinking water (450 mg/l) corresponding to 1.8 mg of diltiazem per day. Short-term studies involved treatment of mice with either enalapril, atenolol, or fludrocortisone added to their drinking water to achieve a dose of 25 mg/kg/d. All mice were maintained according to protocols approved by the Institutional Animal Care and Use Committee of Harvard Medical School.
RNA and protein analyses.
Northern blot analyses were performed as described previously (14). Total RNA was isolated from the left ventricle using Trizol (GIBCO BRL; Life Technologies Inc., Carlsbad, California, USA) and analyzed by standard Northern blot procedures using oligonucleotide probes previously described (14). Myofibrillar-rich protein extracts were made from whole hearts using a high-salt extraction method as previously described (18, 19). In brief, isolated hearts were rapidly transferred to ice-cold extraction buffer containing 50 mM KH2PO4, 70 mM NaF, 5 mM EDTA, and protease inhibitors. The samples were homogenized and centrifuged at 15,000 g for 10 minutes at 4°C. The pellet was resuspended in extraction buffer containing 1% Triton X-100 and centrifuged at 5,000 g for 5 minutes at room temperature. The resultant pellet was resuspended in extraction buffer and protein concentration determined. Samples were analyzed by standard Western blotting procedures (20). SR extracts were also prepared using differential centrifugation (21) and used for Western blot analyses. The following antibodies were used: rabbit polyclonal and mouse monoclonal anti-calsequestrin (both 1:1000; Affinity BioReagents Inc., Golden, Colorado, USA), mouse monoclonal anti-myosin (1:500; Biogenesis Ltd., Poole, United Kingdom), mouse monoclonal anti-triadin (1:500; Affinity BioReagents Inc.), and mouse anti-RyR and rabbit anti-junctin antibodies (dilutions of 1:500 and 1:250, respectively; provided by L. Jones, Indiana University School of Medicine, Indianapolis, Indiana, USA). RNA and protein blots were quantitated by densitometry (NIH Image Analysis software, version 1.60).
Immunoprecipitation of RyR.
Whole-cell homogenates were prepared as previously described (21). After determining the protein concentration, we aliquoted the supernatants and stored them at –80°C until use. Ryanodine receptor-2 (RyR2) was immunoprecipitated from cardiac samples by incubating 500 mg of homogenate with anti-RyR antibody in 0.5 ml of a modified radioimmunoprecipitation buffer (50 mM Tris-HCl [pH 7.4], 0.9% NaCl, 1.0 mM NaF, 1.0 mM Na3PO4, 0.25% Triton X-100, and protease inhibitors) overnight at 4°C. Protein A-Sepharose beads were added, incubated at 4°C for 1 hour. Immunoprecipitated proteins were separated by PAGE and analyzed for RyR2 and FKBP12.6 by immunoblotting.
Back-phosphorylation of RyR2.
RyR2 was immunoprecipitated from cardiac homogenates as described above. The protein A beads were washed with 1× phosphorylation buffer (8 mM MgCl2, 10 mM EGTA, and 50 mM Tris/PIPES, pH 6.8), and resuspended in 10 ml of a 1.5× phosphorylation buffer containing either protein kinase A (PKA) catalytic subunit (Sigma Aldrich Co., St. Louis, Missouri, USA) or PKA plus a PKA inhibitor (PKI5-24, 500 nM; Calbiochem-Novabiochem Corp., San Diego, California, USA). Back-phosphorylation of immunoprecipitated RyR2 was initiated with 33 μM Mg-ATP containing 10% [γ-32P]ATP (NEN Life Sciences, Boston, Massachusetts, USA) and terminated after 5 minutes at room temperature with 5 ml of stop solution (4% SDS and 0.25 M DTT). Samples were heated to 95°C and size-fractionated on 6% SDS-PAGE, and RyR2 radioactivity was quantified using a PhosphorImager (Molecular Dynamics, Sunnyvale, California, USA) and ImageQuant software (Amersham Pharmacia Biotech, Piscataway, New Jersey, USA). Nonspecific phosphorylation (not inhibited by PKA inhibitor) was subtracted, and the resulting value was divided by the amount of RyR2 protein (determined by immunoblotting and densitometry) and expressed as the inverse of the PKA-dependent [γ-32P]ATP signal.
Histopathology.
Cardiac tissue was treated for histologic and morphologic examination as described (13, 14). In brief, the heart was excised from wild-type and αMHC403/+ mice, washed in 37°C Dulbecco’s PBS, and arrested in 50 mM KCl. Sections from fixed, embedded tissues were stained with either hematoxylin and eosin or Masson’s Trichrome. The amount of fibrosis was quantitated using IP Lab Scientific Imaging Software (version 3.5; Scanalytics Inc., Fairfax, Virginia, USA).
Mouse echocardiography.
Echocardiographic studies were performed on mice using a 6- to 15-MHz linear array probe with a Sonos 4500 ultrasonograph (Hewlett-Packard, Andover, Massachusetts, USA) as described (14, 16). Mice were anesthetized with 2.5% tribromoethanol (0.010 ml/g), warmed with a heating pad, and attached to an ECG monitor. Left ventricular end-diastolic (LVDD) and end-systolic (LVSD) chamber dimensions and wall thickness were obtained from M-mode tracings using measurements averaged from three separate cardiac cycles. Left ventricular fractional shortening (%) was derived as follows: (LVDD – LVSD)/LVDD × 100. A single observer, who did not know the mouse’s genotype, made all echocardiographic measurements. Heart rates were determined from electrocardiographic recordings performed during echocardiography.
Cardiac hemodynamics.
Left ventricular hemodynamic studies were performed in mice as described previously (22). In brief, anesthetized mice were intubated and artificially ventilated, and real-time left ventricular pressure-volume relationships were measured using a newly developed miniaturized impedance/micromanometer catheter (Millar Instruments Inc., Houston, Texas, USA). Aortic flow was measured by an ultrasound perivascular probe (Transonic Systems Inc., Ithaca, New York, USA) placed around the thoracic aorta. Pressure-volume signals were recorded at steady-state and during transient reduction of cardiac preload achieved by occlusion of the inferior vena cava. Data were digitized at 2 kHz for subsequent analysis.
Electrocardiogram telemetry.
Electrocardiograms (ECGs) were recorded telemetrically in conscious, freely moving mice by monitoring signals from surgically implanted transmitters (model ETA-F20; Data Sciences International, St. Paul, Minnesota, USA). Studies on wild-type and αMHC403/+ age- and sex-matched mice in a 129/SvEv genetic background were started 48 hours after completion of surgery. Receiver plates and analogue ECG adapters (Data Sciences International) were used to collect data into a MacLab charts program (ADInstruments, Grand Junction, Colorado, USA).
Mouse myocyte preparation and Ca2+ measurements.
Isolated adult myocytes were prepared as described (16) with minor modifications. Hearts were digested with collagenase type I (230 U/ml; Worthington Biochemical Corp., Lakewood, New Jersey, USA) in physiological buffer containing (in mM): 126 NaCl, 4.4 KCl, 1 MgCl2, 4 NaHCO3, 10 HEPES, 11 glucose, pH 7.35, supplemented with 25 μM CaCl2 and 30 mM 2,3-butanedione (BDM). The released myocytes from both ventricles were gradually subjected to increased calcium and decreased BDM concentrations. Cells were loaded with the fluorescent Ca2+ indicator fura-2 (1 mM), and fluorescence was measured using a dual-excitation fluorescence imaging/contractility recording system (IonOptix Corp., Milton, Massachusetts, USA) according to established procedures (16). Caffeine was administered at a dose of 10 mM to produce Ca2+ release from the SR; maximum fluorescence was used as a measure of SR Ca2+ (23).
Immunohistochemistry.
Immunostaining was performed in isolated adult myocytes, which were allowed to settle on polyglycine coverslips and fixed with methanol at –20°C. Calsequestrin primary antibodies used were the same as for Western blotting at a dilution of 1:500 for both. Mouse anti-myosin antibody (Biogenesis Ltd.) was used at a dilution of 1:200. Fluorophore-labeled (Alexa Fluor 488; Molecular Probes Inc., Eugene, Oregon, USA) secondary antibodies at a dilution of 1:500–1:1000 were used for calsequestrin; a rhodamine (red-labeled) secondary antibody at a dilution of 1:200 was used for myosin. Immunostained myocytes were analyzed by both compound and confocal microscopy.
Statistical analysis.
The statistical significance of differences between groups of wild-type and αMHC403/+ mice in continuous variables was determined by unpaired Student t test and ANOVA for determination of drug and genotype effects. Data are expressed as mean ± SD. A P value less than 0.05 was considered significant.
Results
Attenuated Ca2+ responses were previously observed in αMHC403/+ mice treated with two pharmacologically distinct agents, cyclosporin A and minoxidil (16). The hypothesis was therefore made that this deficient response indicated decreased SR Ca2+ stores caused by the sarcomere gene mutation. To test this hypothesis, isolated cardiac myocytes from wild-type and untreated αMHC403/+ mice were fura-2–loaded and exposed to caffeine, which promotes SR Ca2+ release via RyR2. The amount of cytosolic Ca2+ was monitored by emission of fluorescence at specified wavelengths. A 10-mM caffeine bolus produced prompt and significant Ca2+ release into cytoplasm from wild-type myocytes (maximum height = 158.8 ± 55 units; n = 28) but a smaller response (maximum height = 133.1 ± 41 units; n = 25) from αMHC403/+ myocytes (Figure 1a and data not shown; P = 0.007). These data suggest that the SRs of wild-type myocytes contain more Ca2+ than do those of mutant myocytes.
Figure 1.

SR Ca2+ storage and altered Ca2+-related protein expression. (a) Ca2+ changes, assessed in wild-type (+/+) and αMHC403/+ (403/+) myocytes, in response to a bolus administration of 10 mM caffeine (vertical arrows indicate time of administration). (b) Western blot analysis of calsequestrin (CSQ) and components of the quaternary complex in myofibrillar protein extracts from wild-type and αMHC403/+ mice aged 30–50 weeks. Coomassie staining indicates loading of samples in each lane. Treatment of αMHC403/+ mice with diltiazem led to normalization of all four components of the RyR2 complex — calsequestrin (8 days treatment), triadin, junctin, and RyR2 (30 weeks treatment). (c) Time course study of changes in calsequestrin protein expression in equal amounts of myofibrillar extracts from mice aged 2, 4, 6, 8, 12, and 30 weeks.
Ca2+-binding proteins from whole-cell preparations, myofibrillar extracts, and SR extracts were assessed by Western blot analysis. Several proteins were specifically reduced in myofibrillar extracts from mutant myocytes as compared with wild-type extracts. The amounts of calmodulin, calcineurin (PP2B-Aα and -Aβ), SERCA2, and phospholamban in myofibrillar extracts were comparable in wild-type and αMHC403/+ myocytes (data not shown). However, calsequestrin, the primary Ca2+ storage protein in the SR, was markedly reduced in myofibrillar preparations and SR extracts of αMHC403/+ ventricles. A 56% ± 19% reduction of normal calsequestrin levels was observed in myofibrillar preparations (n = 8) from mutant mice (Figure 1b). Comparable low calsequestrin levels were observed in 15- to 20-week-old and 30- to 50-week-old αMHC403/+ mice (data not shown). Calsequestrin protein levels were then examined in younger αMHC403/+ mice. Although calsequestrin levels in myofibrillar extracts from neonates and 2-week-old mutant mice (Figure 1c) were comparable to levels found in wild-type mice, calsequestrin was decreased in extracts from 4-week-old mutant mice. This decrease in calsequestrin level paralleled the augmented expression of the mutant α isoform of cardiac myosin heavy chain, which occurs 1–2 weeks after birth (24). Notably, this timeline precedes the onset of left ventricular hypertrophy by many weeks (14, 25). These data indicate that changes in calsequestrin levels are an early molecular event in the pathogenesis of hypertrophic cardiomyopathy.
Calsequestrin forms a quaternary complex in the SR membrane, with the cardiac RyR2 channel and two other membrane proteins, junctin and triadin (26). Levels of these proteins were examined by Western blotting. Each of these components was significantly decreased in myofibrillar preparations from the SR of αMHC403/+ myocytes compared with wild-type cells (Figure 1b; decrease of 63% ± 20%, 48% ± 4%, and 38% ± 5% for RyR2, triadin, and junctin respectively; n = 3 in each group; P < 0.001 vs. wild-type). RyR2 channel phosphorylation was also assessed. (RyR2 was also decreased in SR extracts from αMHC403/+ hearts; data not shown.) PKA phosphorylation of the RyR2 channel was increased almost threefold in SR preparations from αMHC403/+ mouse hearts compared with wild-type (2.8 ± 0.2 units vs. 1.0 ± 0.1 units in wild-type; n = 3 in each group; P < 0.001). Consistent with the PKA hyperphosphorylation of RyR2 channels, coimmunoprecipitation assays showed that the amount of FKBP12.6 bound to the RyR2 channel was reduced by 40% (0.6 ± 0.1 units vs. 1.0 ± 0.1 units in wild-type; n = 3 in each group; P < 0.001). Collectively these data indicate that the RyR2 channels are both reduced in number and functionally modified in αMHC403/+ mice, such that the sensitivity of the channels to Ca2+ and the channels’ gating properties are altered.
To further evaluate SR binding proteins in αMHC403/+ myocytes, immunohistochemical studies were performed. In wild-type myocytes, antibody-labeled SR calsequestrin exists in close association with the sarcomere (27) (Figure 2a). Immunocytochemistry of calsequestrin, like myosin and other sarcomere proteins, exhibited a highly registered pattern of cross-striations, demarcated by the two nuclei characteristic of cardiac myocytes (Figure 2, a and b). αMHC403/+ myocytes are more irregularly shaped than wild-type cells and exhibited a nonuniform pattern of calsequestrin immunohistochemistry (Figure 2a). The normal alignment of calsequestrin along sarcomere cross-striations observed in wild-type cardiac myocytes was observed in approximately half of αMHC403/+ myocytes (data not shown). The remaining αMHC403/+ myocytes demonstrated no association between calsequestrin and sarcomere cross-striations, despite having normal sarcomere structure based on normal sarcomeric patterns with myosin (Figure 2, a and b) and actin antibodies (data not shown). High-power magnification of αMHC403/+ myocytes confirmed a disordered alignment of SR calsequestrin but did not show sarcomere disarray: cross-striations of the myosin-decorated sarcomere appeared normal in αMHC403/+ myocytes (Figure 2, a and b). Together with the reduction of calsequestrin levels in myofibrillar extracts, this indicates that both structural and functional alteration of SR Ca2+-binding proteins occurs in αMHC403/+ myocytes.
Figure 2.

Calsequestrin localization within myocytes. (a) Immunostaining of isolated wild-type (+/+) and αMHC403/+ (403/+) myocytes with calsequestrin (CSQ) and myosin primary antibodies. Calsequestrin primary antibodies were detected with a fluoro-conjugated secondary antibody, and myosin antibodies were visualized with a rhodamine-red secondary antibody (confocal images at ×40 magnification). (b) Corresponding images analyzed at higher magnification (×200), demonstrating loss of normal distribution of calsequestrin within αMHC403/+ myocytes.
In an attempt to correct aberrant Ca2+ handling in αMHC403/+ myocytes, several pharmacologic agents were administered. Diltiazem, an L-type Ca2+ channel inhibitor; atenolol, a β-adrenergic inhibitor; enalapril, an angiotensin-converting enzyme inhibitor; and fludrocortisone, a mineralocorticoid that alters cardiac volume, were orally administered (see Methods) to young (age 6–8 weeks) male wild-type and αMHC403/+ mice (n = 6 in diltiazem treatment group; n = 2 each for other three agents) for 7–10 days. In addition to specific effects of each drug, these pharmacologic agents alter cardiac hemodynamics. Both diltiazem and atenolol decrease heart rate; these and enalapril can also reduce blood pressure. Fludrocortisone can alter circulatory volume. Only the L-type Ca2+ channel inhibitor diltiazem affected levels of the calsequestrin complex in mutant mice, suggesting that the effect of diltiazem was independent of its effects on heart rate or blood pressure. Levels of calsequestrin, triadin, junctin, and RyR2 from myofibrillar extracts of diltiazem-treated αMHC403/+ mice were higher than those of untreated αMHC403/+ extracts (Figure 1b) and comparable to levels found in myofibrillar extracts from wild-type mice. Diltiazem restoration of calsequestrin levels was brisk, occurring within 8 days of treatment, and sustained during 30 weeks of treatment (Figure 1b). Furthermore, the amount of Ca2+ in the SR of myocytes, as measured by caffeine release (23), from diltiazem-treated wild-type and mutant hearts was not different (n = 16 wild-type myocytes and 29 mutant myocytes; P = 0.23).
Previous studies had demonstrated that diltiazem abrogated the accelerated and exaggerated hypertrophic response of αMHC403/+ mice to cyclosporin A and minoxidil (16). To ascertain the effects of diltiazem on the natural progression of disease, this calcium channel inhibitor was orally administered to male, prehypertrophic αMHC403/+ mice (n = 8 wild-type and 12 αMHC403/+) for 30 weeks. No differences were observed between treatment initiated in mice of age 6–8 weeks and in mice of age 10–12 weeks. Cardiac hemodynamics were profiled in diltiazem-treated wild-type and αMHC403/+ mice. ECG telemetry demonstrated a 13.4% reduction in heart rates of both wild-type and mutant unanesthetized mice (582 ± 25 beats per minute in untreated vs. 504 ± 1 beats per minute in diltiazem-treated; P < 0.05). Diltiazem did not change atrioventricular (PR interval) or intraventricular (QRS) conduction as compared with untreated mice, and no ventricular arrhythmias were observed (data not shown). Tail-cuff blood pressure measurements (n > 20) were compared in four conscious treated and untreated mutant mice; no differences were found in the mean systolic blood pressure (treated, 121 ± 6 mmHg; untreated, 120 ± 7 mmHg).
Invasive cardiac hemodynamic monitoring in diltiazem-treated αMHC403/+ mice demonstrated improvements in contractile (systolic) properties. Increases in end-diastolic and end-systolic volumes compared with untreated αMHC403/+ mice were observed, indicating overall improved cardiac function (Table 1). Maximal change in pressure over change in time (dP/dtmax) values and end-systolic elastance (EES) also indicated enhanced ventricular contraction with diltiazem (P < 0.001 vs. untreated αMHC403/+ mice). Measures of ventricular relaxation, e.g., time to peak filling, dP/dtmin, and tau, were unchanged in diltiazem-treated compared with untreated αMHC403/+ mice.
Table 1.
Left ventricular function of wild-type and αMHC403/+ mice aged 30–50 weeks with or without diltiazem treatment

Ventricular hypertrophy is demonstrable by two-dimensional echocardiogram at 30 weeks in αMHC403/+ mice (14, 25). We performed echocardiography in diltiazem-treated wild-type and αMHC403/+ mice at ages 30 and 39 weeks and compared left ventricular wall measurements in untreated age-, sex-, and strain-matched wild-type and αMHC403/+ mice (n = 23 total untreated). Ventricular chamber volumes, left atrial dimensions, and fractional shortening in αMHC403/+ mice were not affected by diltiazem treatment (data not shown). Left ventricular wall thickness of wild-type mice was not affected by diltiazem treatment (0.89 ± 0.03 vs. 0.87 ± 0.06 mm in untreated; P = not significant; Figure 3a). In contrast, the maximum left ventricular wall thickness of diltiazem-treated αMHC403/+ mice was significantly less than that of untreated age-matched αMHC403/+ mice at 30 weeks and at 39 weeks (1.01 ± 0.05 vs. 1.12 ± 0.07 mm; P < 0.001; Figure 3a). Wall thickness reduction was seen uniformly throughout the left ventricle, with comparable diminution of anterior and posterior left ventricular wall hypertrophy. Molecular markers of cardiac hypertrophy were also assessed. Untreated adult αMHC403/+ mice had a three- to fivefold increase in RNA expression of atrial natriuretic factor (ANF) and α-skeletal actin compared with wild-type mice (Figure 3b). In contrast, diltiazem-treated αMHC403/+ mice had approximately 50% less ANF and α-skeletal actin RNA expression (Figure 3b) than untreated mutant mice.
Figure 3.

Prevention of cardiac hypertrophy in αMHC403/+ mice. (a) Left ventricular anterior wall thickness (LVAW) was evaluated in wild-type and αMHC403/+ mice at 30–50 weeks of age using transthoracic echocardiography. A left ventricular anterior wall thickness greater than 1.00 mm, in mice 30–50 weeks of age, is considered to be hypertrophied. White bars indicate wild-type mice; black bars represent αMHC403/+ mice. *P < 0.01 vs. untreated αMHC403/+ mice. Two mice (one wild-type, one αMHC403/+ mouse) died during the study and were not included in the analysis. (b) Changes in RNA molecular markers of cardiac hypertrophy. RNA expression in hearts from αMHC403/+ mice (30–50 weeks old) compared with expression in age-matched wild-type (WT) hearts, with and without diltiazem treatment. Northern blots of atrial natriuretic factor (ANF) and α-skeletal actin expression are shown. RNA levels were standardized to 28S RNA band stained with ethidium bromide. The intensity of hybridizing species was assessed by densitometry of the Northern blots, and the RNA levels were normalized to the ANF level in wild-type left ventricle.
The pathologic hallmarks of hypertrophic cardiomyopathy — myocyte hypertrophy, disarray, and fibrosis — were also assessed in diltiazem-treated mice. Thirty-week-old untreated αMHC403/+ mice demonstrate typical histopathology of hypertrophic cardiomyopathy (13, 14). Sections that were reacted with Masson’s Trichrome stain exhibited significant fibrosis (Figure 4a). The fractional area of myocardium that reacted with Masson’s Trichrome stain was compared in histologic sections from diltiazem-treated and untreated αMHC403/+ hearts (Figure 4a). Twenty pathologic sections were analyzed from ten anatomic levels in the left ventricles of hearts. Whereas 4.07% ± 0.18% of the left ventricle was fibrotic in untreated αMHC403/+ hearts (n = 3), significantly less fibrosis (0.23% ± 0.04%) was present in diltiazem-treated αMHC403/+ hearts (n = 4; P < 0.001 vs. untreated αMHC403/+ mice). Comparison of hematoxylin and eosin–stained myocardial sections also revealed less myocyte hypertrophy and disarray in 30-week-old diltiazem-treated than in untreated αMHC403/+ mice (Figure 4b).
Figure 4.
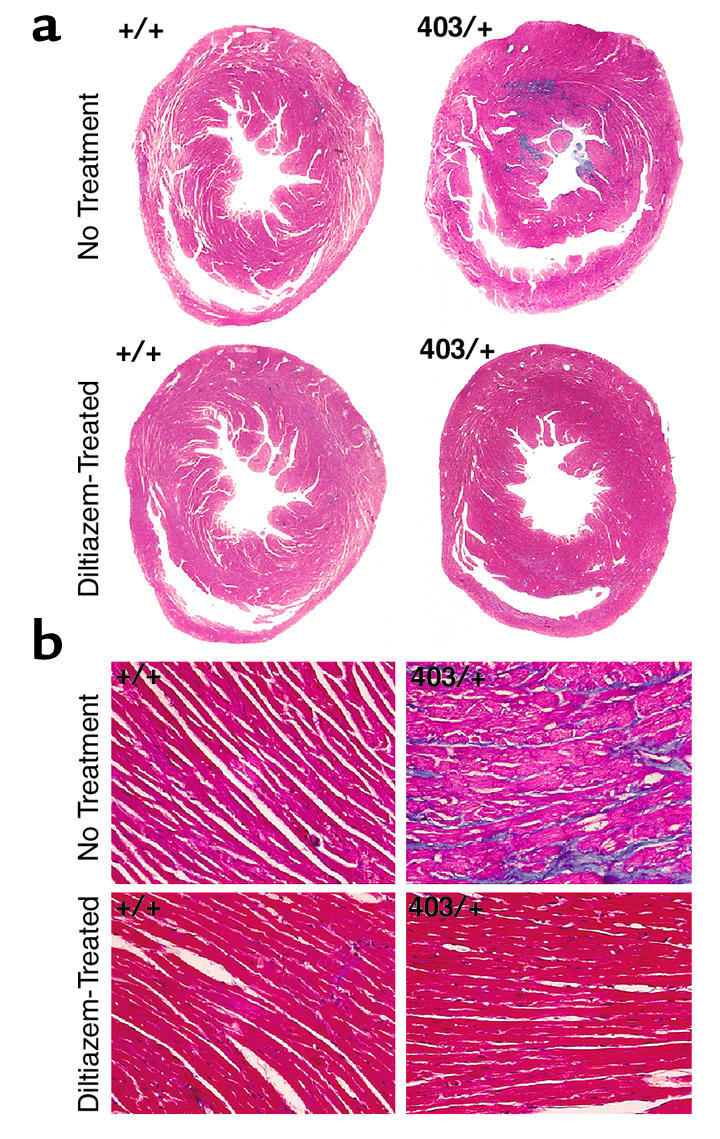
Histopathologic analyses of diltiazem-treated mice. (a) Low-magnification (×2.5) images of whole-heart sections from wild-type and αMHC403/+ mice cut transversely through the left and right ventricles, and stained with Masson’s Trichrome. Areas of fibrosis (blue) are seen in αMHC403/+ hearts that are absent in diltiazem-treated hearts. (b) High-magnification (×100) images of left ventricular sections, stained with Masson’s Trichrome. Myocyte hypertrophy, disarray, and fibrosis are present in untreated αMHC403/+ hearts; all changes are prevented with diltiazem treatment.
Discussion
This study demonstrates that abnormal SR Ca2+ responses and reduced Ca2+-binding proteins are early events in the pathogenesis of hypertrophic cardiomyopathy in αMHC403/+ mice. Caffeine-induced release of SR Ca2+ stores and levels of calsequestrin, the Ca2+ channel RyR2, and associated peptides junctin and triadin were decreased in advance of histopathology or echocardiographic evidence of cardiac hypertrophy in αMHC403/+ mice. Restitution of normal Ca2+ storage protein levels occurred in response to diltiazem-mediated inhibition of L-type Ca2+ channels, and when diltiazem was administered early to αMHC403/+ mice, the cardiac histopathology and hemodynamic features of this sarcomere protein mutation were significantly abated.
Previous studies demonstrated that levels of cytosolic Ca2+ in αMHC403/+ myocytes are comparable to those found in wild-type cells (16). However, responses to pharmacologic agents such as cyclosporin A or minoxidil, which produce a brisk (30%) increase in diastolic Ca2+ levels in wild-type myocytes, are attenuated (<8% increases) in αMHC403/+ myocytes (16). This diminutive response by αMHC403/+ myocytes is explained by the current studies. In response to the potent activation of RyR2 channels by caffeine, the maximum and total Ca2+ release from the SR was less in αMHC403/+ myocytes than in wild-type myocytes. This suggests that the SRs of αMHC403/+ myocytes contain lower levels of Ca2+ than do the SRs of wild-type myocytes (Figures 1 and 2).
Because αMHC403/+ myocyte SRs contained reduced amounts of Ca2+, we hypothesized that SR Ca2+ storage proteins would correspondingly be diminished. Decreased levels of calsequestrin and RyR2 in the SRs of αMHC403/+ myocytes were found. Calsequestrin is a 55-kDa high-capacity Ca2+-binding protein that both serves as a reservoir for SR Ca2+ and, with junctin and triadin, forms a complex that regulates Ca2+ release via RyR2 (26, 28, 29). Downregulation of calsequestrin and the other components of this quaternary RyR2 complex in αMHC403/+ myocytes is consistent with decreased SR Ca2+ levels. SR Ca2+ levels may further be depleted by increased phosphorylation of the RyR2 channel in αMHC403/+ myocytes. Phosphorylation decreases the amount of RyR2-associated FKBP12.6, which makes the channel more sensitive to calcium-induced activation and increases the probability of channel opening (21, 30, 31). Whereas only one of three potential sites was phosphorylated in wild-type myocytes, most RyR2 channels from αMHC403/+ myocytes had two or more phosphorylated sites.
Reduced SR Ca2+ levels but normal cytosolic Ca2+ levels in αMHC403/+ myocytes are consistent with the model that the mutant sarcomere functions like an ion trap (Figure 5). Altered biophysical properties of a mutated myosin heavy chain could enhance Ca2+ retention by the sarcomere and deplete SR Ca2+ stores. Observations of αMHC403/+ muscle physiology support this model: mutant fibers show greater-than-normal isometric tension development at submaximal Ca2+ levels, and mutant hearts exhibit decreased rates of relaxation (15, 32).
Figure 5.

Model of Ca2+ cycling in cardiac myocytes and the effects of a hypertrophic cardiomyopathy–causing mutation. (a) Wild-type myocytes showing normal Ca2+ regulation. Ca2+ enters the myocyte through L-type Ca2+ channels. Small entry of Ca2+ stimulates Ca release (calcium-induced Ca2+ release; CICR) from the SR via cardiac ryanodine receptors (RyR2) to the sarcomere. Ca2+ returns to the SR via the sarcoplasmic/endoplasmic Ca2+ ATPase (SERCA) pump, which is regulated by phospholamban (PLB). Ca2+ cycling is “balanced” between the sarcomere and SR. (b) Mutant (αMHC403/+) myocytes have a mutation in the sarcomere (represented by an asterisk). The defective sarcomere acts as an ion trap, resulting in accumulation of Ca2+. Less Ca2+ returns to the SR, resulting in decreased SR Ca2+ stores, decreased SR calsequestrin (CSQ), and reduced expression of RyR2. The net effect is a Ca2+ shift within the cell, with a relative Ca2+ excess in the sarcomere, and Ca2+ depletion in the SR.
The development of SR Ca2+ dysregulation follows mutant peptide expression and predates the hypertrophic response in αMHC403/+ mice. By 4 weeks after birth, when αMHC403/+ myocytes express equal amounts of mutant and wild-type peptide (24), dysregulation of SR calcium was apparent, and if it went uninterrupted, phenotypic manifestations of hypertrophic cardiomyopathy ensued (13, 14). L-type channel inhibition by diltiazem most likely interrupts this process by chronic attenuation of calcium-induced SR Ca2+ release, so as to limit Ca2+ sequestration by the mutant sarcomere. The concurrent restoration of normal RyR2 and SR Ca2+-binding proteins levels and reduction of cardiac hypertrophy and fibrosis in diltiazem-treated αMHC403/+ mice further support a mechanistic link between SR Ca2+ dysregulation and disease phenotype.
The L-type Ca2+ channel is the primary target of diltiazem. However, diltiazem is known to have other cellular targets, such as the mitochondrial Na+/Ca2+ exchanger. Alteration in mitochondrial Ca2+ concentration might change αMHC403/+ myocyte energetics and thereby alter the hypertrophic response of the heart. Diltiazem might reduce the hypertrophic response by improving cardiac energetics. However, under normal conditions, diltiazem-treated αMHC403/+ hearts appear to do less work (lower heart rate, reduced stiffness [EES], and reduced dP/dtmax; see Table 1) than the untreated αMHC403/+ heart, suggesting that even if cardiac energetics remain unaltered in αMHC403/+ hearts, the hearts should benefit from diltiazem-mediated work reduction.
Other investigators have identified several important intracellular Ca2+ sensors that can activate several signaling pathways to effect myocyte growth and function (for review, see ref. 33). Based on its association with the SR and function in RyR2 channel phosphorylation, calcium/calmodulin–dependent protein kinase (CamKII) is a particularly intriguing candidate for signaling myocyte growth (34). Levels of two well-characterized signaling molecules, calmodulin and the calcium/calmodulin–activated phosphatase calcineurin (PP2B-Aα and -Aβ), were not altered in αMHC403/+ mice (data not shown). Whether transient or compartmentalized changes in these or Ca2+ activation of other signaling molecules triggers hypertrophy in αMHC403/+ mice remains unknown.
An important conclusion from these data is that early restoration of SR Ca2+ decreases the hypertrophic response to a sarcomere mutation. Administration of diltiazem, in doses resulting in only modest hemodynamic effects, to prehypertrophic αMHC403/+ mice improved cardiac function and provided long-term (39 weeks) attenuation of hypertrophic pathology. Improvement in cardiac function was almost exclusively in systolic function parameters, with little effect on diastolic function (Table 1). This may suggest that altering Ca2+ handling is important for contractile function, but that diastolic function is mediated by alternate mechanisms. In humans with clinical evidence of hypertrophic cardiomyopathy, L-type Ca2+ channel inhibitors such as diltiazem are used to treat symptoms. Our findings indicate that greater benefit should occur if these inhibitors are administered in advance of disease expression. In conjunction with gene-based, preclinical diagnosis of human sarcomere protein mutations, evaluation of the potential for diltiazem, and other pharmacologic agents that restore SR Ca2+ levels, to inhibit development of hypertrophic cardiomyopathy appears warranted.
Acknowledgments
We thank Larry R. Jones for providing the RyR2 and junctin antibodies, and Julie Zhang for myocyte isolation. The Howard Hughes Medical Institute supported these studies. C. Semsarian is the recipient of a National Heart Foundation of Australia grant. U. Mende is supported by an NIH Specialized Centers of Research grant and an American Heart Association Scientist Development grant. I. Ahmad is supported by a Sarnoff Fellowship.
References
- 1.Fatkin, D., Seidman, J.G., and Seidman, C.E. 2000. Hypertrophic cardiomyopathy. In Cardiovascular medicine. J.T. Willerson and J.N. Cohn, editors. W.B. Saunders Co. Philadelphia, Pennsylvania, USA. 1055–1074.
- 2.Seidman, C.E., and Seidman, J.G. 2001. Hypertrophic cardiomyopathy. In The metabolic and molecular bases of inherited disease. C.R. Scriver, A.L. Beaudet, W.S. Sly, and D. Valle, editors. McGraw-Hill. New York, New York, USA. 5433–5452.
- 3.Maron BJ, Bonow RO, Cannon RO, III, Leon MB, Epstein SE. Hypertrophic cardiomyopathy. Interrelations of clinical manifestations, pathophysiology, and therapy (1) N Engl J Med. 1987;316:844–852. doi: 10.1056/NEJM198704023161405. [DOI] [PubMed] [Google Scholar]
- 4.Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775–785. doi: 10.1056/NEJM199703133361107. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Epstein SE, Roberts WC. Causes of sudden death in competitive athletes. J Am Coll Cardiol. 1986;7:204–214. doi: 10.1016/s0735-1097(86)80283-2. [DOI] [PubMed] [Google Scholar]
- 6.Maron BJ, et al. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. JAMA. 1996;276:199–204. [PubMed] [Google Scholar]
- 7.Vikstrom KL, Factor SM, Leinwand LA. Mice expressing mutant myosin heavy chains are a model for familial hypertrophic cardiomyopathy. Mol Med. 1996;2:556–567. [PMC free article] [PubMed] [Google Scholar]
- 8.Kittleson MD, et al. Familial hypertrophic cardiomyopathy in maine coon cats: an animal model of human disease. Circulation. 1999;99:3172–3180. doi: 10.1161/01.cir.99.24.3172. [DOI] [PubMed] [Google Scholar]
- 9.Marian AJ, et al. A transgenic rabbit model for human hypertrophic cardiomyopathy. J Clin Invest. 1999;104:1683–1692. doi: 10.1172/JCI7956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oberst L, et al. Dominant-negative effect of a mutant cardiac troponin T on cardiac structure and function in transgenic mice. J Clin Invest. 1998;102:1498–1505. doi: 10.1172/JCI4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel R, et al. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation. 2001;104:317–324. doi: 10.1161/hc2801.094031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lim DS, et al. Angiotensin II blockade reverses myocardial fibrosis in a transgenic mouse model of human hypertrophic cardiomyopathy. Circulation. 2001;103:789–791. doi: 10.1161/01.cir.103.6.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geisterfer-Lowrance AA, et al. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–734. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]
- 14.McConnell BK, et al. Comparison of two murine models of familial hypertrophic cardiomyopathy. Circ Res. 2001;88:383–389. doi: 10.1161/01.res.88.4.383. [DOI] [PubMed] [Google Scholar]
- 15.Blanchard E, Seidman C, Seidman JG, LeWinter M, Maughan D. Altered crossbridge kinetics in the αMHC403/+mouse model of familial hypertrophic cardiomyopathy. Circ Res. 1999;84:475–483. doi: 10.1161/01.res.84.4.475. [DOI] [PubMed] [Google Scholar]
- 16.Fatkin D, et al. An abnormal Ca2+ response in mutant sarcomere protein-mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 18.Kim SJ, et al. An α-cardiac myosin heavy chain gene mutation impairs contraction and relaxation function of cardiac myocytes. Am J Physiol. 1999;276:H1780–H1787. doi: 10.1152/ajpheart.1999.276.5.H1780. [DOI] [PubMed] [Google Scholar]
- 19.McConnell BK, Moravec CS, Bond M. Troponin I phosphorylation and myofilament calcium sensitivity during decompensated cardiac hypertrophy. Am J Physiol. 1998;274:H385–H396. doi: 10.1152/ajpheart.1998.274.2.H385. [DOI] [PubMed] [Google Scholar]
- 20.McConnell BK, et al. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–1244. doi: 10.1172/JCI7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marx SO, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 22.Georgakopoulos D, et al. The pathogenesis of familial hypertrophic cardiomyopathy: early and evolving effects from an α-cardiac myosin heavy chain missense mutation. Nat Med. 1999;5:327–330. doi: 10.1038/6549. [DOI] [PubMed] [Google Scholar]
- 23.Palmer BM, Lynch JM, Snyder SM, Moore RL. Renal hypertension prevents run training modification of cardiomyocyte diastolic Ca2+ regulation in male rats. J Appl Physiol. 2001;90:2063–2069. doi: 10.1152/jappl.2001.90.6.2063. [DOI] [PubMed] [Google Scholar]
- 24.Fatkin D, et al. Neonatal cardiomyopathy in mice homozygous for the Arg403Gln mutation in the α-cardiac myosin heavy chain gene. J Clin Invest. 1999;103:147–153. doi: 10.1172/JCI4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Semsarian C, et al. A polymorphic modifier gene alters the hypertrophic response in a murine model of familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2001;33:2055–2060. doi: 10.1006/jmcc.2001.1466. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389–23397. doi: 10.1074/jbc.272.37.23389. [DOI] [PubMed] [Google Scholar]
- 27.Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones LR, et al. Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J Clin Invest. 1998;101:1385–1393. doi: 10.1172/JCI1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niki I, Yokokura H, Sudo T, Kato M, Hidaka H. Ca2+ signaling and intracellular Ca2+ binding proteins. J Biochem (Tokyo) 1996;120:685–698. doi: 10.1093/oxfordjournals.jbchem.a021466. [DOI] [PubMed] [Google Scholar]
- 30.Szegedi C, Sarkozi S, Herzog A, Jona I, Varsanyi M. Calsequestrin: more than ‘only’ a luminal Ca2+ buffer inside the sarcoplasmic reticulum. Biochem J. 1999;337:19–22. [PMC free article] [PubMed] [Google Scholar]
- 31.Yano K, Zarain-Herzberg A. Sarcoplasmic reticulum calsequestrins: structural and functional properties. Mol Cell Biochem. 1994;135:61–70. doi: 10.1007/BF00925961. [DOI] [PubMed] [Google Scholar]
- 32.Spindler M, et al. Diastolic dysfunction and altered energetics in the αMHC403/+mouse model of familial hypertrophic cardiomyopathy. J Clin Invest. 1998;101:1775–1783. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frey N, McKinsey TA, Olson EN. Decoding calcium signals involved in cardiac growth and function. Nat Med. 2000;6:1221–1227. doi: 10.1038/81321. [DOI] [PubMed] [Google Scholar]
- 34.Passier R, et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]