

Abstract
Water in the hydrophobic cavity of human interleukin 1β, which was detected by NMR spectroscopy but was invisible by high resolution x-ray crystallography, has been mapped quantitatively by measurement and phasing of all of the low resolution x-ray diffraction data from a single crystal. Phases for the low resolution data were refined by iterative density modification of an initial flat solvent model outside the envelope of the atomic model. The refinement was restrained by the condition that the map of the difference between the electron density distribution in the full unit cell and that of the atomic model be flat within the envelope of the well ordered protein structure. Care was taken to avoid overfitting the diffraction data by maintaining phases for the high resolution data from the atomic model and by a resolution-dependent damping of the structure factor differences between data and model. The cavity region in the protein could accommodate up to four water molecules. The refined solvent difference map indicates that there are about two water molecules in the cavity region. This map is compatible with an atomic model of the water distribution refined by using xplor. About 70% of the time, there appears to be a water dimer in the central hydrophobic cavity, which is connected to the outside by two constricted channels occupied by single water molecules ≈40% of the time on one side and ≈10% on the other.
Analysis of NMR data from solutions of human interleukin-1β (hIL-1β) has indicated that the hydrophobic cavity in the center of the protein contains positionally disordered water (1). However, this water has escaped detection by four independent high resolution (≈2 Å) crystallographic studies (2–5). On the basis that no water molecules were observed crystallographically in this or similar hydrophobic protein cavities, Matthews et al. (6) have questioned the interpretation of the observed nuclear Overhauser enhancement cross peaks attributed to water molecules that are within ≈5 Å of protons of the cavity-forming aliphatic residues.
Because the existence of positionally disordered water in hydrophobic protein cavities has been controversial (and because such mobile water can be crucially involved in protein dynamics), we have critically assessed the information available from x-ray crystallography regarding the distribution of disordered solvent in an hIL-1β crystal. Refinement of the phases for all of the low resolution data, from 55- to 4.5-Å spacing, restrained by the information about the well ordered protein structure from the high resolution (2.3 Å) data, demonstrates that crystallographically elusive solvent, which is detectable by NMR, can be visualized quantitatively in a difference electron density map. The solvent density distribution is mapped from the difference between the average structure of the whole crystal, calculated from the completely phased data, and that of the atomic model of the ordered protein structure, refined from the high resolution data.
Atomic coordinates, occupancies, and temperature factors in the ordered part of a crystal are determined from the position, magnitude, and shape of the peaks in an electron density map calculated from the observed high resolution diffraction amplitudes and the refined phases. Fluctuation in the position of an atom smears out its average density distribution; the effect on the diffraction pattern is to progressively reduce the contribution of the atom to the amplitude of Bragg reflections with increasing resolution. Atoms with rms positional fluctuations >1 Å, corresponding to Debye temperature factors B >80 Å2, will make negligible contributions to Bragg reflections at a resolution finer than ≈4.5 Å. High resolution protein crystallographic analysis, which conventionally neglects data below ≈6- to 10-Å resolution, can identify bound water molecules located in well defined positions but would not detect more mobile water localized in cavities or near surface exposed nonpolar groups.
NMR measurements can detect water protons that remain close to protons of a protein, regardless of the exact position of water relative to fixed protein protons, provided that the fixed protons are further than ≈5 Å from rapidly exchangeable ones. Of the 29 ordered water molecules consistently located in all four crystallographic models of hIL-1β (7), 10 of the 19 bound water molecules potentially identifiable by NMR spectroscopy actually were observed (1). In addition, positionally disordered water was detected associated with the surface-exposed methyl groups as well as the crystallographically elusive water in the hydrophobic cavity.
We have refined phases for all of the diffraction data below ≈4.5 Å resolution from an hIL-1β crystal by applying the restraint that the map calculated from the difference between the phased experimental data and the atomic model structure factors should be flat within the envelope of the ordered protein structure (8, 9). The reliability of this refinement strategy depends on the correctness of the atomic model and the accuracy of the measured diffraction data; furthermore, care must be taken to damp spurious peaks in the difference map caused by noise in the higher resolution data, which could lead to an unrealistically overfitted representation of the solvent density distribution (10, 11).
Data and Difference Map Refinement
Diffraction data from a single hIL-1β crystal to 2.3-Å spacing were collected, including all of the low resolution data. From this data (Table 1), two atomic models of the protein, including well ordered bound solvent, were refined with good statistics, starting from two of the published models (2, 3). The differences between these two refinements are comparable to the differences among the re-refinements by Ohlendorf (7) of the four independently determined atomic models (2–5) against a single data set. In accord with the previous crystallographic analyses, there is no indication of ordered water in the cavity of the models of the hIL-1β that we have refined against data from 15- to 2.3-Å resolutions. The volume of the cavity can accommodate up to four water molecules without bad contacts but, including two or three water molecules with B factors of ≈70 Å2, produced a statistically insignificant decrease in the R factor. However, this R factor does not provide a meaningful test of the possible presence of disordered water inside the protein.
Table 1.
Data statistics and atomic model statistics
| Data statistics | |
| Unit cell: a = b = 55.02 Å; c = 77.16 Å, Spacegroup P43 | |
| Resolution range | 55–2.28 Å |
| Observed reflections | 10,313 |
| Completeness 55–2.37 Å | 100% |
| Completeness 2.37–2.28 Å | 86% |
| Rsym 55-4.74 Å | 0.046 |
| Rsym all data | 0.056 |
| Atomic model statistics | ||
| Model A | Model B | |
| Bond length rms | 0.013 Å | 0.013 Å |
| Bond angle rms | 2.13° | 2.16° |
| Water molecules | 55 | 55 |
| Rcrys | 15.7% | 15.9% |
| Rfree | 21.3% | 21.6% |
Human recombinant interleukin 1β was crystallized from 2.4 M ammonium sulfate, 1 mM dithiothreitol, and 100 mM Hepes at pH 8.5 by hanging drop vapor diffusion. Data were collected on an RAXIS-II imaging-plate detector by using copper Kα radiation. All data were collected from a small single crystal (0.3 × 0.3 × 0.2 mm) at room temperature in a single sweep of 45 2° oscillation frames. The relatively small size of the crystal ensured that none of the strong low resolution reflections were saturated, but, consequently, reflections beyond 2.3-Å resolution were too weak to be measured reliably. The data were processed with the denzo/scalepack package (24). The care taken in recording all of the low resolution data provided intensity measurements that were accurately on the same intensity scale as the weaker high resolution measurements. The model coordinates were refined against data from 15 to 2.3-Å resolution by using the tnt least-square package (25, 26) with correlated thermal factor restraints. Because of the availability of independently refined models, we carried out refinements with two different starting models. The coordinates for the initial models were taken from the Protein Data Bank (27). Model A was based on the coordinates from Priestle et al. (2) file 2ILB, and Model b was based on coordinates from Finzel et al. (3) file 1ILB. The number of water molecules was limited to those 55 that appeared clearly in the maps from both starting models. The rms differences between the initial and final coordinates of Cα atoms for the two models refined against our data were 0.3 Å, and the R factor comparing these two models was 13%.
The plot of the R factor comparing calculated structure factors from a refined atomic model of the hIL-1β molecular structure in a vacuum against the complete crystal diffraction data as a function of resolution (Fig. 1) shows a progressive increase in discrepancy for reciprocal spacings smaller than 1/5Å. This disparity below 1/7Å reciprocal spacing is reduced substantially by introducing a uniform density model for the solvent in the space not occupied by protein, as demonstrated with other protein crystals (8–10, 12, 13). Envelopes for the solvent-accessible space were generated with occlusion radii of 0.6, 1.0, and 1.4 Å from the tight mask of the atomic model defined by the van der Waals radii of the protein atoms and 55 well ordered bound water molecules. The different solvent envelopes, when filled with a uniform electron density of 0.38 electrons/Å3 (as calculated for the crystal solvent composition), gave very similar calculated phases for data below 4.5-Å resolution. The envelope filled with the uniform density solvent that gave the best agreement with the estimated number of solvent electrons in the unit cell was the 1.0-Å occlusion radius.
Figure 1.

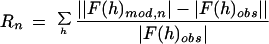 |
To calculate electron density maps on an absolute scale, the experimentally measured structure factors, Fhkl, were scaled to the calculated structure factors from the refined atomic model in the resolution range 4.5–2.3 Å, and the value of F000 was set equal to the estimated total number of electrons in the unit cell. The disordered solvent makes a negligible contribution to the high resolution diffraction data; thus, these Bragg reflections are well represented by the Fourier transform of the atomic model. Because all of the diffraction data were collected in a single sweep and care was taken to avoid saturation of any of the strong low resolution reflections by using a small crystal, the low resolution data were recorded on exactly the same intensity scale as the high resolution data. Thus, the scale factor, which fits the high resolution structure factors to those of the model in units of electrons, puts all of the measured data on an absolute scale.
The value of F000 is equal to the sum of the number of electrons in the ordered structure, represented by the atomic model, plus the electrons in the disordered solvent. The number of solvent electrons was estimated from the calculated solvent density and volume. A more direct experimental measure of the number of solvent electrons was obtained from the refined difference map calculated from the complete phased data minus the model protein structure factors. This difference should be zero within the envelope of the ordered structure when F000 is scaled properly. The total number of electrons in the difference map representing the solvent density distribution was defined by the constraint that the mean value within the envelope of the ordered structure must be zero. The correction to the value of F000 based on the initial estimate of the solvent content was <2% when the refinement of the difference map converged.
The starting model for the refinement traced in Fig. 1 was the electron density map generated from the atomic coordinates of model A (Table 1) with a flat solvent density beyond the envelope enclosing the ordered protein and bound water structure. This envelope was generated by contouring the map of the atomic model [constructed by using the sfall program in the Collaborative Computing Project 4 Suite (14)] to contain 40.7% of the crystal volume, corresponding to the sum of the canonical volumes of the constituent amino acid residues (15, 16) and bound solvent. The total number of electrons within the envelope of the ordered structure was scaled to include the contribution of all hydrogen atoms and to correct for the truncation of the Gaussians representing the density of the individual atoms. Flat solvent of density 0.38 electrons/Å3 was added to pixels in the map that are at least 1 Å beyond the outside envelope of the ordered structure but the cavity inside the protein was left empty.
The first step in the solvent density modification refinement was to compute a smoothed difference map from the damped structure factor differences between the starting model and the experimental data. The starting model structure factors are the Fourier transform (FT) of the initial model density distribution, ρo(r), at coordinates r for all pixels in the unit cell: FT(ρo(r)) = F(h)mod,o exp(iφo(h)), where h denotes the indices hkl of the individual structure factor terms, and φo(h) are the phase angles. Analytically, the initial cycle smoothed difference density distribution is:
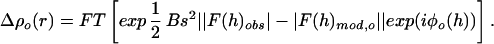 |
B is a Debye damping factor, and s is the reciprocal space coordinate. The phase angles for the initial difference amplitudes ΔFo(h) are φo(h) as calculated from the flat solvent model for Bragg spacings s < 1/4.5 Å, but for s ≥ 1/4.5 Å, φ(h) were fixed for all cycles of solvent density modification at the values for the refined high resolution model.
The phases from the atomic model were maintained beyond 4.5-Å resolution because, as indicated by the R factor measurements (Fig. 1), density fluctuations in the bulk solvent density make little contribution to the measured higher resolution diffraction data, and the damping factor was introduced to reduce the contribution of the higher spacial frequency noise. Without this damping and phase restraint, the difference map flattening refinement (8) overfits the high resolution data (10). For our refinement (Fig. 1), the Debye B factor was set at 100 Å2, which reduces the difference amplitudes, ΔF(h), by 0.36 at 7-Å spacing, by 0.085 at 4.5-Å, and by 0.004 at 3-Å spacing.
In the first cycle of refinement, the difference density Δρo(r) was added to all pixels of the initial model map with coordinates r′ outside the tight envelope enclosing the atomic model of the ordered structure. This generates a first cycle modified map: ρ1(r) = ρ0(r) + Δρo(r′). The modified map then was Fourier transformed to generate a first cycle set of model structure factors, F(h)mod,1, and the differences, |ΔF1(h)| = ||F(h)obs| − |F(h)mod,1||, were damped and used with phases φ1(h) for s < 1/4.5 Å to calculate Δρ1(r). This process then was iterated to generate an nth cycle modified map ρn(r) = ρn-1(r) + Δρn-1(r′), and, at each cycle, the R factor comparing F(h)obs and F(h)mod,n was evaluated as a function of resolution.
In Fig. 1, the R factor is plotted for the atomic model of hIL-1β in a vacuum and for this model with the 1st and 20th cycles of modification of the solvent density. After 15 cycles of refinement, the R factor below 4.5-Å resolution had decreased to <10%, and further iteration produced no significant improvement. In the course of refinement, phases for the low resolution data did not change substantially compared with the initial model with uniform density solvent. For example, only 6 of 42 reflections with s < 1/15 Å had phase differences Δφ > 20°, and the mean Δφ for these reflections was 29°. Solvent density modification refinements starting with an alternate atomic model of the ordered structure generated similar solvent density maps. Thus, the small adjustments in the phases of the measured low resolution amplitudes, which produce a physically plausible map of the coarse solvent density distribution, are not very sensitive to fine details of the starting model.
Refined Solvent Density Distribution
The map of the complete density distribution in the hIL-1β unit cell calculated from our refinement is ρtot(r) = FT[ F(h)obs exp(iφ(h))]. The solvent density distribution is the difference between ρtot(r) and ρatm(r), the map of the atomic model. This difference map encompasses all of the crystal solvent except for the 55 ordered bound water molecules in the atomic model. In calculating this solvent difference map, a temperature factor B = 15 Å2 was applied to the ΔF values to smooth out high spatial frequency ripples caused by truncation at the 2.3-Å spacing of the relatively noisy high resolution data. This temperature factor is much smaller than that used for the solvent density modification refinement to retain the more detailed information about the solvent density distribution, which is implicit in the amplitudes of the moderate and higher resolution diffraction data. The histogram of the densities in the final solvent map confirmed that the damping restraints applied in the phase refinement avoided unrealistically large negative or positive density values. Beyond the surface of the protein envelope, the distinctive features in the solvent density map resemble those in the solvent maps determined for other proteins (8–10, 17). The critical portion of the refined solvent map of hIL-1β for this study is the density distribution within the hydrophobic cavity.
Electrons in the Cavity
To display the density within the cavity, without superposition of bulk solvent outside the protein surface, the solvent map was truncated beyond a radius of 10 Å centered on the middle of the cavity (Fig. 2A). Four phenylalanine, one valine, one isoleucine, and six leucine residues define the cavity, which is connected to the surface by narrow channels. If these channels were rigid, the constrictions would not allow passage of a water molecule. To integrate the total number of solvent electrons inside the protein, the cavity region was defined by that portion of the solvent map within a central sphere of 6-Å radius (Fig. 2 B and C).
Figure 2.

Difference map of the density distribution within the cavity region of the hIL-1β molecule that is not accounted for by the refined atomic model. (A) The difference electron density map within a sphere of 10-Å radius is displayed inside the Cα trace of an hIL-1β molecule viewed from the side containing the C terminus (center) and N terminus (top), with ball and stick representations of the cavity forming residues. The methyl groups of the aliphatic residues are drawn as large tan spheres, and the aromatic carbons are smaller, light blue spheres. Every tenth Cα atom is labeled, except 120, which is obscured behind the cavity density map. The red contours contain 70% of the total of 18 solvent electrons integrated in the cavity region, and the blue contours contain 50% of these electrons. (B and C) Stereopair images of the cavity solvent density within a sphere of 6-Å radius are viewed from the front (B) and back (C) of the orientation shown in A. The six leucine (L) one isoleucine (I), one valine (V), and four phenylalanine (F) residues forming the cavity are labeled. The green contours superimposed on the solvent difference map mark the envelope of the atomic model of the partially occupied water molecules in the cavity, refined with the protein model by xplor. This model, which contains 18.5 electrons, is contoured at the 70% level. The difference in shape of the experimental solvent difference map (red and blue) based on refinement of phases for the low resolution diffraction data and the xplor model of the cavity water (green) based on refinement of a single-conformer protein model may be caused by fluctuations in the cavity shape or, alternatively, to noise in diffraction data. The graphics were created with o (28), molscript (29), and rayshade (30).
After 20 cycles of refinement, there were 18 electrons inside the cavity region of the solvent map. In our initial flat solvent model, there were no electrons in the cavity. After the first cycle of refinement, which adds the calculated Δρo(r′) to all pixels outside the tight envelope of the atomic model, there were 11 electrons in the cavity region. Thus, more than half of the final content of 18 electrons in the cavity obtained after 20 cycles of refinement was accounted for in the first cycle map, calculated by using phases for the low resolution data from the flat solvent model. Starting with a flat solvent map model containing 40 electrons in the cavity region, corresponding to the maximum of four water molecules that could fit in the available space, the refinement procedure reduced this content to 25 electrons on convergence after 20 cycles. These calculations, starting with the cavity empty or overfull, suggest that the number of solvent electrons inside the protein is between 18 and 25, corresponding to an average of about two water molecules. If the cavity were actually empty, it should at least be possible to force the refinement to produce a null map in this region. However, restraining the cavity to be empty at each cycle of refinement only reduced the electron count to 6 in the final difference map, which still showed positive features similar to those displayed in Fig. 2 in the cavity region.
The small number and delocalized distribution of electrons in the cavity make their contribution to the high resolution Fourier transform negligible. Below 4.5-Å resolution, the calculated mean amplitude due to solvent in the cavity is ≈1% of the mean structure factors. The R factors comparing the low resolution data with refined models containing and excluding solvent in the cavity were, respectively, 9.5 and 9.7%. Thus, the overall R factor comparing models with and without electrons in the cavity is an insensitive test of significance. A more critical test is to compute R factors only for the relatively weak low-resolution reflections to which the solvent density in the cavity does make a significant contribution. Comparing structure factors calculated for the 1,383 reflections below 4.5-Å resolution from the refined models containing and excluding solvent in the cavity, 36 structure factors were identified for which the model amplitudes differed from each other by >7%. The R factor comparing these two subsets of model data with each other was 13%, and comparing each model with the experimental data gave R factors of 25% for that with solvent in the cavity and 32% for that without. The noise level is high for these weak reflections, but the noise contribution to the R factor should be uncorrelated with the difference caused by any cavity solvent contribution. The square root of the difference of the squares of the R factors for the two models, which is 20%, may be a statistically more significant measure than the direct 7% difference of the better fit with the data of the model containing solvent in the cavity compared with that without.
Conventionally, most protein crystallographic measurements do not include data below ≈20-Å resolution. To test the importance of the very low resolution data in counting solvent electrons, we carried out a refinement in which the experimental data for the 17 structure factors below 20-Å resolution were replaced with those calculated for the flat solvent model. As previously noted, the phases refined for the experimental data below 15-Å resolution did not differ substantially from those for the initial flat solvent model. However, the calculated amplitudes from the flat solvent model for these 17 terms do differ substantially from the data, as indicated by the initial R factor of 75%. In the course of the refinement, starting with the 17 model low-order structure factors in place of data, flattening the difference map inside the protein envelope had little effect on the calculated amplitudes of these terms, which were allowed to float (ΔF = 0 at each cycle) as in a free R factor calculation. The final R factor for these terms compared with the data was 65%. The shape of the positive density features in the cavity region of the refined map starting with synthetic low resolution data was similar to that obtained with real data, but the integrated cavity density amounted to 35 electrons. This unrealistically large value resulted because the positive contribution of the modeled low spatial frequency Fourier coefficients at the middle of the protein was inflated compared with the data. The sensitivity of the electron counting to the amplitudes of the low order structure factors demonstrates the need for accurate intensity measurement of all of the low resolution reflections to reliably evaluate the magnitude of diffuse features in an experimental electron density map.
The small disconnected density peaks inside the protein envelope of the difference map (Fig. 2), which should be zero, are caused predominantly by noise in the higher resolution diffraction data, whose phases were not adjusted in the refinement procedure. (The largest of the peripheral features at the right side of Fig. 2A corresponds to part of a water-accessible crevice, which is encompassed within the displayed 10-Å radius sphere of density.) Negative features inside the envelope, which are not displayed, are comparable to the small positive peaks. The SD of the electron count inside the protein envelope of the difference map, calculated by integration over volume elements equal to that of the cavity, is ±4 electrons. This noise level provides an estimate of the uncertainty in the value of 18 electrons measured inside the cavity volume.
Simulated Water Arrangement in Cavity
A more conventional crystallographic model of the distribution of water in the cavity formed by the eight aliphatic and four aromatic side chains of the atomic model was mapped by using xplor (18). A starting model was constructed that consisted of 25 partially occupied sterically accessible water sites on a regular grid of 1 Å, each with an occupancy of 0.09 to give a total of 22.5 electrons uniformly distributed in the cavity volume. Occupancies of the water sites were allowed to change during the energy minimization, with all interactions among water sites turned off and with temperature factors maintained at ≈70 Å2. In the course of the energy minimization, the side chains of the protein residues defining the cavity did not move significantly, but the occupancies of the water sites that were in energetically unfavorable positions dropped to zero and those of a few favored sites increased. There were three water-accessible regions left in the central domain of the cavity, which form an approximate equilateral triangle of side-length of ≈2 Å with occupancies of 0.29, 0.64, and 0.43. This distribution most plausibly corresponds to about two-thirds occupancy of a hydrogen-bonded water dimer in two possible orientations, with rms fluctuations in position of ≈1 Å. There were also two auxiliary sites with occupancies of 0.4 and 0.1 and a rms fluctuation ≈1 Å, located in the constricted channels leading to bulk solvent on either side of the cavity. The occupancy of all sites in the cavity region of the simulated water arrangement corresponds to a total of 18.5 electrons, which agrees well with the estimate of 18 ± 4 electrons made from the refined difference map. The simulated water arrangement is illustrated in Fig. 2 B and C, superimposed on the refined solvent difference map in the cavity region.
The xplor model represents an energetically plausible distribution of water molecules in the protein cavity that is consistent with the measured high resolution diffraction data whereas the experimental solvent difference map depends most critically on the amplitudes of the low resolution data and their refined phases. The difference in shape of the solvent distribution calculated experimentally by refining phases for the low resolution data compared with the model obtained by using xplor may result from noise in the data. However, this difference may actually reflect breathing movements of the protein that would allow a larger solvent-accessible volume in the cavity than the refined single conformation model for the atomic structure of the protein.
Discussion
Our results demonstrate that the mobile water located in the hydrophobic cavity of hIL-1β, which was identified by NMR spectroscopy (1), can be assessed quantitatively from x-ray crystallographic data. Reliable counting of the number of electrons in such diffusely distributed density requires accurate measurement and self-consistent phasing of all of the low resolution diffraction data. The count of 18 electrons obtained by the difference map flattening refinement, starting with an empty cavity, represents a plausible estimate of the solvent content. Simulation of the water arrangement by using xplor, starting with 22.5 solvent electrons uniformly distributed in the cavity space, converged to a model with 18.5 electrons, which overlaps the significant domains mapped by the difference map flattening refinement (Fig. 2 B and C).
Cross-validation criteria have been established for testing the accuracy of crystallographic atomic models (19), and the flat solvent model has been shown to provide a good approximation for low resolution phases (9, 10). Model-independent phases for low resolution data can be obtained from multiple isomorphous replacement or multiwavelength anomalous dispersion measurements from crystals containing suitable heavy atoms (10, 17) or by measurement of multiple beam interference for crystals with very small mosaic spread (20). Comparison of multiple isomorphous replacement phases for the penicillopepsin crystal structure (21) with those obtained by iterative difference map density modification has demonstrated self-consistency for data below ≈4.5-Å resolution (10). The complete low resolution data contains critical information about the magnitude of coarse features in an electron density map, and this information can be extracted from the refined difference density map.
The plot of the R factor as a function of resolution (Fig. 1) illustrates that the major improvement in the fit between model and data, produced by the solvent density modification, occurs for structure factors below 5-Å resolution. There are also small decreases in R factors at higher resolution, even though no phase changes were allowed in the modeled data beyond 1/4.5Å spacing. In the course of the iterative refinement, however, damped high spatial frequency features that are persistent in the difference between model and data can build up in the modified solvent map. These features most likely correspond to bound solvent not included in our atomic model, which contained only 55 well ordered water molecules. The number of modeled water molecules in the four independently refined crystal structures (2–5) ranged from 83 to 168, and the two models with the largest number (2, 4) had 74 in common (7). If more water molecules had been included in our atomic model, smaller R factors would have been attained for the high resolution data, and, presumably, the improvement in this data during the difference map density refinement would have been smaller.
Existence of solvent in hydrophobic cavities of proteins has been contentious (6) because of a belief that absence of density in a crystallographic electron density map was evidence for the absence of matter. However, such maps calculated without properly phased low resolution data will not show any atom whose average position is not well localized. Mobile solvent plays a crucial role in the dynamics of all vital macromolecules, and mapping such water making surface contacts can provide insights into the mechanics of the molecular movements. It is evident from the large body of studies on hydrogen exchange (22) that solvent can penetrate into the deepest recesses of protein molecules. Some of this access is through fluctuating channels and some is through local unfolding.
Our models of the solvent density distribution in the interior of hIL-1β suggest the pathways by which water may enter and leave the central cavity (Fig. 2). The small opening in the constricted channel between Leu 18 and Leu 26 apparently contains a water molecule ≈40% of the time, and the even more restricted channel between Leu 10 and Leu 60, on the other side of the central cavity, may contain a water molecule ≈10% of the time. Water in these sites may be exchanging rapidly, going in or out between the central cavity and the bulk solvent. The estimate of 13–14 electrons in the central cavity most likely represents a water dimer that is resident ≈70% of the time and whose hydrogen bond is strengthened in the low dielectric constant environment. NMR data (1) indicate that the residency time for this interior water is >1 ns. Consistent with the idea that water should avoid hydrophobic surfaces, this cavity appears, sometimes, to be empty, and, also consistent with the antithetical idea that nature abhors a vacuum, the cavity is often full. The average density within this cavity volume appears to be about half that of liquid water, which some observers may regard as half empty and others as half full.
Acknowledgments
We thank Eric Fontano for constructing the computer graphics images and John Badger for helpful advice. This work has been supported by U.S. Public Health Service Outstanding Investigator Grant CA47439 to D.L.D.C. from the National Cancer Institute, by U.S. Public Health Service Grant GM54429-01 to M.B., and by the Intramural AIDS Targeted Antiviral Program of the Office of the Director of the National Institutes of Health to A.M.G. and G.M.C.
ABBREVIATION
- hIL-1β
human interleukin 1β
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, Biology Department, Brookhaven National Laboratory, Upton, NY 11973 [PDB ID codes 9ilb (coordinates) and r9ilbsf (structure factors)].
References
- 1.Ernst J A, Clubb R T, Zhou H-X, Gronenborn A M, Clore G M. Science. 1995;267:1813–1817. doi: 10.1126/science.7892604. [DOI] [PubMed] [Google Scholar]
- 2.Priestle J P, Schäer H-P, Grütter M G. Proc Natl Acad Sci USA. 1989;86:9667–9671. doi: 10.1073/pnas.86.24.9667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finzel B C, Clancy L L, Holland D R, Muchmore S W, Watenpaugh K D, Einspahr H M. J Mol Biol. 1990;209:779–791. doi: 10.1016/0022-2836(89)90606-2. [DOI] [PubMed] [Google Scholar]
- 4.Treharne A C, Ohlendorf D H, Weber P C, Wendoloski J J, Salemme F R. In: Molecular and Cellular Biology of IL-1, TNF and Lipocortins in Inflammation and Differentiation. Parente L, editor. New York: Liss; 1990. pp. 309–319. [Google Scholar]
- 5.Veerapandian B, Gilliland G L, Raag R, Svensson A L, Masui Y, Harai Y, Poulos T L. Proteins Struct Funct Genet. 1992;12:1–23. doi: 10.1002/prot.340120103. [DOI] [PubMed] [Google Scholar]
- 6.Matthews B W, Morton A G, Dahlquist F W. Science. 1995;270:1847–1849. doi: 10.1126/science.270.5243.1847. [DOI] [PubMed] [Google Scholar]
- 7.Ohlendorf D H. Acta Crystallogr D. 1994;50:808–812. doi: 10.1107/S0907444994002659. [DOI] [PubMed] [Google Scholar]
- 8.Badger J, Caspar D L D. Proc Natl Acad Sci USA. 1990;88:622–626. doi: 10.1073/pnas.88.2.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badger J. Biophys J. 1993;65:1656–1659. doi: 10.1016/S0006-3495(93)81220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang J-S, Brünger A T. J Mol Biol. 1994;243:100–115. doi: 10.1006/jmbi.1994.1633. [DOI] [PubMed] [Google Scholar]
- 11.Yu B. Ph.D. thesis. Waltham, MA: Brandeis Univ.; 1997. [Google Scholar]
- 12.Phillips S E W. J Mol Biol. 1980;142:531–554. doi: 10.1016/0022-2836(80)90262-4. [DOI] [PubMed] [Google Scholar]
- 13.Blake C C F, Pulford W C A, Artymiuk P J. J Mol Biol. 1983;167:693–723. doi: 10.1016/s0022-2836(83)80105-3. [DOI] [PubMed] [Google Scholar]
- 14.Collaborative Computing Project 4. Acta Crystallogr D. 1994;50:760–763. [Google Scholar]
- 15.Chothia C. Nature (London) 1975;254:304–308. doi: 10.1038/254304a0. [DOI] [PubMed] [Google Scholar]
- 16.Richards F M. Methods Enzymol. 1985;115:440–464. doi: 10.1016/0076-6879(85)15032-9. [DOI] [PubMed] [Google Scholar]
- 17.Burling F T, Weis W I, Flaherty K M, Brünger A T. Science. 1996;271:72–77. doi: 10.1126/science.271.5245.72. [DOI] [PubMed] [Google Scholar]
- 18.Brünger A T. xplor 3.1: A System for X-ray Crystallography and NMR. New Haven, CT: Yale Univ. Press; 1992. [Google Scholar]
- 19.Brünger A T. Nature (London) 1992;355:472–474. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 20.Weckert E, Hümmer K. Acta Crystallogr A. 1997;53:108–143. [Google Scholar]
- 21.James M N G, Sielecki A R. J Mol Biol. 1983;163:299–361. doi: 10.1016/0022-2836(83)90008-6. [DOI] [PubMed] [Google Scholar]
- 22.Englander S W, Kallenbach N R. Q Rev Biophys. 1984;16:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski Q, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 24.Tronrud D E, Ten Eyck L F, Matthews B W. Acta Crystallogr A. 1987;43:489–501. [Google Scholar]
- 25.Tronrud D E. Acta Crystallogr A. 1992;48:912–916. doi: 10.1107/s0108767392005415. [DOI] [PubMed] [Google Scholar]
- 26.Bernstein F C, Koetzle T F, Williams G J B, Meyer E F, Jr, Brice M D, Rogers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 27.Jones T A, Kleywegt G. Proceedings of the CCP4 Study Weekend. Warrington, U.K.: Daresbury Laboratory; 1994. [Google Scholar]
- 28.Kraulis P. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 29.Kolb C, Bogart R. rayshade 4.0. Princeton, NJ: Princeton Univ.; 1991. [Google Scholar]