

Abstract
We have examined the genetic requirements for efficient repair of a site-specific DNA double-strand break (DSB) in Schizosaccharomyces pombe. Tech nology was developed in which a unique DSB could be generated in a non-essential minichromosome, Ch16, using the Saccharomyces cerevisiae HO-endonuclease and its target site, MATa. DSB repair in this context was predominantly through interchromosomal gene conversion. We found that the homologous recombination (HR) genes rhp51+, rad22A+, rad32+ and the nucleotide excision repair gene rad16+ were required for efficient interchromosomal gene conversion. Further, DSB-induced cell cycle delay and efficient HR required the DNA integrity checkpoint gene rad3+. Rhp55 was required for interchromosomal gene conversion; however, an alternative DSB repair mechanism was used in an rhp55Δ background involving ku70+ and rhp51+. Surprisingly, DSB-induced minichromosome loss was significantly reduced in ku70Δ and lig4Δ non-homologous end joining (NHEJ) mutant backgrounds compared with wild type. Furthermore, roles for Ku70 and Lig4 were identified in suppressing DSB-induced chromosomal rearrangements associated with gene conversion. These findings are consistent with both competitive and cooperative interactions between components of the HR and NHEJ pathways.
Keywords: DNA integrity checkpoint/HO-endonuclease/homologous recombination/non-homologous end joining/site-specific DNA double-strand break
Introduction
Double-strand breaks (DSBs) are potentially lethal DNA lesions that can be produced either endogenously during normal DNA metabolism or exogenously through exposure to DNA-damaging agents such as ionizing radiation (IR; reviewed in Pfeiffer et al., 2000). Failure to repair these genomic insults correctly can result in cell death or loss of chromosome integrity, which can contribute to tumorigenesis (reviewed in Pierce et al., 2001a). In eukaryotic cells, DSBs are repaired through homologous recombination (HR) repair mechanisms or non-homo logous end joining (NHEJ). Efficient DNA repair is facilitated further by DNA integrity checkpoint pathways. How these pathways are coordinated in response to DSBs is largely unknown.
HR has been studied extensively in Saccharomyces cerevisiae, where it is the major repair pathway and requires the RAD52 epistasis group of genes, including RAD51, RAD52, RAD54, RAD55 and RAD57. Disruption of these genes results in reduced levels of HR and acute sensitivity to DNA-damaging agents (reviewed in Paques and Haber, 1999). Additionally, the Mre11–Rad50–Xrs2 complex is involved in HR, potentially through the processing of DSB ends (reviewed in D’Amours and Jackson, 2002). A Rad51 nucleoprotein filament is formed that is able to invade an undamaged homologous sequence, usually the sister chromatid (Kadyk and Hartwell, 1992), to initiate HR. This process is facilitated by Rad52, which binds DNA ends (Sugiyama et al., 1998), as well as by the Rad55–Rad57 heterodimer and Rad54 (Sung, 1997; Petukhova et al., 1998; Sugiyama et al., 1998). Following resynthesis of the damaged region, DSB repair is completed by resolution of the cross-stranded intermediates, resulting in gene conversion with or without crossover. Homologues of the S.cerevisiae RAD52 epi stasis group have been identified in the distantly related fission yeast, Schizosaccharomyces pombe. Strains mutated in rhp51+ (rad51 homologue S.pombe) or rhp54+ exhibit acute sensitivity to γ-radiation, sensitivity to methyl methanesulfonate (MMS) and reduced levels of HR (Muris et al., 1993, 1996, 1997). The rhp55 and rhp57 single mutants exhibit γ-radiation sensitivity, which is enhanced further at lower temperatures, thus resembling the rad55 and rad57 mutants in S.cerevisiae (Khasanov et al., 1999; Tsutsui et al., 2000). In contrast to S.cerevisiae, S.pombe encodes two RAD52-like genes, rad22A+ and rad22B+, which function in mitotic and meiotic recombination, respectively (Suto et al., 1999; van den Bosch et al., 2001). rad22A mutants exhibit only a moderate sensitivity to γ-radiation, and the rad22A rad22B double mutant is only slightly more sensitive to γ-radiation than the rad22A single mutant (Ostermann et al., 1993). The rad32+ and rad50+ genes are the respective homologues of MRE11 and RAD50 in S.cerevisiae, and their disruption also results in increased sensitivity to DNA-damaging agents and defects in HR (Tavassoli et al., 1995; Hartsuiker et al., 2001).
NHEJ is the major repair pathway in mammalian cells and coordinates direct re-joining of the DSB ends, which can result in small deletions. The broken ends are protected from excessive processing by the highly conserved Ku70–Ku80 heterodimer (reviewed in Doherty and Jackson, 2001). In mammalian cells, this is bound by DNA–PKcs (protein kinase catalytic subunit) to form the DNA–PK complex (Yaneva et al., 1997; Yoo and Dynan, 1999). This complex is thought to stimulate XRCC4 binding to the DSB, resulting in re-ligation of the DSB through activation of LIG4 (Grawunder et al., 1997; Modesti et al., 1999). Functional homologues of the KU70 (YKU70/HDF1), KU80 (YKU80/HDF2), LIG4 (DNL4) and XRCC4 (LIF1) genes have been identified in S.cerevisiae. Efficient NHEJ in S.cerevisiae additionally requires the Mre11–Rad50–Xrs2 complex (reviewed in Lewis and Resnick, 2000). In S.pombe, ku70+ and lig4+ are required for NHEJ, but in contrast to their counter parts in S.cerevisiae, Rad32 and Rad50 are not required (Manolis et al., 2001).
The DNA integrity checkpoint pathway functions to delay the cell cycle in response to DNA damage, and promotes repair. Disruption of this pathway results in loss of chromosomal integrity and is associated with cancer (reviewed in Zhou and Elledge, 2000). In fission yeast, the damage sensory machinery consists of six highly conserved, non-essential checkpoint proteins, Hus1, Rad1, Rad3, Rad9, Rad17 and Rad26. These proteins are required for cell cycle arrest in response to blocked DNA replication or DNA damage from UV or γ-radiation. Disruption of these proteins results in acute sensitivity to γ-radiation and inappropriate entry into mitosis (reviewed in Humphrey, 2000). Although the cause of acute radiosensitivity of these mutants is unknown, studies in both yeast and mammalian systems indicate a number of links between the DNA integrity checkpoint pathway and HR (reviewed in Carr, 2002).
To begin to understand the relationship between the HR, NHEJ and DNA integrity checkpoint pathways, we examined the role of components of these pathways in the repair responses to a site-specific DSB in fission yeast. We employed the S.cerevisiae HO-endonuclease, together with its MATa target site, to generate a site-specific DSB within a non-essential minichromosome in fission yeast. We have identified a requirement for components of the HR, nucleotide excision repair (NER) and DNA integrity checkpoint pathways in efficient site-specific DSB repair through interchromosomal gene conversion. Moreover, our studies indicate that components of the NHEJ and HR pathways can function both competitively and cooperatively to maintain genome stability in response to a site-specific DSB.
Results
Induction of a site-specific DSB within a minichromosome
To characterize the cellular responses to a site-specific DSB in S.pombe, a strain was constructed in which different site-specific DSB responses could be examined genetically (Figure 1; Materials and methods). The S.cerevisiae HO-endonuclease was utilized to generate a site-specific DSB at its target site, MATa, in S.pombe (Osman et al., 1996). Since failure to repair a DSB can result in cell death, MATa was integrated into a non-essential minichromosome, Ch16, to facilitate genetic analysis of the repair responses to DSB production. Ch16 is a highly stable 500 kb linear minichromosome, derived from chromosome III (ChIII; Niwa et al., 1986). Ch16 encodes an ade6-M216 point mutation, which when present with an ade6-M210 heteroallele on ChIII results in an ade+ phenotype through intragenic complementation (Leupold and Gutz, 1964). The MATa site, together with an adjacent G418 resistance marker (G418R; encoded by the kanMX6 module; see Supplementary data, available at The EMBO Journal Online), was integrated into the rad21+ gene of minichromosome Ch16 by one-step homologous recombination to form Ch16-MG (MATa, G418R). Integrating a G418R marker adjacent to the MATa site within the Ch16-rad21 allele provided the possibility of distinguishing genetically between different mechanisms of DSB repair at the MATa site: prior to HO expression, strains containing Ch16-MG are ade+ and G418R. Following HO expression, DSB repair through HR would be expected to result in loss of the MATa and G418R regions through interchromosomal gene conversion, utilizing the rad21+ gene on ChIII as a template, resulting in cells becoming ade+ and G418 sensitive (G418S). DSB repair through NHEJ would be expected to result in rejoining of the DSB within the MATa sequence (embedded within a 250 bp fragment), thus retaining the G418R marker. Such cells would be ade+ and G418R. Failure to repair the DSB would be expected to result in loss of the minichromosome, resulting in the cells becoming ade– and G418S. Ade– G418S colonies could also result from long-tract gene conversion of Ch16-ade6-M216 to ade6-M210 following DSB induction (see Figure 1).

Fig. 1. Schematic of the Ch16-MG strain (TH805). Ch16-MG, ChIII, centromeric regions (ovals) and the complementary ade6-M216 (or ade6-M210) heteroalleles (dark grey) are indicated. rad21 alleles (white) are located 28 kb distal to the ade6 locus on ChIII, and Ch16. The MATa (black) and kanMX6 sequences (light grey) were cloned into Ch16-rad21, as shown, to form Ch16-MG. Derepression of pREP81X-HO in the absence of thiamine results in DSB production within MATa (scissors). Phenotypes predicted from DSB repair by HR, NHEJ, long-tract gene conversion or non-repair are shown (see text for details).
To examine the responses to an HO-dependent DSB generated within a minichromosome, Ch16-MG pREP81X-HO (TH844) was cultured, in either the presence (+) or absence (–) of the repressor, thiamine, for up to 48 h. Southern blot analysis showed that the MATa probe hybridized to a 6 kb fragment prior to HO expression (Figure 2A, upper panel, lane 2), consistent with the size expected for the Ch16-MG allele. Following derepres sion of the HO-endonuclease, the intensity of the MATa fragment decreased after 30 and 48 h incubation (Figure 2A, upper panel, lanes 6 and 7). In contrast, no significant MATa loss was observed when cells were grown in the presence of thiamine for 48 h (Figure 2A, upper panel, lane 8). Similar results were observed when the Southern blot was re-probed with kanMX6 (Figure 2A, upper middle panel). A 3.5 kb fragment was also detected transiently after longer exposure with a kanMX6 probe, and is maximally present after 24 h following HO derepression (Figure 2A, lower middle panel). This band is the size expected for the MATa-EcoRI fragment encoding kanMX6, and indicates the presence of a DSB at the MATa site (see Supplementary figure A). Southern blot analysis of the rad21 alleles revealed the presence of 4.2 and 6 kb fragments in the absence of HO-endonuclease expression, consistent with detection of both the endogenous rad21+ allele and the Ch16-MG allele, respectively (Figure 2B, upper panel, lanes 2 and 8). Following expression of the HO-endonuclease, the intensity of the 6 kb rad21 fragment decreased at later time points (30 and 48 h), as was observed with the kanMX6 and MATa fragments (Figure 2B). Quantitation of these data is provided in Supplementary figure B, and indicates significant levels of marker loss after 30 and 48 h in the absence of thiamine.

Fig. 2. Site-specific DSB repair results in loss of the MATa and kanMX6 sequences. (A) DSB induced loss of MATa and kanMX6 sequences. Southern blot of EcoRI-digested chromosomal DNA from wild-type cells, grown under the conditions indicated, probed with MATa (upper panel), kanMX6 (middle panels) and a leu1 loading control (lower panel). M indicates the DNA size marker (lanes 1 and 9). (B) DSB-induced loss of rad21::MATa-kanMX6. Southern blot analysis of EcoRI-digested chromosomal DNA samples, as above, probed with rad21 (upper panel) and a leu1 loading control (lower panel). (C) Graph of percentage marker loss in wild-type, grown under the conditions indicated. See Table I.
To determine whether loss of the MATa and kanMX6 (G418R) sequences resulted from DSB repair by gene conversion or minichromosome loss, the levels of G418R and ade+ marker loss were quantitated using a colony assay following HO-dependent DSB induction (Materials and methods). From this genetic analysis, a striking increase in HO-dependent G418R marker loss was observed, from 2% after 16 h to 84% after 48 h incubation in the absence of thiamine (Figure 2C; Table I). In addition, ade+ marker loss increased from 0% at 0 h to 17% after 48 h (Figure 2C; Table I). Since the levels of G418R marker loss were considerably greater than levels of ade+ marker loss, G418R loss could not have resulted primarily from minichromosome loss, but was likely to have resulted from DSB repair. The proportion of cells yielding G418R marker loss through DSB repair in the wild-type strain was calculated to be 64% (Table II). Importantly, there was no stimulation of marker loss without DSB induction (Figure 2C; Table II), and no loss of viability upon DSB induction in Ch16-MG pREP81X-HO (our unpublished data).
Table I. Genetic analysis of HO induction.
| Time (h) | 0 | 16 | 20 | 24 | 30 | 48 | 48 |
| Repressor | – | – | – | – | – | – | + |
| % G418S | 2 | 2 | 13 | 32 | 56 | 84 | 2 |
| % ade– | 0 | 0 | 4 | 9 | 14 | 17 | 0 |
At least 500 colonies were scored for each timepoint.
Table II. Genetic determinants of site-specific DSB-induced marker loss.
| Genetic background | Strain number | h | T | Total scored | % ade–G418S | % ade+ G418S | % ade+ G418R | % DSB-induced gene conversion |
|---|---|---|---|---|---|---|---|---|
| Wild type | TH844 | 0 | – | 2591 | 0.1 ± 0.1 | 2.5 ± 0.6 | 97.4 ± 0.6 | |
| 48 | – | 1810 | 19.8 ± 1.7 | 65.9 ± 2.0 | 14.3 ± 1.6 | 63.7 ± 2.6 | ||
| 48 | + | 2640 | 0.5 ± 0.1 | 2.2 ± 0.6 | 97.3 ± 0.7 | |||
| rhp51Δ | TH895 | 0 | – | 1680 | 8.5 ± 1.7 | 1.1 ± 0.6 | 90.4 ± 2.2 | |
| 48 | – | 1317 | 74.5 ± 2.8 | 3.5 ± 0.9 | 22.0 ± 3.3 | 2.8 ± 0.9 | ||
| 48 | + | 2125 | 15.4 ± 2.9 | 0.7 ± 0.4 | 83.9 ± 2.7 | |||
| rad22AΔ | TH906 | 0 | – | 1813 | 1.1 ± 0.8 | 3.8 ± 3.0 | 95.1 ± 2.5 | |
| 48 | – | 1582 | 42.7 ± 4.7 | 21.3 ± 0.4 | 36.0 ± 4.5 | 19.8 ± 0.6 | ||
| 48 | + | 1912 | 4.5 ± 0.3 | 1.5 ± 0.3 | 94.0 ± 0.5 | |||
| rad32Δ | TH1083 | 0 | – | 2011 | 6.3 ± 1.3 | 3.6 ± 1.3 | 90.1 ± 1.2 | |
| 48 | – | 1079 | 44.7 ± 2.9 | 28.8 ± 6.6 | 26.5 ± 4.2 | 25.6 ± 6.4 | ||
| 48 | + | 2684 | 11.1 ± 1.6 | 3.1 ± 0.5 | 85.8 ± 1.6 | |||
| rad16Δ | TH873 | 0 | – | 1651 | 2.9 ± 1.1 | 1.4 ± 0.3 | 95.7 ± 0.9 | |
| 48 | – | 1150 | 48.2 ± 2.8 | 17.9 ± 2.1 | 33.9 ± 1.2 | 16.0 ± 2.3 | ||
| 48 | + | 1842 | 4.0 ± 1.8 | 1.9 ± 0.1 | 93.7 ± 1.8 | |||
| rhp55Δ | TH871 | 0 | – | 3218 | 2.2 ± 0.5 | 0.4 ± 0.2 | 97.4 ± 0.5 | |
| 48 | – | 3210 | 21.8 ± 1.4 | 5.7 ± 0.5 | 72.5 ± 1.8 | 5.0 ± 0.3 | ||
| 48 | + | 3536 | 4.3 ± 1.0 | 0.7 ± 0.2 | 95.0 ± 1.1 | |||
| rhp55Δ ku70Δ | TH1009 | 0 | – | 1841 | 8.7 ± 1.9 | 13.8 ± 0.7 | 77.6 ± 2.2 | |
| 48 | – | 2117 | 15.9 ± 1.9 | 23.2 ± 1.9 | 60.9 ± 3.6 | 11.6 ± 0.8 | ||
| 48 | + | 2229 | 6.0 ± 0.7 | 11.6 ± 1.2 | 82.4 ± 1.7 | |||
| rhp55Δ rhp51Δ | TH1102 | 0 | – | 2076 | 5.4 ± 1.0 | 0.3 ± 0.3 | 94.3 ± 1.1 | |
| 48 | – | 1842 | 54.8 ± 2.7 | 2.7 ± 0.2 | 42.5 ± 2.6 | 2.1 ± 0.3 | ||
| 48 | + | 2342 | 8.9 ± 0.9 | 0.6 ± 0.2 | 90.5 ± 0.9 | |||
| ku70Δ | TH932 | 0 | – | 4036 | 0.4 ± 0.2 | 1.0 ± 0.3 | 98.6 ± 0.2 | |
| 48 | – | 1185 | 4.3 ± 0.7 | 65.7 ± 4.7 | 30.0 ± 4.2 | 62.5 ± 3.8 | ||
| 48 | + | 4127 | 0.0 ± 0.0 | 3.2 ± 0.9 | 96.8 ± 0.9 | |||
| lig4Δ | TH1216 | 0 | – | 4193 | 1.8 ± 0.3 | 5.8 ± 0.6 | 92.4 ± 0.4 | |
| 48 | – | 4459 | 9.1 ± 0.8 | 52.6 ± 2.2 | 38.3 ± 1.5 | 45.7 ± 2.2 | ||
| 48 | + | 4718 | 1.7 ± 0.7 | 6.9 ± 0.7 | 91.4 ± 0.2 | |||
| ku70Δ rhp51Δ | TH987 | 0 | – | 1777 | 12.7 ± 4.2 | 1.6 ± 0.9 | 85.7 ± 5.0 | |
| 48 | – | 1033 | 59.7 ± 2.2 | 1.1 ± 0.1 | 39.2 ± 2.1 | 0.2 ± 0.3 | ||
| 48 | + | 1530 | 25.3 ± 8.6 | 0.9 ± 0.3 | 73.8 ± 8.7 | |||
| rad3Δ | TH877 | 0 | – | 1672 | 0.7 ± 0.2 | 0.4 ± 0.3 | 98.9 ± 0.4 | |
| 48 | – | 1902 | 32.1 ± 2.7 | 29.2 ± 2.4 | 38.7 ± 4.5 | 28.8 ± 2.7 | ||
| 48 | + | 1686 | 1.2 ± 0.1 | 0.4 ± 0.3 | 98.4 ± 0.4 |
For each genetic background, the genetic assay was repeated independently at least three times, such that >1000 colonies were scored at each time point. The average value for each time point and the standard errors between the independent experiments are shown. The percentage DSB-induced gene conversion was calculated as: [% ade+ G418S (48 h –T) – % ade+ G418S (48 h +T)].
Analysis of site-specific DSB repair in a wild-type background
To examine the predominant mechanism by which the site-specific DSB was repaired within the minichromosome, chromosomal DNA was analysed from a number of individually isolated ade+ G418S colonies derived from Ch16-MG pREP81X-HO following HO expression. Southern blot analysis of five such ade+ G418S colonies indicated that they had all lost the MATa site (Figure 3A, upper panel, lanes 1–5) and the 6 kb rad21 fragment (Figure 3A, middle panel, lanes 1–5). The 4.2 kb rad21 fragment present in all of these ade+ G418S colonies was of a higher intensity than that of the 4.2 kb fragment from strain TH805, in which the HO-endonuclease was absent (Figure 3A, middle and lower panels). These results suggested that the MATa and kanMX6 sequences were removed from Ch16-MG, to re-form a wild-type (4.2 kb) rad21 allele, consistent with DSB repair through interchromosomal gene conversion. Since gene conversion would be expected to re-form a functional rad21 allele on Ch16, the ability of a ‘repaired’ rad21 allele to functionally complement a temperature-sensitive rad21-K1 mutation on ChIII was examined. An ade+ G418S colony derived from TH844, in which the HO-endonuclease had been expressed, was crossed with a temperature-sensitive rad21-K1 mutant (TH1017). Tetrad dissection and subsequent genetic analysis of this cross indicated that the repaired rad21 allele on Ch16 was able to complement an endogenous rad21-K1 temperature-sensitive allele at 35.5°C and therefore was fully functional (Figure 3B; colonies B2, C2 and C4). In contrast, the non-functional Ch16-rad21::MATa-kanMX6 allele present on the minichromosome of strain TH805 could not complement the rad21-K1 allele (our unpublished data).
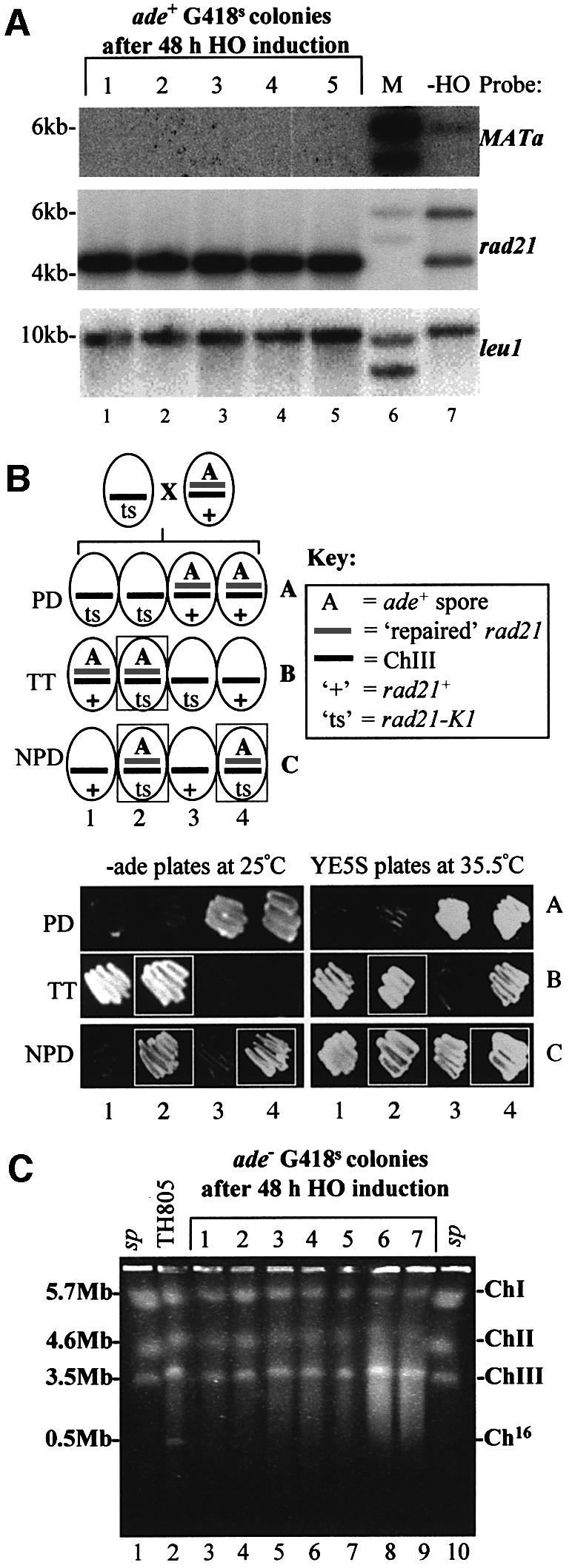
Fig. 3. Analysis of site-specific DSB repair in G418S cells. (A) Southern blot analysis of EcoRI-digested chromosomal DNA from five independent ade+ G418S colonies, obtained after 48 h of HO induction in strain TH844 (lanes 1–5). Chromosomal DNA from TH805 (which lacks pREP81X-HO) was also analysed (lane 7). Southern blot probed with MATa (upper panel), rad21 (middle panel) and leu1 (lower panel). M indicates the DNA size marker (lane 6). (B) A repaired ade+ G418S colony can complement a rad21-K1 mutant. Upper panel: schematic of predicted genotypes from a cross between a ‘ts’ rad21-K1 mutant and an ade+ G418S colony, which contains a repaired rad21 allele on Ch16. The PD (parental ditype), TT (tetra type) and NPD (non-parental ditype) genotypes are shown. Lower panel: examples of dissected PD, TT and NPD tetrads obtained from the above cross with rad21-K1 (TH1017). The ability of the repaired Ch16-rad21 to complement the rad21-K1 mutant was determined by the ability to grow at the restrictive temperature (35.5°C; boxed). Colony coordinates correspond to the predicted genotypes shown above. (C) PFGE analysis of chromosomal DNA from seven individual ade– G418S colonies obtained following DSB induction in a wild-type background (lanes 3–9). Chromosomal DNA from wild-type TH805 (which lacks pREP81X-HO) is shown as a control (lane 2). Schizosaccharomyces pombe commercial markers are shown (lanes 1 and 10).
The genetic assay also showed that 14% of the population were ade+ G418R after 48 h growth without thiamine (Figure 4A; Table II). To test the possibility that DSBs were repaired by NHEJ in this population, the MATa sequence was examined for mutations in these ade+ G418R colonies. No mutations were identified within the MATa site from sequence analysis, and wild-type levels of genetic marker loss were obtained in 50 ade+ G418R colonies re-challenged with HO-endonuclease (our unpublished data). HO induction for longer times (72 h without thiamine) did not result in increased G418R marker loss, indicating that maximal DSB induction had occurred following HO derepression for 48 h (Supplementary figure C). Thus, although DSB induction could occur in all cells tested, we could not distinguish between uncut and accurately re-annealed MATa sites in ade+ G418R cells by sequence analysis.

Fig. 4. Genetic analysis of site-specific DSB repair in wild-type, HR and NER backgrounds. Graphs of percentage marker loss in: (A) wild-type (TH844); (B) rhp51Δ (TH895); (C) rad22AΔ (TH906); (D) rad32Δ (TH1083); and (E) rad16Δ (TH873), grown under the conditions indicated.
The genetic assay further indicated that 20% of the population were ade– G418S after 48 h growth without thiamine (Figure 4A; Table II). Such cells could have arisen through minichromosome loss as a result of failure to repair the DSB, or through long-tract gene conversion. As the Ch16-ade6-M216 allele is located 28 kb from the rad21::MATa-kanMX6 (G418R) allele, co-conversion of these alleles would be expected to be infrequent. Analysis of ade– G418S colonies by pulse-field gel electrophoresis (PFGE) indicated that Ch16 was absent in 24 out of 27 colonies tested (see also Figure 3C). These results indicate that 18% of the total population had lost the minichromosome in an HO-dependent manner, consistent with failure to repair the DSB in these cells. No ade– G418R colonies were detected during the course of these studies.
Interchromosomal DSB repair requires homologous recombination genes rhp51+, rad22A+ and rad32+
To understand site-specific DSB repair better, the genetic determinants for efficient DSB repair in this context were examined. For each of the mutant backgrounds analysed, no loss of viability was observed following DSB induction (our unpublished data).
As Rhp51 is central to HR, it was predicted that it would be required for efficient HO-induced DSB repair in this assay. In contrast to DSB production in a wild-type background, HO induction in an rhp51Δ mutant background resulted in a striking increase in ade+ marker loss, from 6% prior to HO derepression, to 74% following DSB induction (Figure 4B; Table II). The increased level of DSB-dependent ade+ marker loss was consistent with the rhp51Δ mutant being unable to repair this lesion, resulting in loss of the Ch16-MG minichromosome. A concomitant increase in G418R marker loss in the rhp51Δ background (78% following DSB induction; Figure 4B; Table II) can be explained by loss of the Ch16-MG minichromosome. Spontaneous levels of minichromosome loss are elevated in rhp51Δ cells (Muris et al., 1996; Supplementary figure C).
ade+ marker loss was also significantly elevated in rad22AΔ and rad32Δ backgrounds, and gene conversion levels were calculated to be significantly reduced to 20 and 26%, respectively (Figure 4C and D; Table II). These data indicate a role for these genes in HR-dependent interchromosomal gene conversion.
rad16+ is required for DSB-induced interchromosomal gene conversion
Rad16 is involved in NER (Carr et al., 1994), but also has a role in mating-type switching (Schmidt et al., 1989) and UV-induced recombination (Osman et al., 2000). A role for the S.cerevisiae Rad16 homologue, Rad1, has been demonstrated in DSB repair (Fishman-Lobell and Haber, 1992; Kang and Symington, 2000). DSB induction in a rad16Δ background resulted in significantly reduced levels of gene conversion (16%), revealing a role for Rad16 in interchromosomal gene conversion in this context (Figure 4E; Table II). The potential roles of the rad13+, rad15+ and rad2+ NER genes in DSB repair were also examined using the DSB repair assay. However, wild-type repair profiles were observed in these mutant backgrounds, indicating that these genes did not play an important role in interchromosomal DSB repair in S.pombe (our unpublished data).
Alternative site-specific DSB repair in an rhp55Δ mutant
Surprisingly, DSB-induced levels of ade+ marker loss in an rhp55Δ background were almost identical to those observed in a wild-type strain (compare Figures 4A and 5A; Table II). Moreover, HO-dependent G418R marker loss was significantly reduced in an rhp55Δ background (Figures 4A and 5A), with only 5% gene conversion being observed (Table II). These genetic data were confirmed by Southern blot analysis, in which the 6 kb Ch16-MG allele was present at an intensity consistent with reduced loss of the G418R marker in an rhp55Δ background, compared with wild-type and rhp51Δ backgrounds, following DSB induction (Figure 5B, compare lanes 6, 3 and 9, respectively). A possible explanation for these findings is that the HO-induced DSB was repaired through a different mechanism in an rhp55Δ mutant. DSB repair through the NHEJ pathway would be expected to result in re-annealing of the cleaved MATa site and retention of the adjacent G418R marker. The contribution of the NHEJ pathway in an rhp55Δ mutant background was tested by examining levels of marker loss in an rhp55Δ ku70Δ double mutant background in which the NHEJ pathway was disrupted (Manolis et al., 2001). DSB-induced G418R marker loss was increased significantly in this double mutant, with 39% G418R marker loss observed following DSB induction, compared with 27% in rhp55Δ (Figure 5A and C). No significant change in ade+ marker loss was observed in the rhp55Δ ku70Δ double mutant, compared with that of rhp55Δ (Table II). However, the percentage of DSB-induced gene conversion increased significantly from 5% in an rhp55Δ background to 12% in an rhp55Δ ku70Δ background (Table II; P = 0.00099 as determined by Student’s t-test).

Fig. 5. Analysis of site-specific DSB repair in an rhp55Δ background. (A) Graph of percentage marker loss in rhp55Δ (TH871). (B) Southern blot of EcoRI-digested chromosomal DNA from wild-type (TH844; lanes 2–4), rhp55Δ (TH871; lanes 5–7) and rhp51Δ (TH895; lanes 8–10) probed with rad21 (upper panel) and leu1 (lower panel). M indicates the DNA size marker (lane 1). Graphs of percentage marker loss in: (C) rhp55Δ ku70Δ (TH1009) and (D) rhp55Δ rhp51Δ (TH1102), grown under the conditions indicated.
Since DSB repair was still observed when NHEJ was disrupted in the rhp55Δ ku70Δ mutant background, HR might still contribute to DSB repair in an rhp55Δ mutant. DSB induction in an rhp55Δ rhp51Δ double mutant resulted in a significantly elevated level of ade+ marker loss (55%) following DSB induction (Figure 5D; Table II) and negligible levels of gene conversion (Table II). These results indicate that DSB repair in an rhp55Δ mutant background was Rhp51 dependent. However, repair in an rhp55Δ background was not associated with interchromosomal gene conversion, as determined by G418R marker loss in this context (Table II). Thus, an alternative Rhp51-dependent DSB repair pathway, involving Ku70, appears to be utilized in an rhp55Δ mutant background.
A role for NHEJ proteins in suppressing chromosomal rearrangements
In a ku70Δ mutant, a high degree of DSB repair through gene conversion (63%) was observed (Figure 6A; Table II). Surprisingly, ade+ marker loss was significantly reduced to 4% in a ku70Δ background following DSB induction, compared with that of 20% in a wild-type strain (Figures 4A and 6A; Table II; P = 0.00004 as determined by Student’s t-test). ade+ marker loss was associated with minichromosome loss in this strain (Supplementary figure D). The reduced DSB-dependent ade+ marker loss observed in the ku70Δ mutant suggested that the DSB was repaired more efficiently in a ku70Δ background than in a wild-type strain. Given the unusual nature of this result, we considered the possibility that mis-repair might be associated with minichromosome retention in a ku70Δ mutant background following DSB induction. To test this possibility, the integrity of the ‘repaired’ minichromosomes from a number of individually selected ade+ G418S colonies derived from the ku70Δ mutant following HO expression was examined by PFGE. The three S.pombe chromosomes and minichromosome Ch16-MG were apparent in DNA extracted from a wild-type control (Figure 6B, lane 1). Similar chromosomal bands were seen in DNA extracted from individual ade+ G418S colonies, derived from the ku70Δ mutant following HO expression (represented in Figure 6B, lane 2). These results were consistent with DSB repair through interchromosomal gene conversion in these ku70Δ colonies. Surprisingly, an abnormal chromosomal banding pattern was observed, which was common to six out of 60 ade+ G418S colonies derived from the ku70Δ background following HO expression (represented in Figure 6B, lane 3). In these colonies, the ChIII (3.5 Mb) and Ch16 (500 kb) bands were absent, and instead an intense band of ∼2.0 Mb (ChXY) was observed in addition to bands corresponding to ChI (5.7 Mb) and ChII (4.6 Mb). Two chromosomal elements of ∼2.0 Mb would be expected if a crossover event had occurred between ChIII and Ch16 at or near to the DSB site (Supplementary figure E). Quantitative Southern blot analysis using rad21 as a probe revealed band ChXY to encode rad21 and to be approximately twice the intensity of ChIII and Ch16, consistent with this band being a doublet (Figure 6B, right panel and C). Cells in which a DSB-induced crossover had occurred between Ch16 and ChIII would still be expected to be ade+, as was observed. Such chromosomal rearrangements were not observed in a wild-type background, following analysis of 60 ade+ G418S colonies.
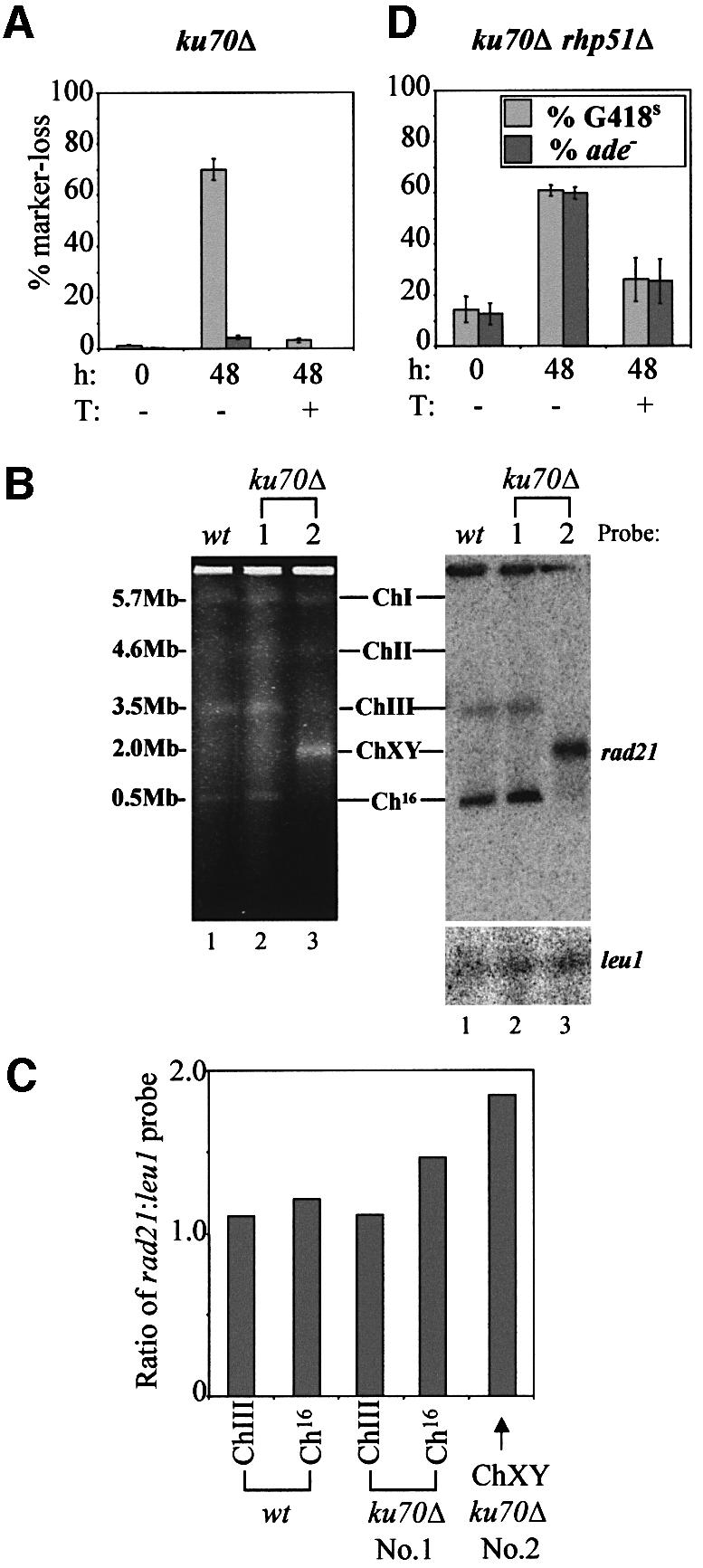
Fig. 6. Analysis of site-specific DSB induction in ku70Δ mutants. (A) Graph of percentage marker loss in ku70Δ (TH932) grown under the conditions indicated. (B) Left panel: PFGE analysis of chromosomal DNA from two representative ade+ G418S colonies obtained following DSB induction in ku70Δ (TH932; lanes 2 and 3). Chromosomal DNA from TH805 (which lacks pREP81X-HO) is shown as a control (lane 1). Sizes and chromosomal bands are indicated. Right panel: Southern blot of the gel in the left panel probed with rad21. (C) Quantitation of the Southern blot above indicating the ratio of the rad21:leu1 signal for the bands indicated. (D) Graph of percentage marker loss in ku70Δ rhp51Δ (TH987), grown under the conditions indicated.
A high degree of gene conversion was observed in a ku70Δ background (Figure 6A; Table II) following derepression of the HO-endonuclease. To determine whether this repair was Rhp51 dependent, marker loss was examined in a ku70Δ rhp51Δ double mutant. DSB-induced gene conversion was abrogated effectively in this strain (Figures 6D; Table II). These data indicate that DSB repair in a ku70Δ mutant is Rhp51 dependent, and is consistent with site-specific DSB repair occurring through interchromosomal gene conversion in a ku70Δ mutant background (Table II).
To determine whether DSB-induced chromosomal rearrangements were common to other NHEJ mutants, DSB repair was examined in a lig4Δ mutant background. DSB-induced ade+ marker loss was significantly reduced to 9% in a lig4Δ background, compared with that of 20% in wild type (Figures 7A and 4A, respectively; Table II; P = 0.00012 as determined by Student’s t-test). However, the level of DSB-induced ade+ marker loss was not significantly different from that observed in a ku70Δ background (Figures 7A and 6A, respectively; Table II; P = 0.080 as determined by Student’s t-test). PFGE analysis was used to determine whether mis-repair was associated with minichromosome retention in a lig4Δ mutant background. A common chromosomal rearrangement pattern was identified in two out of 59 individually selected ade+ G418S colonies derived from lig4Δ mutants. In these colonies, ChIII and Ch16-MG were absent and, in contrast to the chromosomal rearrangements observed in a ku70Δ background, two distinct chromosomal elements of ∼2 Mb could be observed in a lig4Δ background. Southern blot analysis revealed that these two new bands encode rad21, and are therefore derived from ChIII and Ch16-MG (Figure 7B). This chromosomal profile is consistent with loss of the Ch16-MG distal arm during a DSB-induced crossover event (Supplementary figure E).

Fig. 7. Analysis of site-specific DSB induction in lig4Δ mutants. (A) Graph of percentage marker loss in lig4Δ (TH1216). (B) Left panel: PFGE analysis of chromosomal DNA from three ade+ G418S colonies obtained following DSB induction in lig4Δ (lanes 1–3). Chromosomal DNA from TH805 (which lacks pREP81X-HO) is shown as a control (lane 4). The S.pombe commercial markers are shown (lane 5). Right panel: Southern blot of the gel in the left panel probed with rad21.
The DNA integrity checkpoint is required for cell cycle delay and efficient HR following site-specific DSB induction
Cell cycle delay is a hallmark of checkpoint activation in S.pombe and results in cells with an elongated phenotype. We observed a reduction in the rate of cell division following HO induction, whereas cell growth was relatively unaffected, suggesting cells were undergoing a cell cycle delay (Supplementary figure F). Consistent with this, a significant number of elongated cells was found after 24 h following site-specific DSB induction in wild-type cells (Figure 8A and B; Table III). No elongated cells were observed at the 0 and 16 h time points in the absence of DSB induction or in any of the control samples grown with a vector control plasmid (Figure 8B), indicating that the cell cycle delay correlated with DSB production. In contrast, no cell cycle delay was observed in a rad3Δ checkpoint mutant background following DSB induction (Figure 8A and C; Table III), indicating that the HO-induced cell cycle delay was checkpoint dependent. A significantly reduced level of DSB-induced gene conversion was also observed in rad3Δ cells (29%) compared with wild-type cells (64%; Figure 8D; Table II), thus indicating a role for the DNA integrity checkpoint in promoting HR-dependent interchromosomal gene conversion.

Fig. 8. The DNA integrity checkpoint is required for cell cycle delay and efficient HR. (A) Wild-type (+HO; TH844 and vector; TH1010) and rad3Δ (+HO; TH877 and vector; TH876) were grown without thiamine for 24 h, formaldehyde fixed and DAPI stained. The scale bar represents 10 µm. (B and C) Graphs indicating the percentage of cells exhibiting an elongated phenotype (>20 µm) in the strains described above grown without thiamine for the times indicated. See also Table III. (D) Graph of percentage marker loss in rad3Δ (TH877), grown under the conditions shown.
Table III. Percentage of cells exhibiting an elongated phenotype.
| Time | Wild type |
rad3Δ |
||
|---|---|---|---|---|
| (h) | % >20µm | % >20 µm | % >20 µm | % >20 µm |
| +HO | vector only | +HO | vector only | |
| 0 | 2.5 ± 0.6 (959) | 2.4 ± 0.8 (966) | 0.4 (505) | 0.4 (565) |
| 16 | 2.1 ± 0.6 (1072) | 2.8 ± 0.8 (965) | 3.3 (521) | 1.8 (553) |
| 24 | 7.1 ± 1.8 (1024) | 1.8 ± 1.0 (1050) | 0.5 (568) | 0.4 (570) |
| 30 | 18.7 ± 2.5 (989) | 2.9 ± 0.6 (995) | 0.4 (528) | 0.7 (544) |
| 36 | 16.8 ± 1.3 (1066) | 1.7 ± 0.6 (975) | 0.6 (540) | 0.8 (516) |
| 42 | 11.6 ± 2.0 (1203) | 1.2 ± 0.7 (1050) | 0.4 (527) | 0.6 (531) |
| 48 | 9.2 ± 0.8 (1167) | 1.9 ± 0.5 (1001) | 1.1 (550) | 0.5 (565) |
Values in parentheses indicate the total number of cells counted in three independent experiments.
Discussion
Site-specific DSB repair by interchromosomal gene conversion
Site-specific DSB repair within the minichromosome (Ch16-MG) occurred predominantly through interchromosomal gene conversion using the homologous ChIII as a repair template. Evidence to support this conclusion came firstly from the finding that a high degree of G418R marker loss was observed compared with that of ade+ marker loss, thus leading to a calculated 64% G418R marker loss through HO-induced repair rather than Ch16-MG loss (Table II). Secondly, G418R marker loss correlated with the appearance of Ch16-rad21 alleles of wild-type length, in all G418S ade+ cells tested. Thirdly, the repaired Ch16-rad21 allele was fully functional, as determined by its ability to complement the temperature sensitivity of rad21-K1. Since the only source of a functional rad21+ allele was on ChIII, we conclude that this was used as a repair template for the Ch16-rad21::MATa-kanMX6 allele in the ade+ G418S colonies following DSB induction. Ch16-MG was lost in 18% of the cells following HO induction, suggesting that DSB repair had failed in these cells. Three out of 27 ade– G418S cells (2% of the total population) still contained a minichromosome, and may have resulted from long-tract gene conversion. Fourteen percent of the population were either uncut by the HO-endonuclease or the DSB was repaired accurately to reform the MATa site.
The finding that gene conversion was the predominant mechanism of DSB repair in this system is consistent with HR being the principal mechanism of repair in yeast (Pastink et al., 2001). In S.cerevisiae, the sister chromatid is used more commonly as a template for Rad51- dependent HR in haploid cells (Kadyk and Hartwell, 1992). In this study, both sister chromatids would be expected to incur a DSB, and thus the rad21+ allele on ChIII would be used preferentially as a repair template. However, if a single chromatid was cut, HR-dependent repair could use the sister chromatid as a template.
Genetic determinants of interchromosomal gene conversion
Genetic analysis of site-specific DSB repair identified a requirement for the rhp51+, rad32+ and rad22A+ HR genes in efficient interchromosomal gene conversion. Strains in which these genes were deleted exhibited significantly increased levels of DSB-induced Ch16-MG loss. The degree of DSB-induced Ch16-MG loss reflected the relative sensitivity of these mutants to IR, with the highest Ch16-MG loss being observed in the rhp51Δ background (Tavassoli et al., 1995; Khasanov et al., 1999). We identified rad16+ to be required additionally for efficient site-specific DSB repair. In contrast, the rad2+, rad13+ and rad15+ NER genes were not required for HO-induced DSB repair (our unpublished data). These findings strongly resemble those of the rad16+ homologue, RAD1, in S.cerevisiae (Fishman-Lobell and Haber, 1992; Ivanov and Haber, 1995) and indicate a conserved role for the Rad1/Rad10 endonuclease in the removal of non-homologous regions during HR. In this context, Rad16 may function in the removal of the heterologous MATa and kanMX6 (G418R) sequences present within the disrupted Ch16-rad21 allele during interchromosomal gene conversion.
Site-specific DSB repair in an rhp55Δ background
Our data identify an important role for Rhp55 in interchromosomal gene conversion. However, alternative site-specific DSB repair mechanisms were utilized efficiently in an rhp55Δ background. This conclusion was based firstly on the finding that, despite a significant reduction in G418R marker loss, rhp55Δ cells retained wild-type levels of ade+ marker loss. Secondly, levels of DSB-induced gene conversion were increased in an rhp55Δ ku70Δ background. In the absence of Ku70, the HR pathway was utilized to a greater extent, resulting in increased levels of gene conversion in an Rhp55-independent and Rhp51-dependent manner. Thirdly, the levels of HO-dependent Ch16-MG loss were reduced in an rhp51Δ rhp55Δ background, where 55% ade+ marker loss was observed, compared with that of 74% in an rhp51Δ background, following HO derepression. This finding is consistent with an elevated level of Rhp51-independent DSB repair in an rhp55Δ background. These data together suggest a greater role for the NHEJ pathway in DSB repair in an rhp55Δ background, and are consistent with a competitive relationship between components of the HR and NHEJ pathways (Figure 9).

Fig. 9. Model depicting competitive and cooperative relationships between components of the HR and NHEJ repair pathways in response to a site-specific DSB in fission yeast. Rhp55 promotes efficient use of HR in response to a site-specific DSB. In an rhp55Δ background, DSB repair involves both Ku70 and Rhp51, suggesting sequential use of NHEJ and HR pathways (dotted line). In the absence of the NHEJ pathway, elevated levels of Rhp51-dependent DSB repair are observed. DSB repair is associated additionally with chromosomal rearrangements in ku70Δ and lig4Δ mutants, indicating a potential role for the NHEJ pathway in suppressing crossovers. See text for details.
Genetic analysis also indicated an important role for Rhp51 in site-specific DSB repair, although interchromosomal gene conversion was only a minor repair mechanism in an rhp55Δ background. To explain the roles of Ku70 and Rhp51 in repair of a site-specific DSB in an rhp55Δ background, we speculate that in the absence of Rhp55, HR efficiency is compromised, thus favouring DSB repair through the NHEJ pathway. Under circumstances in which both sister chromatids incur a DSB, e.g. during the G2 phase of the cell cycle, loss of Rhp55 may result in repair of one of the sister chromatids through NHEJ. This reannealed sister chromatid subsequently could be utilized as a template for HR-dependent repair of the second sister chromatid. Such a repair mechanism would result in reduced levels of G418R marker loss, and would explain the roles of both Ku70 and Rhp51 in site-specific DSB repair in an rhp55Δ background. This model predicts that the NHEJ and HR pathways may operate sequentially to effect site-specific DSB repair (see Figure 9).
As the DSB generated in an rhp55Δ background could become a substrate for the NHEJ pathway, it follows that the DSB was not processed irreversibly by HR. It is therefore likely that Rhp55 functions at a very early step in HR. Studies with S.cerevisiae Rad55 are consistent with this conclusion (Hays et al., 1995; Fortin and Symington, 2002). However, our data do not exclude an additional role for Rhp55 at a later stage in HR.
Site-specific DSB repair in NHEJ mutants
Levels of DSB-induced ade+ marker loss were significantly reduced in both ku70Δ and lig4Δ backgrounds, compared with wild-type. DSB repair in the ku70Δ background was shown additionally to be Rhp51 dependent. These results are consistent with disruption of the NHEJ pathway resulting in increased levels of homology-directed repair, although we note that gene conversion levels were not increased in these backgrounds. These results led us to speculate that the wild-type efficiency of DSB repair may be compromised through competition between the NHEJ and HR pathways (Figure 9). These results strongly resemble those observed in both S.cerevisiae and mammalian cell studies, in which homology-directed repair was enhanced in cells deficient for NHEJ (Clikeman et al., 2001; Pierce et al., 2001b). The increased repair efficiency associated with the NHEJ mutants was associated with DSB-induced chromosomal rearrangements observed in 10% of the ade+ G418S ku70Δ and 3.4% of the ade+ G418S lig4Δ colonies examined. These findings identify an important role for components of the NHEJ pathway in maintaining chromosomal stability through suppression of DSB-induced chromosomal rearrangements in fission yeast. The simplest explanation for these chromosomal rearrangements is that interchromosomal gene conversion was accompanied by crossing over between ChIII and Ch16-MG following DSB induction in NHEJ mutants. This hypothesis is supported by the following findings: (i) chromosomal rearrangements were detected in ade+ G418S colonies, and thus were likely to have arisen through gene conversion; (ii) chromosomal rearrangements could only be detected between ChIII and its homologue, Ch16-MG, in a DSB-dependent manner, indicating that these rearrangements were associated with homologous recombination and were not random events; and (iii) chromosomal element sizes were consistent with sizes predicted from crossover events. We note that ChX and Y were clearly resolved in a lig4Δ background compared with those obtained in a ku70Δ background, indicating that additional events presumably were associated with HR-induced crossovers in this strain. Although the precise mechanisms of chromosomal rearrangements await further analysis in these mutants, it is possible that the distal arm of Ch16-MG has been lost as a result of a failed ligation step following DSB induction in the lig4Δ background.
As reduced levels of DSB-induced Ch16-MG loss and appearance of novel chromosomal rearrangements appear to be linked in both ku70Δ and lig4Δ backgrounds, this suggests a role for the NHEJ pathway in maintaining genome stability through a mechanism that functions at the cost of overall repair efficiency. In this respect, the NHEJ pathway might function to suppress DSB-induced crossovers (Figure 9). The finding that both Ku70 and Lig4 function to promote error-free HR indicates that components of the NHEJ and HR pathways can function cooperatively as well as competitively. These studies therefore identify a complex relationship between the NHEJ and HR pathways.
Importantly, these findings resemble those obtained in mouse studies, in which chromosomal rearrangements, including translocations, were associated with DSBs in both Ku80–/– p53–/– and Lig4–/– p53–/– mice, resulting in pro-B cell lymphomas (Difilippantonio et al., 2000; Frank et al., 2000). Further analysis of the role of NHEJ genes in suppressing chromosomal rearrangements in fission yeast is therefore likely to contribute to our understanding of tumorigenesis.
The role of the DNA integrity checkpoint in site-specific DSB repair
We demonstrated that induction of a site-specific DSB generated a checkpoint-dependent cell cycle delay. The Rad3 checkpoint protein was additionally shown to facilitate efficient HR, as indicated by the significantly reduced levels of DSB-induced gene conversion observed in a rad3Δ mutant. These findings indicate a role for the DNA integrity checkpoint in promoting HR in S.pombe, consistent with other recent studies (Bashkirov et al., 2000; Caspari et al., 2002; Osman et al., 2002). The ability to generate a checkpoint response through production of a defined lesion provides a means to study the ordered association of checkpoint proteins with the lesion, and also the possibility of identifying a direct role for checkpoint proteins in DSB repair. It is anticipated that further application and development of this technology will provide insights into the relationship between checkpoint, repair and other pathways in the cellular responses to DSBs in fission yeast, and will thus contribute to our understanding of how these conserved pathways are coordinated in eukaryotes.
Materials and methods
Yeast strains, media and genetic methods
The strains used in this study are listed in Table IV. Cells were cultured in complete media (YE5S), synthetic minimal media (EMM2) and sporulation (ME) media, as described in Moreno et al. (1991). pACYCREP81X-HO (termed pREP81X-HO in this study) is a multicopy plasmid, which contained the HO-endonuclease under the control of the rep81X nmt promoter (Osman et al., 1996). Thiamine (8 µM) was added to EMM2 to repress the nmt promoter (Maundrell, 1990). The construction of Ch16-rad21::MATa-kanMX6 is described in the Supplementary data.
Table IV. Schizosaccharomyces pombe strains used for this study.
| Strain | Genotype |
|---|---|
| TH805 | leu1-32 ade6-M210 ura4-D18 his3+ Ch16-MG h+ |
| TH844 | leu1-32 ade6-M210 ura4-D18 his3+ Ch16-MG h+ pREP81X-HO |
| TH871 | rhp55::ura4+ leu1-32 ade6-M210 ura4-D18 his3+ Ch16-MG h+ pREP81X-HO |
| TH873 | rad16::ura4+ leu1-32 ade6-M210 ura4-D18 Ch16-MG pREP81X-HO |
| TH876 | rad3::ura4+ leu1-32 ade6-M210 ura4-D18 Ch16-MG pREP81X |
| TH877 | rad3::ura4+ leu1-32 ade6-M210 ura4-D18 Ch16-MG pREP81X-HO |
| TH895 | rhp51::ura4+ leu1-32 ade6-M210 ura4-D18 his3+ Ch16-MG h+ pREP81X-HO |
| TH906 | rad22::ura4+ leu1-32 ade6-M210 ura4-D18 Ch16-MG pREP81X-HO |
| TH932 | ku70::ura4+ leu1-32 ade6-M210 ura4-D18 Ch16-MG pREP81X-HO |
| TH987 | rhp51::ura4+ ku70::his3+ leu1-32 ade6-M210 ura4-D18 his3-D1 Ch16-MG pREP81X-HO |
| TH1009 | rhp55::ura4+ ku70::his3+ leu1-32 ade6-M210 ura4-D18 his3-D1 Ch16-MG pREP81X-HO |
| TH1010 | leu1-32 ade6-M210 ura4-D18 his3+ Ch16-MG h+ pREP81X |
| TH1083 | rad32::ura4+ leu1-32 ade6-M210 ura4-D18 Ch16-MG pREP81X-HO |
| TH1102 | rhp55::ura4+ rhp51::ura4+ leu1-32 ade6-M210 ura4-D18 his3-D1 Ch16-MG pREP81X-HO |
| TH1017 | rad21-K1 leu1-32 ade6-M210 ura4-D18 h– |
| TH1216 | lig4::ura4+ leu1-32 ade6-M210 ura4-D18 Ch16-MG pREP81X-HO |
Ch16 represents ade6-M216, which is present on minichromosome 16 (Niwa et al., 1986). Ch16-MG represents ade6-M216 rad21::MATa-kanMX6, which is present on Ch16. The rhp55::ura4+ disruptant was not the same strain as that previously described (Khasanov et al., 1999). For strain construction details, see Supplementary data. Unless otherwise indicated, all strains were maintained at 30°C.
Analysis of site-specific DSB responses
Strains transformed with pREP81X-HO were maintained on EMM + UHT, prior to performing the time courses, to select for Ch16-MG and the pREP81X-HO plasmid. The time course was initiated by washing cells twice in phosphate-buffered saline (PBS), followed by culturing in log phase, for up to 48 h, in either EMM + AUHT or EMM + AUH media to maintain plasmid selection but permit Ch16-MG loss. To perform the genetic DSB repair assay, cells were plated onto non-selective (YE5S) plates, incubated at 30°C and the total numbers of colonies calculated. Colonies were replica-plated onto YE5S + G418 (500 mg/l geneticin) and ade– (EMM + UHLT) plates, to calculate the percentage of the population that had become G418S or ade–, respectively. Cell viability was determined through spotting serial dilutions of cultures onto EMM + UHAT or EMM + UHA plates, incubating at 30°C and determining colony-forming ability. Southern blot analysis and PFGE conditions are described in the Supplementary data.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We are grateful to Drs Fekret Osman, Tony Carr, Felicity Watts and Robin Allshire for strains and reagents provided, and to Drs Joel Huberman, Amanda Pearce and David Papworth for helpful comments. This work was supported by the Medical Research Council and Commission of the European Communities.
References
- Bashkirov V.I., King,J.S., Bashkirova,E.V., Schmuckli-Maurer,J. and Heyer,W.D. (2000) DNA repair protein Rad55 is a terminal substrate of the DNA damage checkpoints. Mol. Cell. Biol., 20, 4393–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr A.M., (2002) DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair, 1, 983–994. [DOI] [PubMed] [Google Scholar]
- Carr A.M. et al. (1994) The rad16 gene of Schizosaccharomyces pombe: a homolog of the RAD1 gene of Saccharomyces cerevisiae. Mol. Cell. Biol., 14, 2029–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspari T., Murray,J.M. and Carr,A.M. (2002) Cdc2–cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev., 16, 1195–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clikeman J.A., Khalsa,G.J., Barton S.L. and Nickoloff,J.A (2001) Homologous recombinational repair of double-strand breaks in yeast is enhanced by MAT heterozygocity through yKU-dependent and independent mechanisms. Genetics, 157, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amours D. and Jackson,S.P. (2002) The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell. Biol., 3, 317–327. [DOI] [PubMed] [Google Scholar]
- Difilippantonio M.J., Zhu,J., Chen,H.T., Meffre,E., Nussenzweig,M.C., Max,E.E., Ried,T. and Nussenzweig,A. (2000) DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature, 404, 510–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty A.J. and Jackson,S.P. (2001) DNA repair: how Ku makes ends meet. Curr. Biol., 11, R920–R924. [DOI] [PubMed] [Google Scholar]
- Fishman-Lobell J. and Haber,J.E. (1992) Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science, 258, 480–484. [DOI] [PubMed] [Google Scholar]
- Fortin G.S. and Symington,L.S. (2002) Mutations in yeast Rad51 that partially bypass the requirement for Rad55 and Rad57 in DNA repair by increasing the stability of Rad51–DNA complexes. EMBO J., 21, 3160–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank K.M. et al. (2000) DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell, 5, 993–1002. [DOI] [PubMed] [Google Scholar]
- Grawunder U., Wilm,M., Wu,X., Kulesza,P., Wilson,T.E., Mann,M. and Lieber,M.R. (1997) Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature, 388, 492–495. [DOI] [PubMed] [Google Scholar]
- Hartsuiker E., Vaessen,E., Carr,A.M. and Kohli,J. (2001) Fission yeast Rad50 stimulates sister chromatid recombination and links cohesion with repair. EMBO J., 20, 6660–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays S.L., Firmenich,A.A. and Berg,P. (1995) Complex formation in yeast double-strand break repair: participation of Rad51, Rad52, Rad55 and Rad57 proteins. Proc. Natl Acad. Sci. USA, 92, 6925–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey T. (2000) DNA damage and cell-cycle control in Schizosaccharomyces pombe. Mutat. Res., 451, 211–226. [DOI] [PubMed] [Google Scholar]
- Ivanov E.L. and Haber,J.E. (1995) RAD1 and RAD10, but not other excision repair genes, are required for double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 2245–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadyk L.C. and Hartwell,L.H. (1992) Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics, 132, 387–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L.E. and Symington,L.S. (2000) Aberrant double-strand break repair in rad51 mutants of Saccharomyces cerevisiae. Mol. Cell. Biol., 20, 9162–9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasanov F.K., Savchenko,G.V., Bashkirova,E.V., Korolev,V.G., Heyer,W.D. and Bashkirov,V.I. (1999) A new recombinational DNA repair gene from Schizosaccharomyces pombe with homology to Escherichia coli RecA. Genetics, 152, 1557–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leupold U. and Gutz,H. (1964) Genetics Today: Proceedings of the XIth International Congress on Genetics, Vol. 3, The Hague, The Netherlands, 1963. Pergamon Press, Oxford, UK, pp. 31–35.
- Lewis L.K. and Resnick,M.A. (2000) Tying up loose ends: non homologous end-joining in Saccharomyces cerevisiae. Mutat. Res., 451, 71–89. [DOI] [PubMed] [Google Scholar]
- Manolis K.G., Nimmo,E.R., Hartsuiker,E., Carr,A.M., Jeggo,P.A. and Allshire,R.C. (2001) Novel functional requirements for non-homologous DNA end joining in Schizosaccharomyces pombe. EMBO J., 20, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maundrell K. (1990) nmt1 of fission yeast. A highly transcribed gene completely repressed by thiamine. J. Biol. Chem., 265, 10857–10864. [PubMed] [Google Scholar]
- Modesti M., Hesse,J.E. and Gellert,M. (1999) DNA binding of Xrcc4 protein is associated with V(D)J recombination but not with stimulation of DNA ligase IV activity. EMBO J., 18, 2008–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S., Klar,A. and Nurse,P. (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol., 194, 795–823. [DOI] [PubMed] [Google Scholar]
- Muris D.F., Vreeken,K., Carr,A.M., Broughton,B.C., Lehmann,A.R., Lohman,P.H. and Pastink,A. (1993) Cloning the RAD51 homologue of Schizosaccharomyces pombe. Nucleic Acids Res., 21, 4586–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muris D.F., Vreeken,K., Carr,A.M., Murray,J.M., Smit,C., Lohman,P.H. and Pastink,A. (1996) Isolation of the Schizosaccharomyces pombe RAD54 homologue, rhp54+, a gene involved in the repair of radiation damage and replication fidelity. J. Cell Sci., 109, 73–81. [DOI] [PubMed] [Google Scholar]
- Muris D.F., Vreeken,K., Schmidt,H., Ostermann,K., Clever,B., Lohman,P.H. and Pastink,A. (1997) Homologous recombination in the fission yeast Schizosaccharomyces pombe: different requirements for the rhp51+, rhp54+ and rad22+ genes. Curr. Genet., 31, 248–254. [DOI] [PubMed] [Google Scholar]
- Niwa O., Matsumoto,T. and Yanagida,M. (1986) Construction of a mini-chromosome by deletion and its mitotic and meiotic behaviour in fission yeast. Mol. Gen. Genet., 203, 397–405. [Google Scholar]
- Osman F., Fortunato,E.A. and Subramani,S. (1996) Double-strand break-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Genetics, 142, 341–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman F., Adriance,M. and McCready,S. (2000) The genetic control of spontaneous and UV-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Curr. Genet., 38, 113–125. [DOI] [PubMed] [Google Scholar]
- Osman F., Tsaneva,I.R., Whitby,M.C. and Doe,C.L. (2002) UV irradiation causes the loss of viable mitotic recombinants in Schizosaccharomyces pombe cells lacking the G(2)/M DNA damage checkpoint. Genetics, 160, 891–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann K., Lorentz,A. and Schmidt,H. (1993) The fission yeast rad22 gene, having a function in mating-type switching and repair of DNA damages, encodes a protein homolog to Rad52 of Saccharomyces cerevisiae. Nucleic Acids Res., 21, 5940–5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F. and Haber,J.E. (1999) Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev., 63, 349–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastink A., Eeken,J.C. and Lohman,P.H. (2001) Genomic integrity and the repair of double-strand DNA breaks. Mutat. Res., 480–481, 37–50. [DOI] [PubMed] [Google Scholar]
- Petukhova G., Stratton,S. and Sung,P. (1998) Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature, 393, 91–94. [DOI] [PubMed] [Google Scholar]
- Pfeiffer P., Goedecke,W. and Obe,G. (2000) Mechanisms of DNA double-strand break repair and their potential to induce chromosomal aberrations. Mutagenesis, 15, 289–302. [DOI] [PubMed] [Google Scholar]
- Pierce A.J., Stark,J.M., Araujo,F.D., Moynahan,M.E., Berwick,M. and Jasin,M. (2001a) Double-strand breaks and tumorigenesis. Trends Cell Biol., 11, S52–S59. [DOI] [PubMed] [Google Scholar]
- Pierce A.J., Hu,P., Han,M., Ellis,N. and Jasin,M. (2001b) Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev., 15, 3237–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H., Kapitza-Fecke,P., Stephen,E.R. and Gutz,H. (1989) Some of the swi genes of Schizosaccharomyces pombe also have a function in the repair of radiation damage. Curr. Genet., 16, 89–94. [DOI] [PubMed] [Google Scholar]
- Sugiyama T., New,J.H. and Kowalczykowski,S.C. (1998) DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc. Natl Acad. Sci. USA, 95, 6049–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P. (1997) Yeast Rad55 and Rad57 proteins form a heterodimer that functions with replication protein A to promote DNA strand exchange by Rad51 recombinase. Genes Dev., 11, 1111–1121. [DOI] [PubMed] [Google Scholar]
- Suto K., Nagata,A., Murakami,H. and Okayama,H. (1999) A double-strand break repair component is essential for S phase completion in fission yeast cell cycling. Mol. Biol. Cell, 10, 3331–3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavassoli M., Shayeghi,M., Nasim,A. and Watts,F.Z. (1995) Cloning and characterisation of the Schizosaccharomyces pombe rad32 gene: a gene required for repair of double strand breaks and recombination. Nucleic Acids Res., 23, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui Y., Morishita,T., Iwasaki,H., Toh,H. and Shinagawa,H. (2000) A recombination repair gene of Schizosaccharomyces pombe, rhp57, is a functional homolog of the Saccharomyces cerevisiae RAD57 gene and is phylogenetically related to the human XRCC3 gene. Genetics, 154, 1451–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bosch M., Vreeken,K., Zonneveld,J.B., Brandsma,J.A., Lombaerts,M., Murray,J.M., Lohman,P.H. and Pastink,A. (2001) Characterization of RAD52 homologs in the fission yeast Schizosaccharomyces pombe. Mutat. Res., 461, 311–323. [DOI] [PubMed] [Google Scholar]
- Yaneva M., Kowalewski,T. and Lieber,M.R. (1997) Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy studies. EMBO J., 16, 5098–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo S. and Dynan,W.S. (1999) Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res., 27, 4679–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.B. and Elledge,S.J. (2000) The DNA damage response: putting checkpoints in perspective. Nature, 408, 433–439. [DOI] [PubMed] [Google Scholar]