

Abstract
Mdm2 harnesses the p53 tumor suppressor, yet loss of one Mdm2 allele in Mdm2+/– mice has heretofore not been shown to impair tumor development. Here we report that Mdm2 haplo-insufficiency profoundly suppresses lymphomagenesis in Eµ-myc transgenic mice. Mdm2+/–Eµ-myc transgenics had greatly protracted rates of B cell lymphoma development with life spans twice that of wild-type transgenic littermates. Im paired lymphoma development was associated with drastic reductions in peripheral B cell numbers in Mdm2+/–Eµ-myc transgenics, and primary pre-B cells from Mdm2+/–Eµ-myc transgenics and Mdm2+/– littermates were extremely susceptible to spontaneous apoptosis. Loss of p53 rescued all of the effects of Mdm2 haplo-insufficiency, indicating they were p53 dependent. Furthermore, half of the lymphomas that ultimately emerged in Mdm2+/–Eµ-myc transgenics harbored inactivating mutations in p53, and the majority overcame haplo-insufficiency by overexpressing Mdm2. These results support the concept that Mdm2 functions are rate limiting in lymphomagenesis and that targeting Mdm2 will enhance p53-mediated apoptosis, compromising tumor development and/or maintenance.
Keywords: apoptosis/lymphoma/Mdm2/Myc/p53
Introduction
Mdm2 (murine double minute 2, termed Hdm2 in humans) is amplified in 10% of all human malignancies (Momand et al., 1998), and Mdm2 protein is frequently overexpressed in murine and human lymphomas through other mechanisms (Watanabe et al., 1996; Eischen et al., 1999). Mdm2’s oncogenic properties are due in part to its ability to inactivate the p53 tumor suppressor (reviewed in Momand et al., 2000). Mdm2 is an E3 ubiquitin ligase that inhibits p53 by blocking p53 transcriptional activity (Momand et al., 1992), ubiquitylating p53 (Honda and Yasuda, 1999) and shuttling p53 from the nucleus into the cytoplasm where it is degraded by the proteosome (Freedman and Levine, 1998; Roth et al., 1998). Mdm2-null mice die shortly after implantation due to unregulated p53 activity (de Rozieres et al., 2000), as the lethality of Mdm2-null mice is rescued by crossing them to p53-null mice (Jones et al., 1995; Montes de Oca Luna et al., 1995). Mdm2 is a direct transcriptional target of p53, and therefore Mdm2 is the critical component of a negative feedback loop that regulates p53 activity (Wu et al., 1993). However, Mdm2’s regulation of p53 is also controlled by the ARF tumor suppressor. ARF binds to and can sequester Mdm2 in the nucleolus (Tao and Levine, 1999; Weber et al., 1999). However, nucleolar relocalization of Mdm2 by ARF is not essential to inhibit Mdm2 (Korgaonkar et al., 2002), as ARF also blocks Mdm2 ubiquitin ligase activity, p53 degradation and transcriptional inactivation of p53 (Kamijo et al., 1998; Pomerantz et al., 1998; Stott et al., 1998; Zhang et al., 1998; Honda and Yasuda, 1999).
Both ARF and p53 mediate apoptosis in response to hyperproliferation signals from oncogenes, including Myc, E2F-1, E1A and Ras (reviewed in Sherr, 1998). Consequently, inactivation of ARF or p53 occurs frequently in cancers that overexpress oncogenes, including the majority of B cell lymphomas that arise in Eµ-myc transgenic mice engineered to overexpress Myc in the B cell compartment (Eischen et al., 1999; Schmitt et al., 1999). In Eµ-myc transgenics, a mouse model of human non-Hodgkin’s lymphoma, inactivation of p53 and ARF occurred in 28% and 24%, respectively, of the B cell lymphomas that emerged (Eischen et al., 1999). Furthermore, loss of ARF or p53 confers resistance to Myc-induced apoptosis and accelerates lymphoma development in Eµ-myc transgenics (Eischen et al., 1999; Jacobs et al., 1999; Schmitt et al., 1999), highlighting the importance of ARF and p53 in inhibiting oncogene-induced tumorigenesis.
Despite the essential role ARF and p53 play in Myc-initiated lymphomagenesis, little is known about Mdm2’s role in lymphoma development. A number of reports have shown that lymphomas in both humans and Eµ-myc transgenic mice frequently overexpress Mdm2 protein without gene amplification (Momand and Zambetti, 1997; Eischen et al., 1999, 2001b). Furthermore, enforced expression of Mdm2 in transgenic mice where mdm2 was under the control of its own promoter results in late-onset of lymphomas and sarcomas (Jones et al., 1998). Although these findings suggest that Mdm2 expression can contribute to lymphoma development, it was unclear whether Mdm2 plays an essential role during lymphomagenesis and whether this was linked to Mdm2’s ability to harness p53. Here we show that Mdm2 haplo-insufficiency has profound and surprising effects in impairing lymphoma development in Eµ-myc transgenic mice. The inhibition of B cell lymphomagenesis by Mdm2 haplo-insufficiency was due to a marked increase in p53-dependent apoptosis of B cells, resulting in severely reduced numbers of peripheral B cells in Mdm2+/–Eµ-myc transgenics. Finally, the lymphomas that ultimately did arise in Mdm2+/–Eµ-myc mice preferentially suffered mutations that inactivated p53, and also compensated for Mdm2 haplo-insufficiency by overexpressing Mdm2. Therefore, Mdm2 functions are rate limiting in tumorigenesis and targeting Mdm2 in even a quantitative fashion may prove efficacious in cancer therapy.
Results
Mdm2 haplo-insufficiency inhibits Myc-induced lymphomagenesis
Eµ-myc transgenic mice develop pre-B and/or B cell lymphoma (Adams et al., 1985), and half of these lymphomas overexpress Mdm2 protein (Eischen et al., 1999, 2001b). This observation suggests that Mdm2 overexpression is selected for during lymphoma development and that Mdm2 may be necessary for and/or facilitate Myc-induced lymphomagenesis. Therefore, we postulated that a decrease in Mdm2 expression would inhibit lymphoma development. To test this issue, we crossed Mdm2 heterozygous mice (Jones et al., 1995; Montes de Oca Luna et al., 1995) to Eµ-myc transgenics and evaluated whether there were any effects of Mdm2 haplo-insufficiency on Myc-induced lymphomagenesis. Strikingly, lymphoma development was drastically delayed in Mdm2+/–Eµ-myc transgenics. The Mdm2+/–Eµ-myc transgenics had an average survival of 44.3 weeks, more than twice that of the 20.6 week average survival for the Mdm2+/+Eµ-myc transgenic littermates (log-rank test P < 0.001; Figure 1). Furthermore, 20% (9/45) of the Mdm2+/–Eµ-myc transgenics failed to develop lymphoma (and are still alive), whereas only 5% (3/58) of the Mdm2+/+Eµ-myc transgenics remained disease free. These results support the hypothesis that Mdm2 functions are rate limiting for tumor development during Myc-induced lymphomagenesis.

Fig. 1. Myc-induced lymphomagenesis is inhibited by Mdm2 haplo-insufficiency. Kaplan–Meier survival curves of Mdm2+/–Eµ-myc transgenic, Mdm2+/+Eµ-myc transgenic, p53+/–Eµ-myc transgenic and p53+/–Mdm2+/–Eµ-myc transgenic mice. The average survivals are 44.3, 20.6, 5.6 and 5 weeks, respectively (log-rank test, P < 0.001). n, the number of mice in each group. Vertical lines indicate ages of surviving mice. Three (3/58) Mdm2+/+Eµ-myc transgenic mice and nine (9/45) Mdm2+/–Eµ-myc transgenic mice are still alive, whereas none of the p53+/–Eµ-myc and p53+/–Mdm2+/–Eµ-myc transgenics is alive. The Kaplan–Meier curves are right-censored because the study was terminated before all of the animals were sacrificed. Therefore, although 20% (9/45) of the Mdm2+/–Eµ-myc transgenic mice are still alive at 70 weeks, the Kaplan–Meier estimate at 70 weeks is lower because there were fewer animals at risk of death at 70 weeks. Lymphoma was documented in all of the animals.
The B cell lymphomas that arose in the Mdm2+/–Eµ-myc transgenic mice were typical of those that arise in Eµ-myc transgenics, with the majority having a large diffuse cell lymphoma and expressing IgM (data not shown). Both wild-type Eµ-myc and Mdm2+/–Eµ-myc transgenics also developed splenomegaly, with spleens 10–20 times the normal size, and both genotypes had frequent metastases to the lungs. Surprisingly, many of the lymphomas that ultimately emerged in the Mdm2+/–Eµ-myc transgenics failed to localize to peripheral lymph nodes. In wild-type Eµ-myc transgenics, B cell lymphomas usually arise in the peripheral lymph nodes in the neck, front and back legs and can also be observed in the mesenteric lymph nodes in the abdomen and occasionally in the thymus (Adams et al., 1985; Harris et al., 1988). In contrast, the primary site of lymphoma development in the majority (58%, 21/36) of Mdm2+/–Eµ-myc transgenics appeared to be in the liver and mesenteric lymph nodes, followed by the thymus (data not shown), especially in older mice. The underlying cause for altered sites of lymphomagenesis is unclear, as Mdm2 has not been reported to regulate lymphocyte homing. Nonetheless, although many of the lymphomas arose in atypical regions, they were typical B cell lymphomas as determined by FACS analysis (B220+, CD19+) and/or immunohistochemistry (B220+) (data not shown). Therefore, along with a delay in tumor onset, there was also a difference in the sites of B cell lymphomagenesis in the Mdm2 haplo-insufficient Eµ-myc transgenics.
Mdm2 haplo-insufficiency predisposes B cells to apoptosis
Our previous studies demonstrated a crucial role for ARF- and p53-dependent apoptosis in inhibiting lymphoma development in Eµ-myc transgenic mice (Eischen et al., 1999, 2001b). We therefore addressed whether the profound delay in lymphoma development in Mdm2+/–Eµ-myc transgenic mice was due to an altered susceptibility of Mdm2+/– B cells to Myc-induced apoptosis. We attempted to address this issue by infecting bone marrow-derived primary pre-B cells isolated from Mdm2+/– and wild-type littermates with a retrovirus encoding a 4-hydroxytamoxifen (HT)-inducible myc fusion protein (termed Myc-ERTAM, described in the Materials and methods; Littlewood et al., 1995) that we have previously utilized (Zindy et al., 1998; Eischen et al., 1999, 2001b,c). Surprisingly, however, Mdm2+/– pre-B cells had very high apoptotic indices (<40% viable at day 19) and failed to grow ex vivo, whereas wild-type pre-B cells from littermate controls were viable (>85% at day 19) and readily expanded in culture (Figure 2A). This finding was unexpected, as Mdm2+/– mice have been reported to have normal numbers of B cells and no obvious lymphocyte defects (Jones et al., 1995; Montes de Oca Luna et al., 1995), but clearly Mdm2+/– pre-B cells are highly susceptible to apoptosis when cultured ex vivo.

Fig. 2. Mdm2 heterozygous bone marrow is sensitive to spontaneous and Myc-induced apoptosis. Bone marrow from two Mdm2+/– mice [(A), triangles], two wild-type littermates (A, squares), two Mdm2+/–Eµ-myc transgenics [(B), triangles], and two Mdm2+/+Eµ-myc transgenic littermate controls (B, squares) prior to any detectable lymphoma was placed into IL-7-containing medium (day 0). Cells were counted on the indicated days. Pre-B cell growth was calculated as net population doublings at the indicated intervals. Viability was determined by Trypan Blue dye exclusion and apoptosis was verified by PI staining.
Since overexpression of Myc by retroviral infection of Mdm2+/– B cells was not feasible, we assessed effects of Myc overexpression in bone marrow from 10- to 12-week-old Mdm2+/–Eµ-myc and wild-type Eµ-myc transgenic littermates prior to any detectable disease. The bone marrow cells were cultured in interleukin (IL)-7-containing medium, as described previously (Eischen et al., 1999). Not surprisingly, Mdm2+/–Eµ-myc transgenic pre-B cells were unable to grow ex vivo (Figure 2B), due to very high rates of spontaneous apoptosis. In contrast, pre-B cells from wild-type Eµ-myc transgenics, after a normal initial lag period (Eischen et al., 1999), proliferated over the same time frame (Figure 2B). Therefore, Mdm2 haplo-insufficient B cells are prone to undergo apoptosis ex vivo, particularly when Myc is overexpressed.
To determine whether Mdm2 haplo-insufficiency had similar effects when combined with Myc overexpression in vivo, we directly measured the apoptotic index in splenocytes of Mdm2+/–Eµ-myc transgenics and Mdm2+/+Eµ-myc transgenics prior to any detectable disease, and also in Mdm2+/– and wild-type mice. Splenocytes from both Mdm2+/+Eµ-myc transgenics and Mdm2+/– mice had a higher apoptotic index than splenocytes from wild-type controls (Figure 3). Importantly, there was also a greater percentage of apoptotic splenocytes in Mdm2+/–Eµ-myc transgenics, compared with the percentage of apoptotic splenocytes in wild-type Eµ-myc transgenics (Figure 3). Therefore, loss of one Mdm2 allele alone increases the susceptibility of B cells to apoptosis, and this response is augmented in cells overexpressing Myc.

Fig. 3. Increased apoptosis in splenocytes from Mdm2+/–Eµ-myc transgenics. Disaggregated, ethanol fixed splenocytes from the indicated genotypes prior to any detectable disease were stained with PI and analyzed on a FACScan. Sub-G1 DNA was quantified from three separate mice of each genotype with CellQuest software. Each bar is an average and error bars represent 1 SD.
To address potential effects of Mdm2 haplo-insufficiency on lymphocyte populations, we performed detailed phenotyping of B cells in the spleens of Mdm2+/–Eµ-myc transgenics. Normally, mature B cells (IgM+, CD19+, B220+) account for 45–50% of the cells in a mouse spleen, and the B cell numbers from wild-type and Mdm2+/– spleens were in accord with this value (Figure 4A). As previously reported (Adams et al., 1985; Langdon et al., 1986), there was a population of B cell precursors IgM–/CD19+ (8.8%), and slightly reduced numbers (34%) of mature IgM+/CD19+ B cells, in the spleens of the Eµ-myc transgenics (Figure 4A). In contrast, Mdm2+/–Eµ-myc transgenics had profoundly reduced numbers of splenic B cells. Only 5–18% of the cells in the spleens of Mdm2+/–Eµ-myc transgenics were IgM+ B cells, and these mice completely lacked peripheral IgM–/CD19+ B cell precursors (Figure 4A). There were also modest increases (36% versus 23%) in the percentage of splenic T cells (CD3+) in the Mdm2+/–Eµ-myc transgenic mice (Figure 4B). However, this may simply reflect the decreased levels of B cells in the spleens of Mdm2+/–Eµ-myc transgenics. Therefore, Mdm2 haplo-insufficient Eµ-myc transgenic mice have very few splenic B cells, and this may have also contributed to the delayed lymphoma development in these mice.

Fig. 4. Mdm2+/–Eµ-myc transgenics have drastically reduced numbers of B cells. (A and B) Splenic cells from mice of the indicated genotypes prior to any detectable disease were stained with fluorescent antibodies specific for B cells (IgM, CD19) and T cells (CD3) and subjected to flow cytometry phenotype analysis. The location of the quadrant axes was determined from isotype controls, and the numbers in the quadrants are the percentage of cells in each of those quadrants. Data are representative of five separate experiments.
To determine whether the decreased B cell numbers in the spleens of Mdm2+/–Eµ-myc transgenics was also reflected in the numbers of circulating lymphocytes, we quantitated peripheral lymphocytes from whole blood of mice prior to any detectable disease. Strikingly, Mdm2+/–Eµ -myc transgenic mice had less than half the number of circulating lymphocytes (2750 lymphocytes/µl) compared with wild-type Eµ-myc transgenic controls (6170 lymphocytes/µl), which were similar to wild-type controls (6800 lymphocytes/µl; Figure 5). Notably, the number of peripheral blood lymphocytes in Mdm2+/–Eµ-myc transgenics was also appreciably lower than the peripheral blood lymphocyte numbers in non-transgenic Mdm2+/– mice (5310 lymphocytes/µl; Figure 5). There fore, there is a combined inhibitory effect of Myc overexpression and Mdm2 haplo-insufficiency on peripheral lymphocyte numbers in vivo.

Fig. 5. Mdm2+/–Eµ-myc transgenics have reduced numbers of circulating lymphocytes. Peripheral blood lymphocytes were counted from retro-orbital eye bleeds from wild-type, Mdm2+/–, Mdm2+/+Eµ-myc transgenic and Mdm2+/–Eµ-myc transgenic mice prior to any detectable disease. n, number of animals that were analyzed from each genotype. Error bars represent 1 SD. K/µl = 1000 cells/µl.
The effects of Mdm2 haplo-insufficiency are p53 dependent
Since loss of one allele of Mdm2 leads to B cell apoptosis ex vivo and in vivo, and Mdm2-null embryos appear to die from constitutive p53 activation (de Rozieres et al., 2000), we predicted that the propensity of the Mdm2+/– B cell precursors to undergo apoptosis, particularly when Myc was overexpressed, was due to dysregulated p53. To test this issue, we evaluated pre-B cells from p53–/–, p53–/–Mdm2+/– and p53–/–Mdm2–/– mice. Both Mdm2+/– and Mdm2–/– pre-B cells lacking p53 proliferated ex vivo and grew at rates similar to those of p53-null pre-B cells (Figure 6A). All pre-B cells lacking p53, regardless of their Mdm2 status, were >95% viable and proliferated more rapidly than wild-type pre-B cells (Figure 6A), as previously reported for primary p53-null or p53/Mdm2 double-null cells (McMasters et al., 1996; Eischen et al., 1999). Therefore, the spontaneous apoptosis of the Mdm2+/– pre-B cells (Figure 2A) was p53 dependent.
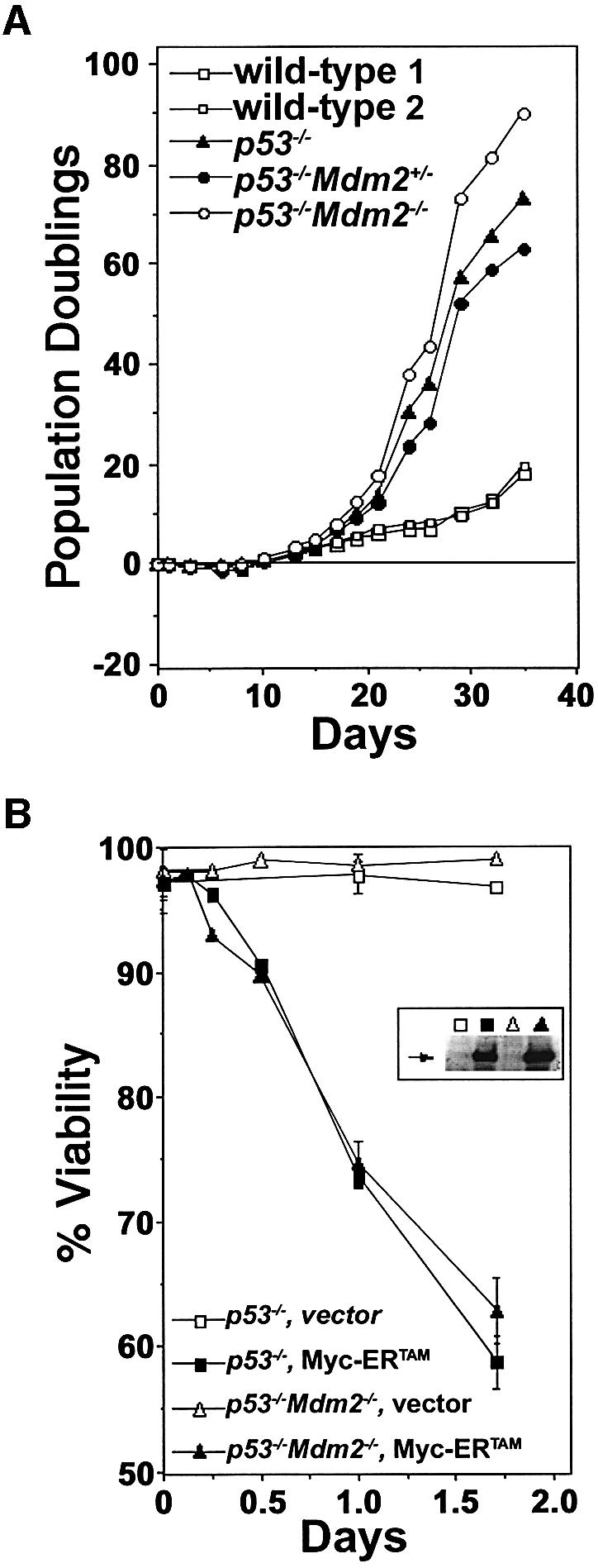
Fig. 6. Loss of p53 inhibits apoptosis of Mdm2+/– pre-B cells. (A) Bone marrow cells from p53–/–, p53–/–Mdm2+/–, p53–/–Mdm2–/– (prior to any detectable disease) and two wild-type littermates were placed into IL-7 containing medium (day 0) and pre-B cell growth, calculated as net population doublings, was determined at the indicated intervals. (B) 4-HT was added to the indicated primary pre-B cell cultures to activate Myc-ERTAM, and their viability was determined at intervals thereafter by Trypan Blue dye exclusion. Apoptosis was confirmed by analysis of subdiploid DNA content after staining with PI. Steady-state levels of apoptosis in the wild-type primary pre-B cells are indicated at the 0 h time point. Data points are an average of at least three separate experiments, and error bars represent 1 SD. (B, inset) The protein levels of Myc-ERTAM in the indicated pre-B cells was determined by immunoblotting with an antibody specific for Myc. Arrow denotes location of Myc-ERTAM.
To determine whether p53 loss also influenced the sensitivity of Mdm2-deficient B cells to Myc-induced apoptosis, we infected pre-B cells lacking p53 alone or also lacking Mdm2 with a retrovirus encoding Myc-ERTAM or vector-only virus. As expected (Eischen et al., 1999), the rates of Myc-induced apoptosis following the addition of 4-HT was markedly impaired in p53-null pre-B cells. Importantly, the rates of Myc-induced apoptosis were comparable in p53-null and Mdm2/p53 double-null pre-B cells following the addition of 4-HT (Figure 6B). Thus, p53 loss blocks the inherently high rates of spontaneous and Myc-induced apoptosis of Mdm2- deficient pre-B cells, and Mdm2 haplo-insufficiency impairs Myc-induced lymphomagenesis by augmenting p53-dependent apoptosis.
p53 is preferentially inactivated in Mdm2+/–Eµ-myc transgenic lymphomas
p53 and ARF are inactivated in 28% and 24%, respectively, of lymphomas arising in Eµ-myc transgenics (Eischen et al., 1999). If Mdm2 haplo-insufficiency augments p53 activation in pre-cancerous transgenic B cells, then one would predict that many of the lymphomas that ultimately arise in Mdm2+/–Eµ-myc transgenics would harbor mutations in p53. To address this issue, we analyzed the p53, ARF and Mdm2 status in the lymphomas that arose in Mdm2+/–Eµ-myc transgenics. Importantly, half (48%, 11/23) of the Mdm2+/–Eµ-myc lymphomas had sustained p53 mutations, which is detected as greatly increased levels of p53 protein and concomitant elevated ARF protein (Figure 7; Table I). The sites of the p53 mutations in these lymphomas, as determined by sequence analysis, were within the DNA binding core domain, a hot spot for p53 mutations (data not shown). p53 is infrequently deleted in wild-type Eµ-myc lymphomas (Eischen et al., 1999), and similarly Southern blot analysis demonstrated that only one lymphoma from an Mdm2+/–Eµ-myc transgenic had deleted p53 (Table I). However, p53 can also be inactivated by Mdm2 overexpression (Momand et al., 2000), and Mdm2 protein is overexpressed in half of all lymphomas that arise in wild-type Eµ-myc transgenics (Eischen et al., 1999). The percentage of lymphomas overexpressing Mdm2 protein was even higher in the lymphomas that arose in Mdm2+/–Eµ-myc transgenics (65%, 15/23), and Mdm2 was also overexpressed in many of the lymphomas that had mutated p53 (Figure 7; Table I). Therefore, Mdm2 overexpression is selected for during Myc-induced lymphomagenesis, and this occurs at a higher frequency when Mdm2 is haplo-insufficient. Notably, biallelic deletion of ARF, as detected by Southern blots, was only observed in one of the Mdm2+/–Eµ-myc transgenic lymphomas (Table I). However, one-quarter of the lymphomas overexpressed ARF and had reduced levels of wild-type p53 (Figure 7; Table I), suggesting that the regulation of ARF expression and/or the feedback control of p53–Mdm2–ARF pathway was affected in these tumors. Overall, these findings point to a key role for Mdm2 in regulating the rate of Myc-induced tumorigenesis, and bypass of haplo-insufficiency effects selects for events that preferentially disable p53 and/or result in Mdm2 overexpression.

Fig. 7. Analysis of p53, ARF and Mdm2 status in lymphomas arising in Mdm2+/–Eµ-myc transgenic mice. Western blot analysis: levels of p53 (top panel), p19ARF (second panel), Mdm2 (third panel) and β-actin (bottom panel) protein in whole-cell extracts of lymphomas from the indicated Eµ-myc transgenic mice were assessed by immunoblotting with antibodies specific to each protein. The three arrows next to Mdm2 indicate the location of the three isoforms of Mdm2 protein. Lymphomas from Mdm2+/+Eµ-myc transgenics were run as controls for p53, ARF and/or Mdm2 protein expression.
Table I. p53, ARF and Mdm2 expression in lymphomas from Mdm2+/–Eµ-myc transgenics.
| Genetic alteration | Percent of lymphomasa |
|---|---|
| p53 mutationb | 48 (11) |
| p53 deletionc | 4 (1) |
| ARF deletionc | 4 (1) |
| ARF overexpressiond, p53 wild type | 26 (6) |
| Mdm2 overexpressiond only | 4 (1) |
| No detectable alteration in Mdm2, p53 or ARF | 13 (3) |
| Mdm2 overexpressionc, and p53b,c or ARFc inactivation | 60 (14) |
| Alteration in Mdm2d, p53b,c or ARFc expression | 87 (20) |
aNumbers in parentheses indicate numbers of tumors out of 23 total lymphomas analyzed.
bMutations were determined by sequencing p53 cDNA.
cDeletions were determined by Southern blot.
dOverexpression was determined by western blot.
To further test whether the inhibition of lymphomagenesis in Mdm2+/–Eµ-myc transgenics was due to augmenting the p53 apoptotic response, we also generated Mdm2+/–Eµ-myc transgenics deficient in p53. Lymphoma rapidly developed in p53+/–Mdm2+/–Eµ-myc transgenics, which had an average survival of 5 weeks, a rate similar to that of p53+/–Eµ-myc transgenics, which had an average survival of 5.6 weeks (Figure 1). The B cell lymphomas that arose in both of these genotypes were typical and arose in the usual peripheral lymph nodes (data not shown). Furthermore, the second allele of p53 was deleted in 90% (19/21) of the p53+/–Mdm2+/–Eµ-myc transgenic lymphomas analyzed by Southern blot (data not shown), which is characteristic of lymphomas arising in p53+/–Eµ-myc transgenics (Schmitt et al., 1999). Interestingly, the remaining allele of p53 was mutated in the other two (2/21) p53+/–Mdm2+/–Eµ-myc transgenic lymphomas analyzed, which has not previously been reported for p53+/– Eµ-myc transgenic lymphomas. Therefore, p53 was inactivated in 100% (21/21) of the p53+/–Mdm2+/– Eµ-myc transgenic lymphomas. In conclusion, in Eµ-myc transgenic mice, the combined haplo-insufficiency of p53 and Mdm2 has the same effect as haplo-insufficiency of p53 alone. p53–/–Eµ-myc transgenics with or without Mdm2 could not be generated, presumably due to their death in utero, as previously reported (Hsu et al., 1995). Thus, a deficiency in p53 is dominant and overrides any negative effects on cell growth or survival that Mdm2 haplo-insufficiency has upon disease progression in Eµ-myc transgenic mice.
Discussion
The p53 tumor suppressor pathway is a critical checkpoint that inhibits lymphoma development by triggering apoptosis. This is underscored by the fact that Eµ-myc trans genics lacking the tumor suppressor p53 or ARF develop B cell lymphomas at an accelerated rate and that there are inhibitory effects of p53 or ARF loss on Myc-induced apoptosis (Eischen et al., 1999; Schmitt et al., 1999). Furthermore, 80% of the B cell lymphomas that arose in Eµ-myc transgenics have alterations in p53, ARF and/or Mdm2 (Eischen et al., 1999). The current study reveals even more complexity on the regulation of the p53 pathway during Myc-induced lymphomagenesis, and extends what was previously known about Mdm2’s regulation of p53. Strikingly, the findings presented here establish that one allele of Mdm2 is insufficient to inhibit p53 activity induced by Myc overexpression in vivo. Specifically, the results demonstrate that a certain threshold of Mdm2 is essential for B cell survival, as loss of a single allele of Mdm2 renders B cells susceptible to p53-dependent apoptosis and inhibits Myc-induced B cell transformation. The profound effects of Mdm2 haplo-insufficiency on B cell survival and tumor development in Eµ-myc transgenics therefore establishes the concept that a specific threshold of Mdm2 protein is essential for Mdm2 to harness p53-dependent apoptosis. Thus, it appears likely that Mdm2’s enzymatic activity as a ubiquitin ligase is rate limiting and is required for tumor development.
Mdm2 haplo-insufficiency targets the p53 pathway during Myc-induced lymphomagenesis
The profound effects of Mdm2 haplo-insufficiency on B cell apoptosis and transformation were surprising, as Mdm2 heterozygous mice have no obvious phenotype and have normal life spans, and their frequency of tumor development is not different than wild-type mice (Jones et al., 1995; Montes de Oca Luna et al., 1995). Moreover, all cells in Mdm2+/– mice should still have one functional Mdm2 allele, and lymphocyte defects in Mdm2+/– mice have not previously been reported (Jones et al., 1995; Montes de Oca Luna et al., 1995). Nonetheless, Mdm2 haplo-insufficiency clearly augments the apoptotic program in precursor B cells ex vivo, even when Myc was not overexpressed. However, in vivo there were only modest increases in the apoptotic index of B cells in Mdm2+/– mice and little or no difference in peripheral B cell numbers. These observations suggest that the in vivo environment must provide survival signals for Mdm2+/– B cells that are lacking in tissue culture. The culture of primary cells is inherently stressful, and consequently the ARF–p53 pathway is activated (Sherr and DePinho, 2000). Therefore, the culture of cells that are more susceptible to p53-mediated apoptosis, such as Mdm2+/– pre-B cells, should result in the apoptosis of these cells, and this scenario was indeed evident.
In B cells, Myc triggers at least two apoptotic pathways and both of these are inactivated during Myc-induced lymphomagenesis (Eischen et al., 1999, 2001c). First, Myc provokes p53-dependent apoptosis by inducing the expression of ARF, which disables Mdm2 function and thus augments p53 activity (Wagner et al., 1994; Zindy et al., 1998; Eischen et al., 1999). Secondly, Myc suppresses the expression of the anti-apoptotic Bcl-2 family members, Bcl-2 and Bcl-XL, and this occurs even in cells lacking ARF and/or p53 (Eischen et al., 2001a,c). Importantly, 80% of the lymphomas that arise in wild-type Eµ-myc transgenics harbor alterations in ARF, Mdm2 or p53, whereas half of these lymphomas also disable the Bcl-2/Bcl-XL pathway (Eischen et al., 2001a,c). How ever, there are links between these two apoptotic pathways. For example, the loss of bax, a pro-apoptotic Bcl-2 family member that is induced by p53 (Miyashita and Reed, 1995) and antagonizes the function of Bcl-2 and Bcl-XL (Korsmeyer 1999), selectively eliminates the need for p53 mutations that usually arise in one-quarter of all lymphomas of Eµ-myc transgenics, without altering the percentage of tumors bearing deletions in ARF (Eischen et al., 2001b). Here we report the opposite scenario occurs in transgenics that are Mdm2 haplo-insufficient, as lymphomas arising in these mice had an increased frequency of p53 mutations and Mdm2 overexpression, whereas ARF deletions were only rarely observed. Specifically, half of the Mdm2+/–Eµ-myc lymphomas had sustained p53 mutations, twice the percentage of p53 mutations in lymphomas that arise in wild-type Eµ-myc transgenics (24%) (Eischen et al., 1999), and 65% (15/23) of lymphomas overexpressed Mdm2 protein, which is usually only overexpressed in half of the lymphomas of wild-type Eµ-myc transgenics. In contrast, biallelic deletion of ARF, which occurs in one-quarter of Eµ-myc lymphomas, was only detected in one of the lymphomas that emerged in Mdm2+/–Eµ-myc transgenics, suggesting that loss of ARF was not able to inhibit p53-mediated apoptosis in Mdm2+/–Eµ-myc transgenic B cells. Thus, Mdm2 haplo-insufficient B cells bypassed apoptosis during Myc-induced lymphomagenesis by selectively provoking mutations that directly or indirectly disable p53.
Mdm2 functions are rate limiting for lymphomagenesis
Mdm2+/– mice do not have an increased frequency of cancer development and T cell lymphoma development in p53/Mdm2-double null mice is comparable to those just lacking p53 (Jones et al., 1995; Montes de Oca Luna et al., 1995). However, Mdm2/p53/ARF-triple null mice do have an altered tumor spectrum when compared with p53/ARF-double null mice (Weber et al., 2000), indicating that Mdm2 loss can indeed affect tumor development. Further, Mdm2 haplo-insufficiency also influences thymoma development in the context of p53 deficiency, as Mdm2+/–p53–/– mice develop fewer thymomas and more sarcomas than mice null for both Mdm2 and p53, or p53 alone (McDonnell et al., 1999). Thus, Mdm2 haplo-insufficiency, rather than total loss of Mdm2, also impairs T cell lymphomagenesis, a scenario akin to what we observe in Eµ-myc transgenics, where there are little effects of Mdm2 loss when combined with loss of p53 on B cell growth and tumor latency, but there are striking inhibitory effects on B cell lymphomagenesis when Mdm2 is haplo-insufficient. Moreover, Mdm2 haplo-insufficiency also somehow alters the sites of lymphoma development in Eµ-myc transgenics. Therefore, reductions in Mdm2 expression can influence both the site and rate of tumor development, and this is particularly manifest during lymphomagenesis.
Mdm2 protein is overexpressed in half of all B cell lymphomas arising in Eµ-myc transgenic mice (Eischen et al., 1999, 2001b). Furthermore, Hdm2 (the human Mdm2 homolog) expression is elevated in up to half of human non-Hodgkin’s lymphomas (Watanabe et al., 1996), and Hdm2 overexpression correlates with disease progression (Pagnano et al., 2001). Collectively, these results suggest that Mdm2/Hdm2 overexpression is selected for during lymphomagenesis. In support of this notion, the percentage of lymphomas overexpressing Mdm2 is even higher (65%) in tumors derived from Mdm2+/–Eµ-myc transgenics, where Mdm2 haplo-insufficiency is such a formidable barrier to tumor development. Mdm2 overexpression in this scenario and others is usually not due to amplification, as Mdm2 is rarely amplified in leukemia or lymphoma (Momand et al., 1998), and we have never observed Mdm2 amplification in any of the lymphomas of Eµ-myc transgenic mice (our unpublished data). Therefore, the data strongly support the concept that Mdm2 expression is necessary to block the p53-dependent apoptotic effects of Myc, and Mdm2 overexpression is an important regulator that allows tumor cells that overexpress Myc to survive. However, our findings here and in wild-type Eµ-myc transgenics (Eischen et al., 1999, 2001b) have shown that Mdm2 is also overexpressed in many of the lymphomas that have mutant p53. This strongly suggests that Mdm2 may also have targets other than p53 that are harnessed during lymphomagenesis, and other important targets of Mdm2 that have been implicated in cancer include E2F-1, DP-1, p300/CBP and/or pRb (reviewed in Momand et al., 2000), TSG101 (Li et al., 2001), Numb (Juven-Gershon et al., 1998), and/or MTBP (Boyd et al., 2000). Therefore, although these studies certainly reveal the need for a proper threshold of Mdm2 protein to inhibit p53-dependent apoptosis, Mdm2 appears to also provide benefits to tumor cells by inactivating other targets that impair tumorigenesis.
Materials and methods
Transgenic and knockout mice
The Eµ-myc transgenic mouse strain (congenic C57Bl/6) was kindly provided by Drs Alan Harris (Walter & Eliza Hall Institute, Melbourne, Australia) and Charles Sidman (University of Cincinnati, Cincinnati, OH). The p53+/–Mdm2+/– (C57Bl/6 × 129/Sv) (Montes de Oca Luna et al., 1995) mice were generously provided by Dr Guillermina Lozano (M.D. Anderson Cancer Center, Houston, TX). The p53+/–Mdm2+/– mice were crossed to the Eµ-myc transgenics to generate F1s. The F1s were then crossed to generate F2 Mdm2+/–Eµ-myc, Mdm2+/+Eµ-myc, p53+/– Mdm2+/+Eµ-myc and p53+/–Mdm2+/–Eµ-myc transgenics, and p53–/– Mdm2+/+, p53–/–Mdm2+/–, p53–/–Mdm2–/–, Mdm2+/– and wild-type mice. These mice were followed and used for all analyses. A Kaplan–Meier analysis was performed. The Kaplan–Meier plot (Figure 1) includes mice that were still alive at the time the plot was generated, and therefore some of the cases are right-censored because the study was terminated before death occurred. A log-rank test was performed to determine the statistical significance of the survival between the different genotypes of Eµ-myc transgenic mice.
Immunohistochemistry
Tissues and tumors from Mdm2+/–Eµ-myc transgenics were fixed in 10% buffered formalin, paraffin embedded and sectioned (5 µm). Immunohistochemistry on sections of spleen and/or tumor was performed, as described previously (Eischen et al., 2002) with an antibody against B220 (PharMingen, San Diego, CA).
Isolation, culture, and infection of primary pre-B cells
Primary pre-B cell cultures were generated from the bone marrow of 9- to 15-week-old wild-type, Mdm2+/–, p53–/–, p53–/–Mdm2+/–, p53–/–Mdm2–/–, Mdm2+/–Eµ-myc transgenic and Mdm2+/+Eµ-myc transgenic mice, as described previously (Eischen et al., 1999). Briefly, within 14 days bone marrow cultured in IL-7 containing medium results in >98% pure populations of pre-B cells (CD19+, B220+, CD43–, IgM–) as determined by phenotype analysis using fluorescent B-cell-specific antibodies and FACS analysis. All antibodies used for phenotypic analyses were from Southern Biotechnology (Birmingham, AL) or PharMingen. For retroviral infections, MSCV-Myc-ERTAM-IRES-GFP or control MSCV-IRES-GFP retroviruses were generated and used to infect primary pre-B cells as described previously (Eischen et al., 1999). [Myc-ERTAM was previously called Myc-ER™ (Littlewood et al., 1995), but due to the confusion that TM denotes trademark instead of tamoxifen, TM was changed to TAM.] Myc-ERTAM was activated in MSCV-Myc-ERTAM-IRES-GFP-infected pre-B cells by adding 1 µM 4-HT (Sigma, St Louis, MO), which binds to the modified estrogen receptor hormone binding domain (ER) of the Myc-ERTAM fusion protein (Littlewood et al., 1995). This results in the release of Myc-ERTAM from heat shock protein complexes and translocation of Myc-ERTAM into the nucleus, where it activates transcription (Littlewood et al., 1995). Addition of 4-HT to uninfected or GFP control virus infected cells had no effect on pre-B cell growth or viability.
Viability and apoptosis assays
Cell viability following explantation of bone marrow into IL-7 containing medium or after the addition of 1 µM 4-HT to the culture medium of pre-B cells infected with the retrovirus encoding Myc-ERTAM was determined at specific intervals by Trypan Blue dye exclusion. For apoptosis measurements, whole spleens were disaggregated, filtered through a 100 µm nylon filter and then fixed in 70% ethanol prior to propidium iodide (PI) staining of DNA, whereas pre-B cells were PI stained without ethanol fixation. All PI-stained samples were analyzed on a FACScan and quantitation of fragmented (sub-G1) DNA was performed with CellQuest software (BD Immunocytometry Systems, San Jose, CA).
Peripheral blood lymphocyte counts
For peripheral blood lymphocyte counts, blood was collected by retro-orbital eye bleeds from wild-type, Mdm2+/–, Mdm2+/+Eµ-myc transgenic and Mdm2+/–Eµ-myc transgenic mice into EDTA treated tubes to inhibit coagulation. Each blood sample (20 µl) was analyzed on a Hemavet 3700 (Drew Scientific, Oxford, CT) cell counter.
Phenotype analysis
Whole spleens from wild-type, Mdm2+/–, Mdm2+/+Eµ-myc transgenic and Mdm2+/–Eµ-myc transgenic mice were harvested, minced and strained through a 100 µm nylon filter. Approximately 500 000 splenocytes per sample were incubated with one to three fluorescently labeled antibodies (CD19-PE, IgM-FITC, B220-CyChrome, CD3-SRPD and/or fluorescent isotype controls) for 30–45 min at 4°C. Cells were washed with phosphate-buffered saline and immediately analyzed on a MoFlo (Cytomation, Fort Collins, CO) or FACSCalibur (BD Immunocyto metry Systems) instrument. CellQuest software was used to analyze data generated on the FACSCalibur.
Western blotting
Primary pre-B cells or pre-B/B cell lymphomas (3–5 mm3 chunk) were lysed as described previously (Zindy et al., 1998). Briefly, cell pellets or small tumor chunks were sonicated (2× 7 s) in ice cold lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 0.1% Tween-20, 1 mM phenylmethylsulfonyl fluoride, 0.4 U/ml aprotinin, 1 mM NaF, 10 mM β-glycerophosphate and 0.1 mM sodium orthovanadate). Following sedimentation of undissolved cellular material by centrifugation (4°C, 7 min, 14 000 r.p.m.; Sorvall Biofuge), the protein in the supernatant was quantified with the Bio-Rad Protein Assay Reagent (Hercules, CA). Equal amounts of protein (200 µg per lane) were then separated by 10% SDS–PAGE, transferred to nitrocellulose membranes (Protran; Schleicher & Schuell, Dassel, Germany), and blotted with antibodies specific for p19ARF (GeneTex, San Antonio, TX; Quelle et al., 1995), p53 (Ab-7; Calbiochem, La Jolla, CA), Mdm2 (C-18; Santa Cruz, Inc., Santa Cruz, CA), Myc (06-340; Upstate Biotechnology, New York, NY) and β-actin (Sigma). Membranes were incubated with HRP-linked secondary antibodies (Amersham Pharmacia Biotech, Piscataway, NJ) followed by either ECL (Amersham) or Supersignal (Pierce, Rockford, IL) to detect bound immunocomplexes.
Southern blotting
Following isolation from lymphomas emerging in Mdm2+/–Eµ-myc transgenic mice, 15 µg of genomic DNA was digested with AflII or BamHI. Digested DNAs were electrophoretically separated in 0.7–1.0% agarose gels, transferred to Nytran (Scheicher & Schuell) membranes, and then probed with radioactive cDNAs coding ARF (exon 1β) (AflII digested) and p53 (exons 2–10) (BamHI digested).
Acknowledgments
Acknowledgements
The authors would like to thank Dawn Pauli and Chunying Yang for expert technical support, Dr Jane Meza for the Kaplan–Meier curves and statistical analysis, Dr Jonas Nilsson for assistance with Southern blots, Anita Jennings for immunohistochemistry, Rob Jeffers for PCR genotype analysis, Drs Robert Hawley and Derek Persons for retrovial vectors, Drs Hua Xiao and Gerry Zambetti for reviewing the manuscript, the personnel in Eppley’s animal facility, and Karol Kaminski and Carolyn Bratcher at St Jude Children’s Research Hospital’s animal facility. This work was supported by the Eppley Institute for Cancer Research and the Wanda Rizzo memorial fund (C.M.E), NIH training grant T32 CA09476 (J.R.A.), grants CA76379 and DK44158 (J.L.C), and by the American Lebanese Syrian Associated Charities (ALSAC) (J.L.C.).
References
- Adams J.M., Harris,A.W., Pinkert,C.A., Corcoran,L.M., Alexander,W.S., Cory,S., Palmiter,R.D. and Brinster,R.L. (1985) The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature, 318, 533–538. [DOI] [PubMed] [Google Scholar]
- Boyd M.T., Vlatkovic,N. and Haines,D.S. (2000) A novel cellular protein (MTBP) binds to MDM2 and induces a G1 arrest that is suppressed by MDM2. J. Biol. Chem., 275, 31883–31890. [DOI] [PubMed] [Google Scholar]
- de Rozieres S., Maya,R., Oren,M. and Lozano,G. (2000) The loss of mdm2 induces p53-mediated apoptosis. Oncogene, 19, 1691–1697. [DOI] [PubMed] [Google Scholar]
- Eischen C.M., Weber,J.D., Roussel,M.F., Sherr,C.J. and Cleveland,J.L. (1999) Disruption of the ARF–Mdm2–p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev., 13, 2658–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen C.M., Packham,G., Nip,J., Fee,B.E., Hiebert,S.W., Zambetti,G.P. and Cleveland,J.L. (2001a) Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene, 20, 6983–6993. [DOI] [PubMed] [Google Scholar]
- Eischen C.M., Roussel,M.F., Korsmeyer,S.J. and Cleveland,J.L. (2001b) Bax loss impairs Myc-induced apoptosis and circumvents the selection of p53 mutations during Myc-mediated lymphomagenesis. Mol. Cell. Biol., 21, 7653–7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen C.M., Woo,D., Roussel,M.F. and Cleveland,J.L. (2001c) Apoptosis triggered by Myc-induced suppression of Bcl-XL or Bcl-2 is bypassed during lymphomagenesis. Mol. Cell. Biol., 21, 5063–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen C.M., Rehg,J.E., Korsmeyer,S.J. and Cleveland,J.L. (2002) Loss of Bax alters tumor spectrum and tumor numbers in ARF-deficient mice. Cancer Res., 62, 2184–2191. [PubMed] [Google Scholar]
- Freedman D.A. and Levine,A.J. (1998) Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol. Cell. Biol., 18, 7288–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A.W., Pinkert,C.A., Crawford,M., Langdon,W.Y., Brinster,R.L. and Adams,J.M. (1988) The Eµ-myc transgenic mouse. A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J. Exp. Med., 167, 353–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R. and Yasuda,H. (1999) Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J., 18, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu B., Marin,M.C., el-Naggar,A.K., Stephens,L.C., Brisbay,S. and McDonnell,T.J. (1995) Evidence that c-myc mediated apoptosis does not require wild-type p53 during lymphomagenesis. Oncogene, 11, 175–179. [PubMed] [Google Scholar]
- Jacobs J.J., Scheijen,B., Voncken,J.W., Kieboom,K., Berns,A. and van Lohuizen,M. (1999) Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev., 13, 2678–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.N., Roe,A.E., Donehower,L.A. and Bradley,A. (1995) Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature, 378, 206–208. [DOI] [PubMed] [Google Scholar]
- Jones S.N., Hancock,A.R., Vogel,H., Donehower,L.A. and Bradley,A. (1998) Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl Acad. Sci. USA, 95, 15608–15612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juven-Gershon T., Shifman,O., Unger,T., Elkeles,A., Haupt,Y. and Oren,M. (1998) The Mdm2 oncoprotein interacts with the cell fate regulator Numb. Mol. Cell. Biol., 18, 3974–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T., Weber,J.D., Zambetti,G., Zindy,F., Roussel,M.F. and Sherr,C.J. (1998) Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc. Natl Acad. Sci. USA, 95, 8292–8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korgaonkar C., Zhao,L., Modestou,M. and Quelle,D.E. (2002) ARF function does not require p53 stabilization or Mdm2 relocalization. Mol. Cell. Biol., 22, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsmeyer S.J. (1999) BCL-2 gene family and the regulation of programmed cell death. Cancer Res., 59, 1693–1700. [PubMed] [Google Scholar]
- Langdon W.Y., Harris,A.W., Cory,S. and Adams,J.M. (1986) The c-myc oncogene perturbs B lymphocyte development in E-µ-myc transgenic mice. Cell, 47, 11–18. [DOI] [PubMed] [Google Scholar]
- Li L., Liao,J., Ruland,J., Mak,T.W. and Cohen,S.N. (2001) A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control. Proc. Natl Acad. Sci. USA, 98, 1619–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood T.D., Hancock,D.C., Danielian,P.S., Parker,M.G. and Evan,G.I. (1995) A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res., 23, 1686–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell T.J., Montes de Oca Luna,R., Cho,S., Amelse,L.L., Chavez-Reyes,A. and Lozano,G. (1999) Loss of one but not two mdm2 null alleles alters the tumour spectrum in p53 null mice. J. Pathol., 188, 322–328. [DOI] [PubMed] [Google Scholar]
- McMasters K.M., Montes de Oca Luna,R., Pena,J.R. and Lozano,G. (1996) mdm2 deletion does not alter growth characteristics of p53-deficient embryo fibroblasts. Oncogene, 13, 1731–1736. [PubMed] [Google Scholar]
- Miyashita T. and Reed,J.C. (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell, 80, 293–299. [DOI] [PubMed] [Google Scholar]
- Momand J. and Zambetti,G.P. (1997) Mdm-2: ‘big brother’ of p53. J. Cell. Biochem., 64, 343–352. [PubMed] [Google Scholar]
- Momand J., Zambetti,G.P., Olson,D.C., George,D. and Levine,A.J. (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell, 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Momand J., Jung,D., Wilczynski,S. and Niland,J. (1998) The MDM2 gene amplification database. Nucleic Acids Res., 26, 3453–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J., Wu,H.H. and Dasgupta,G. (2000) MDM2—master regulator of the p53 tumor suppressor protein. Gene, 242, 15–29. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R., Wagner,D.S. and Lozano,G. (1995) Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature, 378, 203–206. [DOI] [PubMed] [Google Scholar]
- Pagnano K.B., Vassallo,J., Lorand-Metze,I., Costa,F.F. and Saad,S.T. (2001) p53, Mdm2 and c-Myc overexpression is associated with a poor prognosis in aggressive non-Hodgkin’s lymphomas. Am. J. Hematol., 67, 84–92. [DOI] [PubMed] [Google Scholar]
- Pomerantz J. et al. (1998) The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell, 92, 713–723. [DOI] [PubMed] [Google Scholar]
- Quelle D.E., Zindy,F., Ashmun,R.A. and Sherr,C.J. (1995) Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell, 83, 993–1000. [DOI] [PubMed] [Google Scholar]
- Roth J., Dobbelstein,M., Freedman,D.A., Shenk,T. and Levine,A.J. (1998) Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J., 17, 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt C.A., McCurrach,M.E., de Stanchina,E., Wallace-Brodeur,R.R. and Lowe,S.W. (1999) INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev., 13, 2670–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr C.J. (1998) Tumor surveillance via the ARF–p53 pathway. Genes Dev., 12, 2984–2991. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and DePinho,R.A. (2000) Cellular senescence: mitotic clock or culture shock? Cell, 102, 407–410. [DOI] [PubMed] [Google Scholar]
- Stott F.J. et al. (1998) The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J., 17, 5001–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W. and Levine,A.J. (1999) p19ARF stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc. Natl Acad. Sci. USA, 96, 6937–6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A.J., Kokontis,J.M. and Hay,N. (1994) Myc-mediated apoptosis requires wild-type p53 in a manner independent of cell cycle arrest and the ability of p53 to induce p21waf1/cip1. Genes Dev., 8, 2817–2830. [DOI] [PubMed] [Google Scholar]
- Watanabe T., Ichikawa,A., Saito,H. and Hotta,T. (1996) Overexpression of the MDM2 oncogene in leukemia and lymphoma. Leuk. Lymphoma, 21, 391–397. [DOI] [PubMed] [Google Scholar]
- Weber J.D., Taylor,L.J., Roussel,M.F., Sherr,C.J. and Bar-Sagi,D. (1999) Nucleolar ARF sequesters Mdm2 and activates p53. Nature Cell Biol., 1, 20–26. [DOI] [PubMed] [Google Scholar]
- Weber J.D., Jeffers,J.R., Rehg,J.E., Randle,D.H., Lozano,G., Roussel,M.F., Sherr,C.J. and Zambetti,G.P. (2000) p53-independent functions of the p19ARF tumor suppressor. Genes Dev., 14, 2358–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X., Bayle,J.H., Olson,D. and Levine,A.J. (1993) The p53–mdm-2 autoregulatory feedback loop. Genes Dev., 7, 1126–1132. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Xiong,Y. and Yarbrough,W.G. (1998) ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell, 92, 725–734. [DOI] [PubMed] [Google Scholar]
- Zindy F., Eischen,C.M., Randle,D.H., Kamijo,T., Cleveland,J.L., Sherr,C.J. and Roussel,M.F. (1998) Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev., 12, 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]