

Abstract
Signal transducer and activator of transcription 3 (Stat3) is a latent cytoplasmic transcription factor that can be activated by cytokines and growth factors. Stat3 plays important roles in cell growth, anti-apoptosis and cell transformation, and is constitutively active in various cancers. We examined its potential regulators by yeast two-hybrid screening. GRIM-19, a gene product related to interferon-β- and retinoic acid-induced cancer cell death, was identified and demonstrated to interact with Stat3 in various cell types. The interaction is specific for Stat3, but not for Stat1 and Stat5a. The interaction regions in both proteins were mapped, and the cellular localization of the interaction was examined. GRIM-19 itself co-localizes with mitochondrial markers, and forms aggregates at the perinulear region with co-expressed Stat3, which inhibits Stat3 nuclear translocation stimulated by epidermal growth factor (EGF). GRIM-19 represses Stat3 transcriptional activity and its target gene expression, and also suppresses cell growth in Src-transformed cells and a Stat3-expressing cell line. Our data suggest that GRIM-19 is a novel negative regulator of Stat3.
Keywords: co-localization/GRIM-19/interaction/repression/Stat3
Introduction
Cytokines exert multiple biological responses through interaction with their specific receptors that results in the activation of JAK–STAT pathways. STATs (signal transducers and activators of transcription) are a family of latent cytoplasmic transcription factors which are activated by recruitment to the cytokine receptors and subsequent phosphorylation by the receptor-associated Janus kinases (JAKs). Stat proteins form homo- or heterodimers by reciprocal interaction between SH2 domains and phosphorylated tyrosine residues, translocate into the nucleus, bind to DNA and regulate their target gene expression (Darnell et al., 1994). Seven mammalian STAT genes have been identified to be activated by various cytokines and certain growth factors, and exhibit distinct and overlapping functions (Schindler and Darnell, 1995; Ihle, 2001).
Stat3 originally was cloned as an acute-phase response factor activated by interleukin-6 (IL-6) in mouse liver, and also by homology to Stat1 (Akira et al., 1994; Zhong et al., 1994a). Growth factors, such as epidermal growth factor (EGF), platelet-derived growth factor (PDGF) and colony-stimulating factor-1 (CSF-1), can also stimulate Stat3 activity (Sadowski et al., 1993; Zhong et al., 1994b). Stat3 plays crucial roles in early embryonic development (Takeda et al., 1997), as well as in other biological responses including cell growth and anti-apoptosis (Akira, 2000). Stat3 is constitutively activated in oncogenic tyrosine kinase v-Src- or v-abl-transformed cells (Danial et al., 1995; Yu et al., 1995; Cao et al., 1996), and various primary tumors and cell lines. Stat3 itself acts as an oncogene in NIH-3T3 cells (Bromberg et al., 1999; Bowman et al., 2000). Stat3 plays an anti-apoptotic role in gp130-mediated survival in B cells, as well as in myeloma and head and neck tumor cells (Fukada et al., 1996; Catlett-Falcone et al., 1999; Grandis et al., 2000). In the latter cases, activation of Stat3 may contribute to the loss of cell growth control, therefore leading to carcinogenesis by preventing apoptosis. Therefore, the control of both the activation and inactivation of Stat3 is equally important to maintain normal cell growth.
Interferons (IFNs) regulate antiviral, antitumor and immune responses in vertebrates by activating Jak1, Tyk2, Stat1 and Stat2, and a number of IFN-stimulated genes (Stark et al., 1998). On the other hand, all-trans-retinoic acid (RA), a metabolite of vitamin A, binds to specific nuclear receptors (RARs) and activates the expression of various genes. RA inhibits growth of certain types of cancer and is effective in the prevention of primary cancers, and is used in clinical therapy (Love and Gudas, 1994). A combination of IFN-β and RA induces cell death in several breast cancer cell lines. A few genes that possibly are involved in this process have been identified and named as GRIM (genes associated with retinoid–IFN-induced mortality) (Hofman et al., 1998). One of these genes, GRIM-19, was reported to encode a small protein primarily distributed in the nucleus and was able to promote cell death induced by IFN-β and RA (Angell et al., 2000). Recently, a bovine homolog of GRIM-19 was co-purified with mitochondrial NADH:ubiquinone oxido reductase (complex I) in bovine heart (Fearnley et al., 2001). Therefore, its exact cellular localization and function are unclear.
To understand further the regulation and function of Stat3, we investigated Stat3-interacting proteins using yeast two-hybrid screening. We have identified GRIM-19 as a specific interacting protein which negatively regulates Stat3 activity.
Results
Identification of GRIM-19 as a Stat3-interacting protein by yeast two-hybrid screening
To study the regulation and function of Stat3, we sought to identify the proteins that interact with Stat3 using a yeast two-hybrid system (Durfee et al., 1993). Full-length Stat3 resulted in the constitutive activation of the reporter genes. We therefore used the coiled-coil domain of Stat3, known to be important for protein–protein interaction, as bait (Becker et al., 1998). Four positive clones were identified to encode the mouse homolog of human GRIM-19, whose homologs have been reported in bovine, mouse and Caenorhabditis elegans (Lai et al., 2000). The cDNAs we obtained in the yeast two-hybrid screening exhibited 100% identity in amino acid sequence with the reported murine GRIM-19, which shares 75% amino acid identity and 88% similarity to the human GRIM-19. The specificity of Stat3 and GRIM-19 interaction in yeast was confirmed further by survival assay in selection medium (see Supplementary figure 1, available at The EMBO Journal Online).
Association of Stat3 and GRIM-19 in vivo and in vitro
To verify the interaction of Stat3 and GRIM-19 in mammalian cells, we transfected COS-1 cells that express low levels of Stat3 with Flag-tagged Stat3 and Myc-tagged GRIM-19, or control plasmids. The cell lysates were immunoprecipitated with anti-Myc antibody, and subjected to western blotting analysis to detect bound Stat3 using anti-Flag antibody. As shown in Figure 1, Stat3 was detected in anti-Myc immunoprecipitates when GRIM-19, but not the control plasmid, was co-transfected (top left panel). A reciprocal immunoprecipitation/blotting experiment also showed GRIM-19 co-precipitating with Stat3 (top right panel). We further demonstrated that the coiled-coil domain alone could interact with GRIM-19 in COS-1 cells, and Stat3 and GRIM-19 were able to interact directly in vitro (see Supplementary figures 2 and 3).

Fig. 1. Association of Stat3 and GRIM-19 in transfected cells. COS-1 cells were transfected with Flag-tagged Stat3 and/or Myc-tagged GRIM-19. The cell lysates were immunoprecipitated with either anti-Myc or anti-Flag as indicated. The precipitates were fractionated by 12.5% SDS–PAGE, and blotted with anti-Flag (top left panel) or anti-Myc (top right panel). The respective co-precipitated Stat3 and GRIM-19 are indicated. The blots were stripped and re-probed with anti-Myc or anti-Flag (middle panels). Total cell lysates (TCL) were subjected to western blot analysis with anti-Flag or anti-Myc to monitor the expression of Stat3 and GRIM-19 (bottom panels). The molecular mass markers are indicated in kDa (Bio-Rad Laboratories). The strong bands are the heavy chain of rabbit IgG (top left panel), or the heavy and light chains of mouse IgG (top right panel).
Physiological association of endogenous Stat3 and GRIM-19 in various cell types
To study the possible physiological significance, we investigated the interaction between endogenous Stat3 and GRIM-19 in mammalian cells. For this purpose, a rabbit antiserum against bacterially produced mouse GRIM-19 was generated. The antibody specifically recognizes GST–GRIM-19 expressed in COS-1 cells (Figure 2A). A protein with a molecular mass of ∼18 kDa that was likely to be endogenous GRIM-19 in COS-1 cells was also detectable. In contrast, the pre-immune serum and the anti-GRIM-19 antibody pre-absorbed with recombinant GRIM-19 protein reacted with neither the endogenous nor the transfected GRIM-19. The GRIM-19 antibody also recognized endogenous human GRIM-19 in a breast cancer cell line, MCF-7, induced by a combination of IFN-β and RA treatment for various durations (Figure 2B), which exhibited similar kinetics to those previously reported (Angell et al., 2000).

Fig. 2. Association of endogenous Stat3 and GRIM-19. (A) Evaluation of the GRIM-19 antibody. COS-1 cells were transfected with GST vector (lanes 1, 3 and 5) or GST–GRIM-19 (lanes 2, 4 and 6). Cells were lysed and subjected to western blot analysis using pre-immune serum (PI, lanes 1 and 2), purified GRIM-19 antibody (α-G-19; lanes 3 and 4) or GRIM-19 antibody pre-absorbed by bacterially produced GRIM-19 (lanes 5 and 6). (B) Western blot analysis of endogenous GRIM-19 in MCF-7 cells induced by IFN-β (500 U/ml) and RA (1 µM) for various times. The blot was re-probed with anti-β-actin as a control. (C) Association of endogenous Stat3 and GRIM-19 in various cell types. MCF-7 cells were left untreated (lanes 1 and 2) or treated with IFN-β and RA for 48 h (lane 3). C2C12, SH-SY5Y and PC12 cells were grown under normal conditions. Cell lysates were incubated with either pre-immune serum (PI) or anti-GRIM-19 (α-G-19) as labeled on top of the figures, and blotted with anti-Stat3 (upper panels). The blots were stripped and re-probed with anti-GRIM-19 (lower panels).
We then evaluated the interaction of Stat3 and GRIM-19 in MCF-7 cells. A low amount of Stat3 was co-immuprecipitated with GRIM-19 in unstimulated cells (Figure 2C, lane 2), which was increased in IFN-β/RA-treated cells (lane 3). As a control, Stat3 did not co-immunoprecipitate with pre-immune serum (lane 1). Since GRIM-19 is expressed at a high level in skeletal muscle (Angell et al., 2000) and rat brain (data not shown), we further tested the association between endogenous GRIM-19 and Stat3 in mouse myoblast C2C12, human neuroblastoma SH-SY5Y and rat pheochromocytoma PC-12 cells. Stat3 co-immunoprecipitated with GRIM-19 antiserum, but not pre-immune serum, in these cell lines (Figure 2C). These data indicate a physiological interaction between GRIM-19 and Stat3, which is not limited to human breast cancer cells but extends to other cell types and species.
GRIM-19 does not associate with Stat1 and Stat5a
The interaction between GRIM-19 and other Stat proteins was also examined. In contrast to Stat3, only trace amounts of Stat1 co-precipitated with GST–GRIM-19, whereas no association was detected between Stat5a and GRIM-19 in the GST pull-down experiment (Figure 3). In agreement with these results, no co-localization between GRIM-19 and Stat1 or Stat5a in cells was observed by immunofluorescence staining (see Figure 6B). These data suggested a specific functional link between GRIM-19 and Stat3.

Fig. 3. Lack of interaction between GRIM-19 and other Stat proteins. Flag-tagged Stat1, 3 or 5a were co-transfected with GST or GST– GRIM-19 expression plasmids in COS-1 cells. The cell lysates were incubated with glutathione–Sepharose beads. The bound proteins were subjected to western blot analysis with anti-Flag antibody (top panel). Precipitated GST and GST–GRIM-19 were blotted with anti-GST (middle panel). The expression of Stat1 (lanes 1 and 2), Stat3 (lanes 3 and 4) and Stat5a (lanes 5 and 6) was determined with anti-Flag (bottom panel).

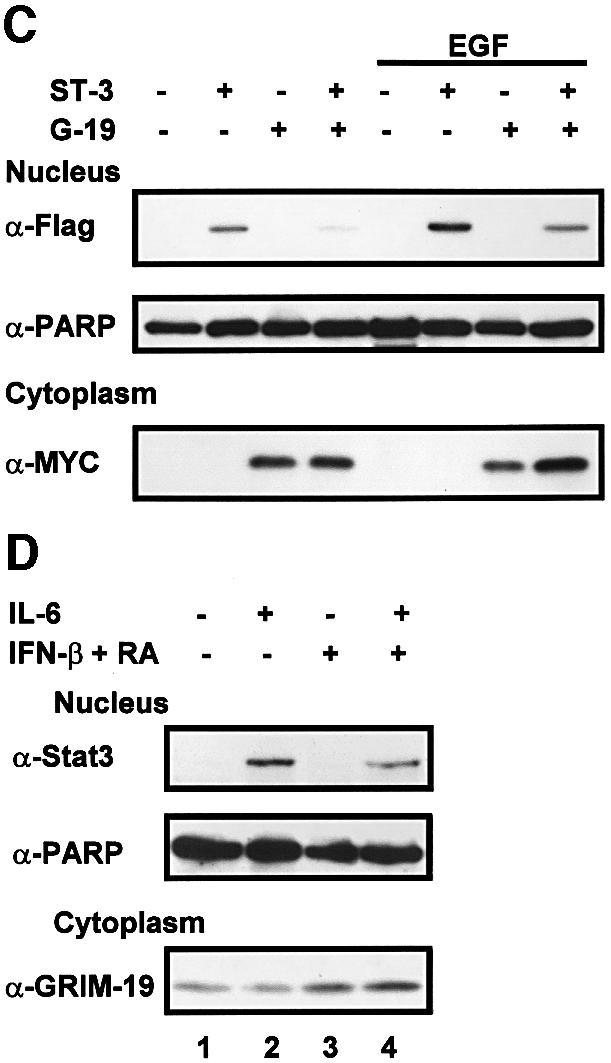
Fig. 6. Co-localization of Stat3 with GRIM-19. (A) MCF-7 cells were transfected with Flag-tagged Stat3 or truncation mutants with or without Myc-tagged GRIM-19. The distribution of Stat3 or GRIM-19 was detected using monoclonal anti-Flag or polyclonal anti-Myc as primary antibody, followed by Cy3-conjugated anti-mouse IgG (Sigma) or FITC-conjugated anti-rabbit IgG. (B) COS-1 cell were transfected with Flag-tagged Stat3, Stat1 or Stat5a in the absence (–) or presence of Myc-GRIM-19. The cells were induced by EGF for 15 min and the cellular localization of Stat proteins and GRIM-19 was detected as described in (A). (C) The effect of GRIM-19 on Stat3 nuclear translocation. COS-1 cells were transfected with vector, GRIM-19 and/or Stat3. The cells were untreated or induced with EGF for 15 min. Cells were lysed and fractionated into cytoplasmic and nuclear portions as described previously (Jain et al., 1999). The nuclear distribution of Stat3 was examined by western blotting. The blot was re-blotted with anti-PARP antibody as a control. The expression of GRIM-19 in the cytoplasmic portion was detected by blotting with anti-Myc antibody. (D) Induction of endogenous GRIM-19 reduced IL-6-stimulated Stat3 nuclear translocation. HepG2 cells were either left untreated or treated with IFN-β and RA for 48 h followed by IL-6 induction for 15 min. Nuclear Stat3 was measured with anti-Stat3 as described in (C).
Mapping the regions that mediate the interaction between Stat3 and GRIM-19
Stat3 contains several functional domains, as illustrated in Figure 4A (Becker et al., 1998). In addition to the coiled-coil domain, other regions may also interact with GRIM-19. To explore this possibility, we generated a series of Flag-tagged Stat3 truncation mutants, which were co-transfected with GST-tagged GRIM-19. GST pull-down experiments indicated that GST–GRIM-19 associated with ST3-DB.LK, a Stat3 protein consisting of the DNA-binding and linker domains. No association was detected with the N-terminal domain, the SH2 domain or the C-terminal domain (Figure 4B). We further delineated the interacting domains by subcloning the individual DNA-binding domain and linker domain. The immunoprecipitation/blotting experiment results showed that both domains were able to interact with GRIM-19 (Figure 4C).

Fig. 4. Mapping of the interacting regions of Stat3 and GRIM-19. (A) Schematic diagram of Stat3 indicating its functional domains specified according to the amino acids (aa). (B and C) Identification of interacting regions of Stat3. (B) COS-1 cells were co-transfected with GST–GRIM-19 and Flag-tagged Stat3, either full-length (ST3-FL) or deletion mutants containing the N-domain (ND), DNA-binding and linker domains (DB.LK), SH2 domain or C-terminal domain (CT) as labeled on top of the figure. GST pull-down was performed, and the associated proteins were probed with anti-Flag antibody (top panel). The middle panel indicates the GST–GRIM-19 that has been pulled-down by beads. The bottom panel shows the expression of the Flag-tagged Stat3 constructs in total cell lysates. (C) For further mapping, Flag-tagged Stat3 mutant expressing either the linker domain (ST3-LK) or the DNA-binding domain (ST3-DB) was co-transfected with Myc-tagged GRIM-19 (+) or vector alone (–). The cell lysates were immunoprecipitated with anti-Myc and blotted with anti-Flag. The blot was stripped and re-blotted with anti-Myc (middle panel). Expression of ST3-LK and ST3-DB in total cell lysate (TCL) was examined by western blot analysis using anti-Flag (bottom panel). (D) Identification of interacting regions on GRIM-19. GST–GRIM-19 fusions harboring various segments of GRIM-19 (as indicated by the aa numbers) were co-transfected with Flag-Stat3, and GST pull-down experiments were performed. The top panels show Stat3 co-precipitated with GST–GRIM-19. The middle panels show GST and the various GST–GRIM-19 fusion proteins pulled-down by glutathione–Sepharose beads, and the bottom panels indicate the expression of Stat3. The schematic diagram of GRIM-19 is displayed on top of the figure, in which the interacting region (aa 36–72) of GRIM-19 is illustrated as a black box.
There is little structural information available for GRIM-19. To determine the interacting region of GRIM-19, several deletion mutations were randomly selected (amino acids 1–35, 1–101, 36–144, 73–144 and 102–144). GST fusion constructs containing these fragments were generated and co-transfected with Flag-tagged Stat3. The GST pull-down experiments indicated that Stat3 co-precipitated with full-length GST–GRIM- 191–144, GST–GRIM-191–101 and GST–GRIM-1936–144, suggesting that amino acids 36–101 in the middle of the protein harbor the interacting region. Further subcloning revealed that the section containing amino acids 36–72 mediated its association with wild-type Stat3 (Figure 4D) and the individual coiled-coil domain, DNA-binding domain and linker domain (data not shown).
Cellular localization of GRIM-19
To characterize further the nature of the interaction between Stat3 and GRIM-19, we attempted to determine where this interaction occurs. The cellular localization of Myc-tagged human GRIM-19 was first analyzed by immunofluorescence in MCF-7 cells. GRIM-19 was detected exclusively in the cytoplasm as punctate structures, which co-localized with two endogenous mitochondrial proteins, cytochrome oxidase subunit IV (COX) (Figure 5A) and cytochrome c (data not shown), but did not co-localize with EEA1 or CD63, markers for early endosome and late endosome/lysosome, respectively (Metzelaar et al., 1991; Mu et al., 1995). Similar results were obtained with either a Myc-tagged mouse GRIM-19 or a hemagglutinin (HA)-tagged human GRIM-19 in MCF-7, as well as in COS-1 cells (data not shown). To determine further the region for mitochondrial localization, Myc-tagged truncation mutants were generated. GRIM-191–101, deleting the C-terminus of the full-length GRIM-19, showed punctate staining in the cytoplasm which co-localized with COX, whereas the mutant GRIM-1936–144, deleting the N-terminal domain, or GRIM-1936–101, containing the middle region of GRIM-19, exhibited a loss of mitochondrial localization (Figure 5B). Therefore, the N-terminus of GRIM-19 seems to harbor the mitochondrial localization sequence.

Fig. 5. Cellular localization of GRIM-19 in MCF-7 cells. (A and B) Cells were transfected with Myc-tagged GRIM-19 in (A), or the truncation mutants in (B). Cells were fixed and incubated with polyclonal anti-Myc, and stained with Cy3-conjugated anti-rabbit IgG (Amersham Pharmacia Biotech). The localization of the GRIM-19 protein was visualized using a laser scanning confocal microscope. Endogenous COX, EEA1 and CD63 were detected with monoclonal anti-COX subunit IV, anti-EEA1 or anti-CD63 antibody, as labeled, followed by staining with FITC-conjugated anti-mouse IgG (Molecular Probes). Merged images are shown on the right. (C) Expression of endogenous GRIM-19 in MCF-7 cells, either untreated or treated with IFN-β and RA for 72 h, was detected with purified rabbit anti-GRIM-19 serum, followed by Cy3-conjugated anti-rabbit IgG. (D) MCF-7 cells were fractionated using the ApoAlert Cell Fractionation Kit from Clontech following the manufacturer’s instructions. The fractions were collected and subjected to western blot analysis with antibodies, as indicated.
We further analyzed the cellular localization of endogenous GRIM-19 in MCF-7 cells using the purified GRIM-19 antibody. Weak staining of GRIM-19 was observed as punctate structures predominantly in the cytoplasm of untreated cells, which was intensified as small dot-like structures aggregating at the perinuclear region and coincided with COX protein upon IFN-β/RA treatment. In addition, a weak staining was also noticeable in the nucleus, which was slightly increased after the treatment (Figure 5C). To verify these results further, the MCF-7 cells were fractionated into cytoplasmic, mitochondrial and nuclear portions, and GRIM-19 expression in these fractions was determined in the absence or presence of IFN-β/RA treatment. Consistent with the immunofluorescence results, the majority of GRIM-19 was detected in the mitochondrial fraction, which was increased upon IFN-β/RA treatment. A trace amount was detected in the nuclear portion, which was also increased slightly after treatment (Figure 5D). Together, these results suggest that in MCF-7 cells, GRIM-19 protein seems to be localized primarily in the mitochondria, with a minor distribution in the nucleus.
Co-localization of GRIM-19 with Stat3 and its effect on Stat3 nuclear translocation
Cellular localization of Stat3 and its interaction with GRIM-19 were examined subsequently. We observed that in unstimulated MCF-7 cells, Stat3 protein was distributed in the cytoplasm and the nucleus, which was in agreement with a previous report in other cell lines (Meyer et al., 2002). Surprisingly, in cells co-expressing GRIM-19, the majority of Stat3 disappeared from the original locations and formed the dot-like dense structures at the nuclear periphery, which co-localized with GRIM-19 (Figure 6A). Since a few domains of Stat3 can interact with GRIM-19, we next investigated their co-localization with GRIM-19. Strikingly, the Stat3 mutant containing the N-terminal and the coiled-coil domains (ST3-ND.4H) was detected exclusively in the nucleus, and co-expression with GRIM-19 resulted in its disappearance from the nucleus, and formation of perinuclear aggregates similar to those observed in full-length Stat3. On the other hand, Stat3 proteins containing either the DNA-binding and linker domains (ST3-DB.LK), or the SH2 and C-terminal domains (ST3-SH2.CT) localized mainly in the cytoplasm. Their cellular locations were not affected by GRIM-19, and no aggregates were detected, although ST3-DB.LK seemed to be co-localized with GRIM-19 (Figure 6A), which was consistent with its ability to interact with GRIM-19 (Figure 4B). These results indicated that the N-terminal and coiled-coil domains of Stat3 may harbor a nuclear localization signal (NLS), and the coiled-coil domain is required for the formation of the unique perinuclear aggregates which may block Stat3 nuclear translocation.
We further addressed this issue in COS-1 cells stimulated by EGF. In untreated cells, the majority of Stat3 was localized in the cytoplasm, and was translocated into the nucleus in response to EGF in the absence of GRIM-19. However, it formed perinuclear aggregates in the presence of GRIM-19, and co-localized with GRIM-19. Stat1 and Stat5a were also localized in the nucleus in response to EGF stimulation. However, they remained in the nucleus and did not co-localize or form aggregates with the co-transfeced GRIM-19 (Figure 6B).
We further confirmed these results with cellular fractionation in COS-1 cells. A small amount of Stat3 was present in the nucleus in untreated cells, which was increased upon EGF treatment. However, the amount of nuclear Stat3 decreased when co-transfected with GRIM-19 in both cases, whereas expression of the nuclear protein poly(ADP-ribose) polymerase (PARP) (D’Amours et al., 1999) remained constant (Figure 6C). To study the physiological effect of GRIM-19, the nuclear translocation of endogenous Stat3 was evaluated further in HepG2 cells stimulated by IL-6. Stat3 translocated into the nucleus after IL-6 stimulation. However, the amount of nuclear Stat3 decreased upon treatment with IFN-β/RA (Figure 6D, top panel, compare lanes 2 and 4), which correlated with a concomitant increase in endogenous GRIM-19 expression (bottom panel), suggesting a physiological role for GRIM-19 in the regulation of Stat3 nuclear localization.
GRIM-19 represses Stat3-dependent transcription
The co-localization and impaired nuclear translocation of Stat3 by GRIM-19 may result in negative regulation of Stat3 transcriptional activity. To confirm this, we next in vestigated whether GRIM-19 affected Stat3 transcriptional activity using a reporter gene containing three copies of hSIE, the high affinity binding site of Stat3, upstream of a CAT gene. A basal level of transcriptional activity was detected in unstimulated COS-1 cells, which was enhanced by EGF treatment. However, ∼86% inhibition of this EGF-induced activity was observed with co-transfection of GRIM-19. The basal activity in the unstimulated cells was also decreased, to a lesser extent (Figure 7A). The effect of GRIM-19 was also examined in MCF-7 cells. Consistent with its nuclear localization in MCF-7 cells, a constitutive transcriptional activity of Stat3 was observed without any stimulation. However, co-expression of GRIM-19 resulted in an ∼85% inhibition of this activity. Induction of endogenous GRIM-19 by IFN-β and RA also decreased the constitutively activated transcriptional activity of Stat3 (∼15%). This inhibition was repressed further to 91% by co-transfected GRIM-19 (Figure 7B). The repression by GRIM-19 was delineated further with the GRIM-19 mutants. The C-terminal deletion mutant GRIM-191–101, but not the N-terminal deletion mutant GRIM-1936–144, showed inhibition of Stat3 activity (Figure 7C). These data indicate that GRIM-19 represses Stat3 transcriptional activity, and this suppression ability is correlated with its mitochondrial localization.

Fig. 7. Effect of GRIM-19 on the transcriptional activity of Stat3 and target gene expression. (A–C) COS-1 and MCF-7 cells were co-transfected with a vector control, Stat3 and/or GRIM-19, the truncation mutant GRIM-191–101 or GRIM-1936–144 together with a reporter plasmid pSIE-CAT and pCMV-β-gal. Cells were either left uninduced or stimulated with EGF (100 ng/ml) for 6 h in (A) or IFN-β and RA for 24 h in (B). Cells were harvested and the lysates used for CAT assays were normalized with equivalent β-galactosidase activity, and CAT assay was performed as described (Jain et al., 1998). The averages of the relative CAT activities from three independent experiments are shown in the graph, with error bars representing the standard deviation of the means. (D and E) GRIM-19 inhibits α2-macroglobulin report expression. HepG2 cells were co-transfected with pGL3-α2M-215luc or the empty luciferase vector (pGL3), GRIM-19, antisense GRIM-19 plasmid or the empty vector as labeled, together with pRL-TK for normalization. Cells were left non-stimulated (–) or were induced overnight with IL-6 (+). Relative luciferase values are indicated, which represent the means of six samples with the standard deviation given as the error bars. Cells were pre-treated for 55 h with IFN-β (500 U/ml) and RA (1 µM) before stimulation by IL-6 in (E).
We further examined the effect of GRIM-19 on a Stat3 target gene, α2-macroglobulin (α2-M), one of the acute-phase proteins that is upregulated via activation of Stat3 in IL-6-stimulated hepatocytes (Heinrich et al., 1998). HepG2 cells were transfected with an α2-M promoter– luciferase gene construct (Terstegen et al., 2001) or a control plasmid in the presence or absence of GRIM-19. The cells were either left untreated or treated with IL-6. IL-6 resulted in high transcriptional activity of α2-M which was inhibited up to 70% by co-expression of GRIM-19. The basal activity in unstimulated cells was also repressed by GRIM-19 (Figure 7D). The IL-6-inducible α2-M promoter activity was also suppressed by treatment of the cells with IFN-β and RA, which was partially restored by introduction of a plasmid encoding an antisense GRIM-19 (Figure 7E). These data provide evidence that GRIM-19 represses Stat3-dependent target gene expression.
GRIM-19 inhibits cell growth
Stat3 is constitutively activated in v-Src-transformed cells (Yu et al., 1995; Cao et al., 1996), and is essential for Src-mediated cell transformation (Bromberg et al., 1998; Turkson et al., 1998). To determine whether the negative regulation by GRIM-19 of Stat3 activity leads to a physiological consequence, we tested the effect of GRIM-19 on the oncogenic function of Stat3 by examing cell proliferation. We found that cell growth of v-Src-transformed cells was inhibited by GRIM-19, either introduced exogenously by transfection or induced by IFN-β and RA, in a dose-dependent manner (Figure 8A). Furthermore, inhibition was also observed in a stable cell line that expresses an active form of Stat3 mutant, ST3-ΔN (Zhang et al., 2000). These cells proliferated ∼25% more rapidly than the control cells, which can be inhibited by GRIM-19, whereas GRIM-19 showed little effect on control cells (Figure 8B).

Fig. 8. Inhibition of Stat3-mediated cell proliferation by GRIM-19. (A) v-Src-transformed NIH-3T3 cells were grown in a 96-well micro titer plate and transfected with two doses of GRIM-19 (low, 0.1 µg; or high, 0.3 µg) or empty vector (–, 0.3 µg) (left panel), or treated with various concentrations of IFN-β and RA for 48 h (right panel). The cell proliferation assay was performed using the Cell Proliferation Kit II (XTT). Spectrophotometric absorbance was measured at 485 nm wavelength, and reference wavelength 650 nm, which presents a mean of three samples, with the standard deviation given as the error bars. (B) NIH-3T3 cells with stable expression of ST3-ΔN (bar with grid) or control vector (empty bar) were transfected with control vector (–) or with 0.1 µg of GRIM-19 (+), and cell proliferation was measured as described in (A).
Discussion
Stat proteins play central roles in transducing signals from cytokine receptors to the nucleus in a rapid and transient manner. Stat activities are switched off subsequently via several negative regulatory mechanisms (O’Shea et al., 2002). For instance, dephosphorylation of the critical tyrosine residue by specific tyrosine phosphatase leads to the inactivation of Stat proteins (Haspel et al., 1996). The inactivation can also be achieved through protein degradation mediated by the ubiquitin–proteasome pathway (Kim and Maniatis, 1996). Furthermore, two families of negative regulators for JAK–STAT pathways have been reported. One was identified and named as suppressors of cytokine signaling (SOCS)/JAK-binding protein (JAB)/STAT-induced STAT inhibitor (SSI)/cytokine-inducible SH2-containing proteins (CIS) by several groups using different strategies (Yoshimura et al., 1995; Endo et al., 1997; Naka et al., 1997; Starr et al., 1997). The expression of these negative regulators is inducible by cytokine stimulation, which inhibits the JAK–STAT pathway in a negative feedback loop (Yasukawa et al., 2000). The other family is named protein inhibitors of activated STAT (PIAS), which consists of several homologous proteins including PIAS1 and PIAS3, which are constitutively expressed and interact only with tyrosine-phosphorylated Stats to inhibit their DNA binding activities (Chung et al., 1997). In this report, we have identified GRIM-19 as an interacting protein of Stat3 in different mammalian cell types and species. GRIM-19 suppresses Stat3 transcriptional activity and its target gene expression. Furthermore, the interaction of GRIM-19 is specific for Stat3, but not for Stat1 or Stat5a. These data suggest that GRIM-19 functions as a negative regulator of Stat3. To explore the mechanism of the negative effect of GRIM-19 further, tyrosine phosphorylation of Stat3 stimulated by EGF was examined, and no obvious change was observed in the presence of GRIM-19 (data not shown). We subsequently analyzed the cellular localization of these two proteins. The results not only verified the physical interaction between Stat3 and GRIM-19, but also revealed the co-localization of these two proteins in unique aggregates distributed around the nucleus. Although the nature of the aggregates remains elusive, these unique structures partially co-localized with COX and may represent mitochondrial aggregation as examined by electron micro scopy (A.Hao and X.Cao, unpublished data). One of the possible consequences and the physiological roles of this aggregate formation is preventing Stat3 from entering the nucleus. Our data suggest that GRIM-19 is a novel type of inhibitor that specifically inhibits Stat3 activity by blocking its nuclear translocation.
The cellular localization of GRIM-19 was unclear. Originally, it was observed predominantly in the nucleus of IFN-β- and RA-treated HeLa cells (Angell et al., 2000), and recently shown to be in both the nucleus and the cytoplasm (Hu et al., 2002). However, its bovine homolog was co-purified with mitochondrial electron transport complex I, implying a mitochondrial localization (Fearnley et al., 2001). In our study, both transfected human and mouse GRIM-19 were found localized predominantly in the mitochondria in MCF-7 and COS-1 cells. However, although the majority of endogenous GRIM-19 was observed in the mitochondria, it was also detectable in the nucleus in MCF-7 cells (Figure 5C and D). These data suggest that GRIM-19 may distribute in the mitochondria and/or nucleus depending on the cell type. Alternatively, its cellular localization may be regulated by post-translational modifications such as phosphorylation and acetylation. Lastly, GRIM-19 may exist in different isoforms that may be distributed in distinct cellular locations.
To examine the nature of the interaction, we defined the interaction regions in both Stat3 and GRIM-19. The results showed that in addition to the coiled-coil domain, the DNA-binding and linker domains of Stat3 are also capable of binding to GRIM-19. However, further study shows that the coiled-coil region, but not the N-terminal domain (data not shown), DNA-binding domain or linker domain, is involved in the formation of aggregates with GRIM-19 (Figure 6A), which exerts a negative regulation on Stat3 nuclear translocation. These data suggest that interaction with the coiled-coil domain may play a physiological role. Surprisingly, we also found that the truncation mutant of Stat3 containing the N-terminal domain and the coiled-coil domain is constitutively localized in the nucleus. Our preliminary results defined an NLS in the coiled-coil domain (J.Ma and X.Cao, unpublished data). The coiled-coil domain contains four antiparallel α-helices, and has often been inferred to be involved in protein–protein interaction (Becker et al., 1998; Chen et al., 1998). Indeed, the coiled-coil domains of Stat proteins have been reported to mediate association with other transcription factors and coactivators, such as p48, a protein from an IFN response factor (IRF) family, c-Jun, and Nmi, an N-Myc interactor, which regulate Stat transcriptional activity (Horvath et al., 1996; Zhang et al., 1999; Zhu et al., 1999). Recently, we have demonstrated a novel role for the coiled-coil domain of Stat3 in the regulation of its SH2 domain-mediated binding activity to the cytokine receptor (Zhang et al., 2000). This regulation is probably achieved via interdomain interaction between the coiled-coil domain and the C-terminal domain of Stat3 (Zhang et al., 2002). In this study, we provide evidence for new roles for the coiled-coil domain. Collectively, these data indicate that the coiled-coil domain actively participates in the regulation of Stat activities in multiple processes by interacting with distinct proteins.
There is evidence correlating Stat3 activation and cancer. Stat3 is found to be constitutively activated in cells transformed by oncogenes, such as v-Src and v-abl (Danial et al., 1995; Yu et al., 1995; Cao et al., 1996), as well as in various cancers including multiple myeloma, breast cancer, and head and neck cancer (Bowman et al., 2000). Furthermore, expression of a constitutively dimerized Stat3 (Stat3-C) is oncogenic (Bromberg et al., 1999). Constitutively activated Stat3 elevates the expression of its target genes that are key regulators in the control of proliferation, cell cycle and programmed cell death, all of which may contribute to oncogenesis (Bowman et al., 2000). In contrast, a combination of IFN-β and RA inhibits cell growth and promotes cell death in cancer cells which does not involve known IFN-stimulated genes, such as RnaseL and protein kinase R, or a change of the level of p53 and the phosphorylation status of pRb (Hofman et al., 1998). It is obvious that Stat3 and GRIM-19 play opposite roles in cell growth, transformation and cell death. Our finding that GRIM-19 is a negative regulator of Stat3 is consistent with this observation. In this report, we have shown that GRIM-19 inhibits Stat3-mediated cell proliferation in both Src-transformed cells and Stat3-ΔN-stable-expressing cells (Figure 8). The next step will be to study whether GRIM-19 suppresses Stat3-mediated cell transformation, and the mechanisms involved. It will also be interesting to investigate whether GRIM-19 exerts its death effect by repressing Stat3 activity via inhibition of the expression of Stat3-regulated anti-apoptotic genes.
Materials and methods
Construction of expression plasmids
Murine Stat3 and its deletion mutants were cloned into pXJ40-FLAG, as described previously (Zhang et al., 2002). Stat1 and Stat5a, obtained from J.E. Darnell,Jr and A.Miyajima, respectively, were also cloned into vector pXJ40-FLAG. The clones containing the cDNA sequence of murine GRIM-19 were isolated from the MATCHMAKER library containing 17th day mouse embryonic cDNAs by yeast two-hybrid screening (Clontech). Full-length GRIM-19 was generated by PCR, and the respective PCR product was sequenced and cloned into pDMYCneo, a double Myc-tagged expression plasmid (Seet and Hong, 2001). The human homolog of GRIM-19 was isolated from a cDNA library of MCF-7 cells (a gift from V.Yu from our institute). The deletion mutants of GRIM-19 were generated by PCR, and the PCR products were cloned into pXJ40-GST, a plasmid expressing GST in mammalian cells (Manser et al., 1998).
Yeast two-hybrid screening
The coiled-coil domain (amino acids 138–318) of Stat3 was cloned into plasmid pGBKT7 containing the GAL4 DNA-binding domain as bait. This plasmid was transformed into Saccharomyces cerevisiae strain AH109.
The evaluation and screening procedures were performed according to the manufacturer’s instructions. Briefly, S.cerevisiae strain Y187 pre-transformed with the MATCHMAKER library containing 17th day mouse embryonic cDNAs fused to the GAL4 activation domain (AD) (Clontech) was mated with the AH109 strain harboring the bait, at 30°C for 24 h. The mating mixture was spread on plates containing selection medium lacking Trp, His, Leu and Ade, and incubated at 30°C for ∼2 weeks. A β-gal colony-lift filter assay was performed to eliminate the false-positive colonies. The plasmids were isolated from the positive colonies and transformed into Escherichia coli strain DH5α by electroporation. Plasmid DNA was purified from E.coli and subjected to sequencing analysis.
Generation of antibody against mouse GRIM-19
GRIM-19 cDNA was amplified by PCR and cloned into the bacterial expression vector pET32B (Novagen) which was transformed into E.coli BL21DE3. The expression of the His-tagged fusion protein was induced by 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h, and the fusion protein was purified with nickel-nitrilotriacetic acid–agarose resin (Qiagen) under urea-denaturing conditions. Bead-bound GRIM-19 was then refolded in phosphate-buffered saline (PBS) and used to immunize rabbits. The antiserum was purified further as described (Olmsted, 1981).
Cell culture and DNA transfection
COS-1, SH-SY5Y, NIH-Src and ST-3-ΔN cells were grown in Dulbecco’s modified Eagle’s medium (DMEM), and MCF-7 in RPMI with 10% fetal bovine serum (FBS) purchased from Gibco-Invitrogen. Mouse C2C12 myoblasts from the ATCC were grown in DMEM with 20% FBS, and PC-12 cells were cultured in DMEM with 10% FBS and 5% horse serum. Transient transfections were performed with LipofectAMINE or LipofectAMINE 2000 (Gibco-Invitrogen) following the manufacturer’s instructions.
Immunoprecipitation, GST pull-down and western blotting
The lysates containing 600 µg of total proteins were subjected to immunoprecipitation/blotting as described previously (Cao et al., 1996). For GST pull-down experiments, the cell lysates containing 600 µg of proteins were incubated with 40 µl of glutathione–Sepharose 4B beads (Amersham Pharmacia Biotech). The precipitates were washed, separated by SDS–PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane, followed by western blotting analysis with the respective antibodies.
Immunofluorescence
Cells grown on coverslips were transfected with various expression plasmids. After transfection for 24 h, cells were fixed with 1% paraformaldehyde in PBS, and washed with PBS. After permeabilizing with methanol for 5 min, the cells were washed and incubated with FDB (PBS with 1 mM CaCl2, 1 mM MgCl2, 5% normal goat serum, 10% fetal bovine serum and 2% bovine serum albumin pH 7.6), followed by incubation with primary antibodies in FDB. After washing with PBS containing 0.1% Triton X-100, cells were incubated with the appropriate secondary antibodies conjugated with fluorescein isothiocyanate (FITC) or Cy3. Cells were then washed three times with PBS containing 0.1% Trixon X-100, mounted with FluoroGuard™ Antifade Reagent (Bio-Rad Laboratories), and examined using a confocal microscope (Bio-Rad MRC 1024). The figures were processed with Adobe Photoshop software.
Luciferase assay
HepG2 cells were seeded at 0.75 × 105 cells per well in a 24-well dish. The firefly luciferase reporter gene construct (pGL3-α2M-215luc) was co-transfected with expression plasmid and pRL-TK (thymidine kinase promoter-dependent Renilla luciferase construct) used as an internal control for transfection efficiency (Promega) using LipofectAMINE 2000 (Invitrogen). Cells were serum starved for 24 h prior to stimulation with IL-6 (40 ng/ml) overnight. Cells were washed three times with PBS, lysed with 100 µl of passive lysis buffer (Promega) and subjected to one freeze–thaw cycle at –80°C. A Dual Luciferase Reporter assay (Promega) was performed according to the manufacturer’s instructions using the Ascent luminoskan (ThermoLabsystems).
Cell growth assay
The assay was performed using the Cell Proliferation Kit II (XTT) from Roche Diagnostics following the manufacturer’s instructions. Briefly, cells were plated in 96-well plates and either transfected with proper plasmids, or treated with IFN-β/RA without transfection. Cells were incubated with XTT labeling mixture at 37°C for the appropriate time to allow the development of the color. Spectrophotometric absorbance of the samples was measured at 485 nm wavelength and reference wavelength 650 nm using SPECTRAFLuo Plus reader from Tecan.
Stable transfection of Stat3
NIH-3T3 cells were co-transfected with pXJ40-flag-ST3-ΔN, a mutant lacking the N-terminal domain of Stat3 with a constitutive activity with pXJ41-neo containing the G418 resistance in a ratio of 20:1. After 48 h, the cells were trypsinized and seeded in different densities with DMEM containing 375 µg/ml G418 (Sigma). After 2 weeks, single colonies were isolated and tested for protein expression by western blot analysis using Flag-M2 antibody (Sigma). A control cell line that expresses the empty vector was established with a similar method.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank J.E.Darnell,Jr and A.Miyajima for Stat1 and Stat5a cDNA, respectively, P.C.Heinrich for luciferase reporter, R.Jove for NIH-Src cells, M.Inoue for IFN-β, V.Yu for the MCF-7 cell line and cDNA library, Z.Feng and A.G.Porter for the SH-SY5Y cell line, W.Hong for pDMYCneo vector and valuable suggestions, and A.G.Porter and C.P.Lim for critically reading the manuscript. X.Cao is also an adjunct member of staff in the Department of Biochemistry, National University of Singapore. This work was supported by the Agency for Science, Technology and Research of Singapore.
References
- Akira S. (2000) Roles of STAT3 defined in tissue-specific gene targeting. Oncogene, 19, 2607–2611. [DOI] [PubMed] [Google Scholar]
- Akira S. et al. (1994) Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell, 77, 63–71. [DOI] [PubMed] [Google Scholar]
- Angell J.E., Lindner,D.J., Shapiro,P.S., Hofmann,E.R. and Kalvakolanu,D.V. (2000) Identification of GRIM-19, a novel cell death-regulatory gene induced by the interferon-β and retinoic acid combination, using a genetic approach. J. Biol. Chem., 275, 33416–33426. [DOI] [PubMed] [Google Scholar]
- Becker S., Groner,B. and Müller,C.W. (1998) Three-dimensional structure of the Stat3b homodimer bound to DNA. Nature, 394, 145–151. [DOI] [PubMed] [Google Scholar]
- Bowman T., Garcoa,R., Turkson,J. and Jove,R. (2000) STATs in oncogenesis. Oncogene, 19, 2474–2488. [DOI] [PubMed] [Google Scholar]
- Bromberg J.F., Horvath,C.M., Besser,D., Lathem,W.W. and Darnell,J.E.,Jr (1998) Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol., 18, 2553–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg J.F., Wrzeszczynska,M.H., Devgan,G., Zhao,Y., Pestell,R.G., Albanese,C. and Darnell,J.E.,Jr (1999) Stat3 as an oncogene. Cell, 98, 295–303. [DOI] [PubMed] [Google Scholar]
- Cao X., Tay,A., Guy,G.R. and Tan,Y.H. (1996) Activation and association of Stat3 with Src in v-Src-transfromed cell lines. Mol. Cell. Biol., 16, 1595–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catlett-Falcone R. et al. (1999) Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity, 10, 105–115. [DOI] [PubMed] [Google Scholar]
- Chen X., Vinkemeier,U., Zhao,Y., Jeruzalmi,D., Darnell,J.E.,Jr and Kuriyan,J. (1998) Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell, 93, 827–839. [DOI] [PubMed] [Google Scholar]
- Chung C.D., Liao,J., Liu,B., Rao,X., Jay,P., Berta,P. and Shuai,K. (1997) Specific inhibition of Stat3 signal transduction by PIAS3. Science, 278, 1803–1805. [DOI] [PubMed] [Google Scholar]
- D’Amours D., Desnoyers,S., D’Silva,I. and Poirier,G.G. (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J., 342, 249–268. [PMC free article] [PubMed] [Google Scholar]
- Danial N.N., Pernis,A. and Rothman,P.B. (1995) Jak–STAT signaling induced by the v-abl oncogene. Science, 269, 1875–1877. [DOI] [PubMed] [Google Scholar]
- Darnell J.E. Jr, Kerr,I.M. and Stark,G.R. (1994) Jak–STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science, 264, 1415–1421. [DOI] [PubMed] [Google Scholar]
- Durfee T., Becherer,K., Chen,P.L., Yeh,S.H., Yang,Y., Kilburn,A.E., Lee,W.H. and Elledge,S.J. (1993) The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev., 7, 555–569. [DOI] [PubMed] [Google Scholar]
- Endo T.A. et al. (1997) A new protein containing an SH2 domain that inhibits JAK kinases. Nature, 387, 921–924. [DOI] [PubMed] [Google Scholar]
- Fearnley I.M., Carroll,J., Shannon,R.J., Runswick,M.J., Walker,J.E. and Hirst,J. (2001) GRIM-19, a cell death regulatory gene product, is a subunit of bovine mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Biol. Chem., 276, 38345–38348. [DOI] [PubMed] [Google Scholar]
- Fukada T., Hibi,M., Yamanaka,Y., Takahashi-Tezuka,M., Fujitani,Y., Yamaguchi,T., Nakajima,K. and Hirano,T. (1996) Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity, 5, 449–460. [DOI] [PubMed] [Google Scholar]
- Grandis J.R. et al. (2000) Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc. Natl Acad. Sci. USA, 97, 4227–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspel R.L., Salditt-Georgieff,M. and Darnell,J.E.,Jr (1996) The rapid inactivation of nuclear tyrosine phosphorylated Stat1 depends upon a protein tyrosine phosphatase. EMBO J., 15, 6262–6268. [PMC free article] [PubMed] [Google Scholar]
- Heinrich P.C., Behrmann,I., Muller-Newen,G., Schaper,F. and Graeve,L. (1998) Interleukin-6-type cytokine signaling through the gp130/Jak/STAT pathway. Biochem. J., 334, 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofman E.R., Boyanapalli,M., Lindner,D.J., Weihua,X., Hassel,B.A., Jagus,R., Gutierrez,P.L. and Kalvakolanu,D.V. (1998) Thioredoxin reductase mediates cell death effects of the combination of β interferon and retinoic acid. Mol. Cell. Biol., 18, 6493–6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath C.M., Stark,G.R., Kerr,I.M. and Darnell,J.E.,Jr (1996) Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex. Mol. Cell. Biol., 16, 6957–6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Angell,J.E., Zhang,J., Ma,X., Seo,T., Raha,A., Hayashi,J., Choe,J. and Kalvakolanu,D.V. (2002) Characterization of monoclonal antibodies against GRIM-19, a novel IFN-β and retinoic acid-activated regulator of cell death. J. Interferon Cytokine Res., 22, 1017–1026. [DOI] [PubMed] [Google Scholar]
- Ihle J.N. (2001) The Stat family in cytokine signaling. Curr. Opin. Cell Biol., 13, 211–217. [DOI] [PubMed] [Google Scholar]
- Jain N., Zhang,T., Fong,S.L., Lim,C.P. and Cao,X. (1998) Repression of Stat3 activity by activation of mitogen-activated protein kinase (MAPK). Oncogene, 17, 3157–3167. [DOI] [PubMed] [Google Scholar]
- Jain N., Zhang,T., Kee,W.H., Li,W. and Cao,X. (1999) Protein kinase Cδ associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J. Biol. Chem., 274, 24392–24400. [DOI] [PubMed] [Google Scholar]
- Kim T.K. and Maniatis,T. (1996) Regulation of interferon-γ-activated STAT1 by the ubiquitin–proteasome pathway. Science, 273, 1717–1719. [DOI] [PubMed] [Google Scholar]
- Lai C.H., Chou,C.Y., Ch’ang,L.Y., Liu,C.S. and Lin,W. (2000) Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res., 10, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love J.M. and Gudas,L.J. (1994) Vitamin A, differentiation and cancer. Curr. Opin. Cell Biol., 6, 825–831. [DOI] [PubMed] [Google Scholar]
- Manser E., Loo,T.H., Koh,C.G., Zhao,Z.S., Chen,X.Q., Tan,L., Tan,I., Leung,T. and Lim,L. (1998) PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell, 1, 183–192. [DOI] [PubMed] [Google Scholar]
- Metzelaar M.J., Wijngaard,P.L., Peters,P.J., Sixma,J.J., Nieuwenhuis,H.K. and Clevers,H.C. (1991) CD63 antigen. A novel lysosomal membrane glycoprotein, cloned by a screening procedure for intracellular antigens in eukaryotic cells. J. Biol. Chem., 266, 3239–3245. [PubMed] [Google Scholar]
- Meyer T., Gavenis,K. and Vinkemeier,U. (2002) Cell type-specific and tyrosine phosphorylation-independent nuclear presence of STAT1 and STAT3. Exp. Cell Res., 272, 45–55. [DOI] [PubMed] [Google Scholar]
- Mu F.T. et al. (1995) EEA1, an early endosome-associated protein. EEA1 is a conserved α-helical peripheral membrane protein flanked by cysteine ‘fingers’ and contains a calmodulin-binding IQ motif. J. Biol. Chem., 270, 13503–13511. [DOI] [PubMed] [Google Scholar]
- Naka T. et al. (1997) Structure and function of a new STAT-induced STAT inhibitor. Nature, 387, 924–929. [DOI] [PubMed] [Google Scholar]
- Olmsted J.B. (1981) Affinity purification of antibodies from diazotized paper blots of heterogeneous protein samples. J. Biol. Chem., 256, 11955–11957. [PubMed] [Google Scholar]
- O’Shea J.J., Gadina,M. and Schreiber,R.D. (2002) Cytokine signaling in 2002. New surprises in the Jak/STAT pathway. Cell, 109, S121–S131. [DOI] [PubMed] [Google Scholar]
- Sadowski H.B., Shuai,K., Darnell,J.E.,Jr and Gilman,M.Z. (1993) A common nuclear signal transduction pathway activated by growth factor and cytokine receptors. Science, 261, 1739–1743. [DOI] [PubMed] [Google Scholar]
- Schindler C. and Darnell,J.E. (1995) Transcriptional responses to polypeptide ligands: the JAK–STAT pathway. Annu. Rev. Biochem., 64, 621–651. [DOI] [PubMed] [Google Scholar]
- Seet L.F. and Hong,W. (2001) Endofin, an endosomal FYVE domain protein. J. Biol. Chem., 276, 42445–42454. [DOI] [PubMed] [Google Scholar]
- Stark G.R., Kerr,I.M., Williams,B.R., Silverman,R.H. and Schreiber, R.D. (1998) How cells respond to interferons. Annu. Rev. Biochem., 67, 227–264. [DOI] [PubMed] [Google Scholar]
- Starr R. et al. (1997) A family of cytokine-inducible inhibitors of signaling. Nature, 387, 917–921. [DOI] [PubMed] [Google Scholar]
- Takeda K., Noguchi,K., Shi,W., Tanaka,T., Matsumoto,M., Yoshida,N., Kishimoto,T. and Akira,S. (1997) Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl Acad. Sci. USA, 94, 3801–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terstegen L., Gatsios,P., Ludwig,S., Pleschka,S., Jahnen-Dechent,W., Heinrich,P.C. and Graeve,L. (2001) The vesicular stomatitis virus matrix protein inhibits glycoprotein 130-dependent STAT activation. J. Immunol., 167, 5209–5216. [DOI] [PubMed] [Google Scholar]
- Turkson J., Bowman,T., Garcia,R., Caldenhoven,E., De Groot,R.P. and Jove,R. (1998) Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol., 18, 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H., Sasaki,A. and Yoshimura,A. (2000) Negative regulation of cytokine signaling pathways. Annu. Rev. Immunol., 18, 143–164. [DOI] [PubMed] [Google Scholar]
- Yoshimura A., Ohkubo,T., Kiguchi,T., Jenkins,N.A., Gilbert,D.J., Copeland,N.G., Hara,T. and Miyajima,A. (1995) A novel cytokine-inducible gene CIS encodes an SH2-containing protein that binds to tyrosine-phosphorylated interleukin 3 and erythropoietin receptors. EMBO J., 14, 2816–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C.-L., Meyer,D.J., Campbell,G.S., Larner,A.C., Carter-Su,C., Schwartz,J. and Jove,R. (1995) Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science, 269, 81–83. [DOI] [PubMed] [Google Scholar]
- Zhang T., Kee,W.H., Seow,K.T., Fung,W. and Cao,X. (2000) The coiled-coil domain of Stat3 is essential for its SH2 domain-mediated receptor binding and subsequent activation induced by epidermal growth factor and interleukin-6. Mol. Cell. Biol., 20, 7132–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Seow,K.T., Ong,C.T. and Cao,X. (2002) Interdomain interaction of Stat3 regulates its SH2 domain-mediated receptor binding activity. J. Biol. Chem., 277, 17556–17563. [DOI] [PubMed] [Google Scholar]
- Zhang X., Wrzeszczynska,M.H., Horvath,C.M. and Darnell,J.E.,Jr (1999) Interacting regions in Stat3 and c-Jun that participate in cooperative transcriptional activation. Mol. Cell. Biol., 19, 7138–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z., Wen,Z. and Darnell,J.E.,Jr (1994a) Stat3 and Stat4: members of the family of signal transducers and activators of transcription. Proc. Natl Acad. Sci. USA, 91, 4806–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z., Wen,Z. and Darnell,J.E.,Jr (1994b) Stat3: a STAT family member activated by tyrosine phosphorylation in respone to epidermal growth factor and interleukin-6. Science, 264, 95–98. [DOI] [PubMed] [Google Scholar]
- Zhu M.-H., John,S., Berg,M. and Leonard,W.J. (1999) Functional association of Nmi with Stat5 and Stat1 in IL-2- and IFNγ-mediated signaling. Cell, 96, 121–130. [DOI] [PubMed] [Google Scholar]