

Abstract
Protein synthesis and protein degradation are highly regulated cellular processes that are essential to maintaining cell viability. Numerous studies now indicate that protein synthesis and protein degradation are significantly altered in Alzheimer's disease (AD), with impairments in these two processes potentially contributing to AD pathogenesis. Alterations in steady state protein regulation may be a particularly important factor in regulating whether cells maintain homeostasis in response to oxidative damage, or conversely whether oxidative stress is induced by oxidative damage. The focus of this review is to discuss recent findings on each of these topics, and to discuss their importance to the onset and progression of AD.
INTRODUCTION
Alzheimer's disease and mild cognitive impairment
Alzheimer's disease (AD) is the leading cause of dementia in the elderly and is characterized by the presence of extensive senile plaque deposition and neurofibrillary pathology [10, 20]. Despite the numerous advances in our understanding of AD, the neurochemical alterations which are responsible for the onset and progression of AD have not been identified. A major obstacle to identifying such factors is the fact that studies involving AD brain tissue have been difficult to interpret. In particular, it has been difficult to distinguish which neurochemical events are playing a causal role in mediating neurodegeneration and neuropathology, and which events are occurring in response to the extensive neurodegeneration and neuropathology observed in AD brain. Because of this, there has been considerable interest in conducting studies in individuals who are in the earliest stages of AD, or individuals who are at the highest risk to develop AD, where the presence of little-to-no neuropathology allows for a clearer experimental interpretation of neurochemical studies.
A number of elderly individuals, who do not have dementia, develop cognitive deficits which are atypical of those observed in normal aging. Longitudinal analysis of these individuals has demonstrated that they convert to AD at a much higher rate than the at large elderly population [12, 13]. It is now thought that these individuals may represent a transitional state between dementia and normal aging, with such subjects being in the earliest stages of AD pathogenesis [12, 13]. Neuropathological studies in these individuals have revealed that these subjects exhibit an extensive overlap with the autopsy findings in older cognitively intact individuals. Because of this, it is believed that these individuals can be used to identify the neurochemical alterations which occur in the earliest stages of AD pathogenesis, and neurochemical alterations which precede the development of dementia and extensive AD neuropathology. Such neurochemical alterations may therefore represent the substrates for pathological and cognitive alterations observed in AD. Individuals in this cohort are now commonly referred to as having mild cognitive impairment (MCI) [12, 13].
Oxidative damage in AD and MCI
Studies from our laboratory and others have demonstrated that increased levels of protein oxidation [7, 16], lipid oxidation [14], and nucleic acid oxidation [2, 14] are all evident in AD. Recent studies have also found that increases in each of these forms of oxidative damage are present in MCI subjects [2, 7, 11, 19]. Interestingly, increases in each of these oxidative modifications preferentially occur in the brain regions involved in regulating cognition. Such an observation is consistent with oxidative damage contributing to the development of dementia. In support of this hypothesis, in studies involving MCI and early AD subjects the increases in protein oxidation were observed to be significantly and inversely correlated with word recall performance [7].
Elevations in oxidative damage in MCI and AD subjects are significantly higher than those of age-matched control subjects. This suggests that the elevations in oxidative damage within MCI and AD either represent an acceleration of the normal age-related generation of oxidative damage, or may indicate that alternative pathways for the generation of oxidative damage occur in MCI and AD subjects. Clarification of this issue is vital to our understanding of AD, and is likely important for the design of therapeutic interventions for AD.
It is well established that while elevations in oxidative damage occur in AD [14, 16], the amount of oxidative damage in an individual does not predict the presence or severity of AD. This same observation holds true for other agerelated neurodegenerative disorders [3, 6], such as Parkinson's disease. This suggests that the ability of elevated levels of oxidative damage to induce oxidative stress is not solely dependent upon the gross levels of oxidative damage. It is likely that the development of oxidative stress is regulated in large part by the ability of cells to replace those macromolecules which have been damaged by oxidation, and the ability of cells to generate sufficient levels of inhibitory macromole cules, which inhibit the ability of oxidative damage to induce oxidative stress.
Protein oxidation, synthesis, and degradation in AD
While there are many different forms of oxidative damage in AD and MCI, protein oxidation is the one most likely to directly impact cellular homeostasis. This is based on the fact that proteins are directly responsible for the various enzymatic processes, and structural support, necessary for cellular homeostasis [17]. Oxidative modification of proteins is capable of inhibiting their normal function, inducing deleterious protein fragmentation, and promoting the ability of proteins to form promiscuous interactions which can lead to the development of protein aggregates [17]. The formation of highly oxidized or cross-linked proteins is likely to have negative effects on the proteolytic pathways (proteasomal and lysosomal), impairing the ability of these proteases to mediate bulk protein turnover [1, 4]. Maintaining low levels of protein oxidation is therefore likely to be a key and important part of maintaining the overall steady state protein kinetics in the cell.
As outlined above, the ability of protein oxidation to induce deleterious effects on a cell is ultimately regulated by the ability of cells to synthesize new proteins. Specifically, proteins are needed to replace those which have been oxidized, and to inhibit the initiation of oxidized protein toxicity. Failure to generate these two types of proteins would be expected to result in a progressive accumulation of aberrantly functional proteins, which would be expected to have direct and complex effects on the different cells of the brain (Figure 1).
Figure 1.
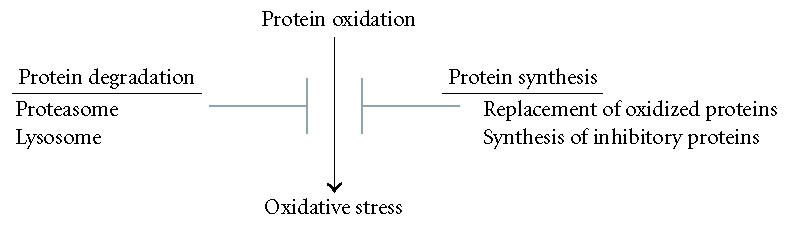
Interplay between protein synthesis, protein degradation, protein oxidation, and oxidative stress. Oxidized proteins are capable of inducing oxidative stress, with oxidative stress a potential mediator of AD pathogenesis. The ability of oxidized proteins to induce oxidative stress is inhibited by protein synthesis and protein degradation pathways. The generation of proteins to replace those which have been oxidized, and the synthesis of proteins which inhibits oxidized protein toxicity (ie, heat shock proteins), inhibits the induction of oxidative stress. Proteolytic pathways (lysosomal, proteasomal) degrade oxidized proteins and thereby ameliorate their ability to induce oxidative stress.
Numerous studies indicate that in both AD and MCI the levels of protein synthesis are impaired [2, 9]. This inhibition appears to be due to deleterious oxidation of RNA molecules, as well as gross disturbances of the ribosome complex [2, 5, 9]. As outlined above, inhibition of protein synthesis would be expected to rapidly exacerbate the ability of oxidized proteins to induce oxidative stress. For example, the inability to replace oxidized proteins could increase the percentage of inactive or malfunctioning proteins in the cell to a level sufficient to induce cellular stress. Similarly, the threshold of oxidized proteins required to form protein inclusions and aggregates would be expected to be lower in cells that are impaired in their ability to synthesize inhibitory proteins (heat shock proteins, proteases). This potentially lethal combination of increased levels of oxidized proteins, and decreased levels of protein synthesis, almost certainly contributes to the oxidative stress believed to occur in AD.
It is possible that variability in the inhibition of protein synthesis helps to explain the inability of gross amounts of protein oxidation to predict the presence or severity of AD. For example, cells which have increasing levels of protein oxidation but are able to maintain sufficient levels of protein synthesis, there may be no induction of deleterious oxidative stress. Conversely, in cells where there is an inhibition of protein synthesis and an increasing amount of protein oxidation, there is likely to be the development of lethal oxidative stress. Experimental clarification of each of these issues, including the identification of which proteins are the most important to inhibiting the ability of oxidative damage to induce oxidative stress, is still very much needed.
Potential therapeutics
Based on the aforementioned studies we propose that interventions which increase protein synthesis, in particular the synthesis of beneficial proteins, may be useful in the treatment of AD. Two promising classes of pharmaceuticals are those which inhibit histone deacetylase (HDAC) activity, and compounds which regulate the mTOR pathway. Each of these types of compounds is known to potently stimulate protein synthesis [8, 18], and has been demonstrated to be neuroprotective. Interestingly, HDAC inhibitors have been demonstrated to not only suppress oxidative stress toxicity in vitro and in vivo, but have also been demonstrated to suppress the formation of neuropathology [8, 15, 18]. Identifying which proteins are increased in response to these experimental treatments, and which of these proteins are responsible for mediating neuroprotection, is important for our understanding of AD and the generation of potentially useful interventions for the treatment of AD and other age-related neurodegenerative disorders.
ACKNOWLEDGMENTS
I would like to thank Drs Qunxing Ding, Quinghua Chen, Harry Levine, Annadora Bruce-Keller, and William Markebery for helpful discussions. This work was supported by Grants from the National Institutes of Aging.
References
- 1.Bahr BA, Bendiske J. The neuropathogenic contributions of lysosomal dysfunction. Journal of Neurochemistry. 2002;83(3):481–489. doi: 10.1046/j.1471-4159.2002.01192.x. [DOI] [PubMed] [Google Scholar]
- 2.Ding Q, Markesbery WR, Chen Q, et al. Ribosome dysfunction is an early event in Alzheimer's disease. Journal of Neuroscience. 2005;25(40):9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giasson BI, Ischiropoulos H, Lee VM, Trojanowski JQ. The relationship between oxidative/nitrative stress and pathological inclusions in Alzheimer's and Parkinson's diseases. Free Radical Biology and Medicine. 2002;32(12):1264–1275. doi: 10.1016/s0891-5849(02)00804-3. [DOI] [PubMed] [Google Scholar]
- 4.Grune T, Merker K, Sandig G, et al. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochemical and Biophysical Research Communications. 2003;305(3):709–718. doi: 10.1016/s0006-291x(03)00809-x. [DOI] [PubMed] [Google Scholar]
- 5.Honda K, Smith MA, Zhu X, et al. Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. Journal of Biological Chemistry. 2005;280(22):20978–20986. doi: 10.1074/jbc.M500526200. [DOI] [PubMed] [Google Scholar]
- 6.Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Annals of Neurology. 1998;44(3 suppl 1):S72–S84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- 7.Keller JN, Schmitt FA, Scheff SW, et al. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64(7):1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 8.Langley B, Gensert JM, Beal MF, et al. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Current Drug Targets. 2005;4(1):41–50. doi: 10.2174/1568007053005091. [DOI] [PubMed] [Google Scholar]
- 9.Langstrom NS, Anderson JP, Lindroos HG, et al. Alzheimer's disease-associated reduction of polysomal mRNA translation. Molecular Brain Research. 1989;5(4):259–269. doi: 10.1016/0169-328x(89)90060-0. [DOI] [PubMed] [Google Scholar]
- 10.Markesbery WR. Neuropathological criteria for the diagnosis of Alzheimer's disease. Neurobiology of Aging. 1997;18(4 suppl):S13–S19. doi: 10.1016/s0197-4580(97)00064-x. [DOI] [PubMed] [Google Scholar]
- 11.Markesbery WR, Kryscio RJ, Lovell MA, et al. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Annals of Neurology. 2005;58(5):730–735. doi: 10.1002/ana.20629. [DOI] [PubMed] [Google Scholar]
- 12.Morris JC. Mild cognitive impairment and preclinical Alzheimer's disease. Geriatrics. 2005;60(6 suppl):9–14. [PubMed] [Google Scholar]
- 13.Petersen RC, Doody R, Kurz A, et al. Current concepts in mild cognitive impairment. Archives of Neurology. 2001;58(12):1985–1992. doi: 10.1001/archneur.58.12.1985. [DOI] [PubMed] [Google Scholar]
- 14.Picklo MJ, Montine TJ, Amarnath V, et al. Carbonyl toxicology and Alzheimer's disease. Toxicology and Applied Pharmacology. 2002;184(3):187–197. doi: 10.1006/taap.2002.9506. [DOI] [PubMed] [Google Scholar]
- 15.Ryu H, Smith K, Camelo SI, et al. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. Journal of Neurochemistry. 2005;93(5):1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- 16.Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Current Medicinal Chemistry. 2001;8(7):721–738. doi: 10.2174/0929867013372922. [DOI] [PubMed] [Google Scholar]
- 17.Sohal RS. Role of oxidative stress and protein oxidation in the aging process. Free Radical Biology and Medicine. 2002;33(1):37–44. doi: 10.1016/s0891-5849(02)00856-0. [DOI] [PubMed] [Google Scholar]
- 18.Soliman GA. The mammalian target of rapamycin signalling network and gene regulation. Current Opinion in Lipidology. 2005;16(3):317–323. doi: 10.1097/01.mol.0000169352.35642.06. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Xiong S, Xie C, et al. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. Journal of Neurochemistry. 2005;93(4):953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 20.Wisniewski HM, Silverman W. Diagnostic criteria for the neuropathological assessment of Alzheimer's disease: current status and major issues. Neurobiology of Aging. 1997;18(4 suppl):S43–S50. doi: 10.1016/s0197-4580(97)00068-7. [DOI] [PubMed] [Google Scholar]