

Abstract
In vitro selection, or directed molecular evolution, allows the isolation and amplification of rare sequences that satisfy a functional-selection criterion. This technique can be used to isolate novel ribozymes (RNA enzymes) from large pools of random sequences. We used in vitro evolution to select a ribozyme that catalyzes a novel template-directed RNA ligation that requires surprisingly few nucleotides for catalytic activity. With the exception of two nucleotides, most of the ribozyme contributes to a template, suggesting that it is a general prebiotic ligase. More surprisingly, the catalytic core built from randomized sequences actually contains a 7-nt manganese-dependent self-cleavage motif originally discovered in the Tetrahymena group I intron. Further experiments revealed that we have selected a dual-catalytic RNA from random sequences: the RNA promotes both cleavage at one site and ligation at another site, suggesting two conformations surrounding at least one divalent metal ion-binding site. Together, these results imply that similar catalytic RNA motifs can arise under fairly simple conditions and that multiple catalytic structures, including bifunctional ligases, can evolve from very small preexisting parts. By breaking apart and joining different RNA strands, such ribozymes could have led to the production of longer and more complex RNA polymers in prebiotic evolution.
Keywords: 2′,5′-link; prebiotic; manganese; RNA editing; ribozyme
The in vitro selection of functional RNA molecules from random sequence pools has successfully isolated far more classes of ribozymes than have yet been found in nature. The classic experiment (1) revealed an abundance of RNA ligase ribozymes in a random sampling of 1015 unique sequences. The first experiments to probe in vitro evolution of ribozymes searched for improved or altered versions of the naturally occurring Tetrahymena group I ribozyme. For example, Green et al. (2) used in vitro evolution to restore and to improve activity of a pool of molecules based on the Tetrahymena group I intron, whereas Joyce and colleagues subsequently isolated variants of the group I ribozyme that performed subtly different tasks, such as cleavage of a DNA substrate (3) or acquisition of a novel dependency on a specific divalent metal cation (4). The ability of directed evolution and ribozyme engineering to evoke secondary activities of group I variants such as cleavage of aminoacyl ester or amide bonds (5–7; reviewed in ref. 8) suggests that RNA has an intrinsic capacity to adapt to novel contexts or substrates, although in these cases the acquisition of new catalytic activity was aided by careful design of the substrate to mimic the natural phosphodiester bond.
In this paper we report the in vitro selection and evolution of a very small ribozyme that catalyzes the ligation of two substrate RNAs but also possesses the unexpected ability to undergo a separate self-cleavage reaction. Substitution of a divalent metal ion in the absence of further sequence evolution triggers the switch from selected ligase to unselected self-cleaving RNA. The facile construction of a minimal RNA ligase ribozyme from random sequences and the simple conversion between two RNA metalloenzymes (9) by a single metal switch imply a surprising plasticity of RNA evolution.
METHODS
Nucleic Acids.
The initial DNA pool was prepared as a 132-base oligonucleotide (16 nucleotides of constant region flanking 100 bases of random sequence) on a Millipore Expedite synthesizer, confirmed for even degeneracy by cycle sequencing, gel purified, and amplified by using large-scale PCR (1) with the 5′ and 3′ primers to extend the constant regions (5′ PCR primer including T7 promotor 5′-TTCTAATACGACTCACTATAGGCTCACACTTGTATAGATCTACT-3′; 3′ constant region PCR primer: 5′-(CUA)4CGGGATCCTAATGAC-3′). Pool RNA for each round was gel-purified and eluted by a crush-and-soak method after T7 transcription [with T7 RNA polymerase either purchased from Epicentre Technologies (Madison, WI) or prepared as described in ref. 11] from phenol-extracted, ethanol-precipitated PCR templates. Double-stranded amplified DNA was also gel-purified after the first round of amplification. Approximately 20 nmol (1.5 mg) of pool RNA was used in the first round, 0.1 nmol was used in the second round, and 20 pmol was used in subsequent rounds.
One of three substrate RNAs was used in each alternating selection round (RNA1, 5′biotin-(cat)4agGUACAAGUGUAGAAAAUCAGUCUUUUUU-3′; RNA2, 5′biotin-(cat)4ctcgagaaUUAUACAAGAAAGAUUACUCUUUUU-3′; RNA3, 5′ggga(cau)4agguaCAAGAAAGAUACAGUCUUUUU-3′; tag sequences are lowercase. RNA1 and RNA2 were constructed on a Millipore Expedite synthesizer [with DNA sequence tags italicized and biotin-dT (Glen Research, Sterling, VA) at the 5′ end] and purified by anion exchange HPLC followed by gel purification. RNA3, used in largest quantity in the first round (1 mg, 90 nmol) was prepared by T7 transcription (11, 12) and gel purified. Other RNA substrates and small ribozymes were either prepared by T7 transcription of oligonucleotide or PCR templates or purchased from Yale University (Keck Oligonucleotide Facility).
In Vitro Selection Strategy.
The selection protocol we followed was similar to the one described in refs. 1 and 10. In the first seven rounds, 1 μM pool RNA and 4-fold excess substrate were incubated overnight at high concentrations of K+ and Mg2+ (400 mM KCl/60 mM MgCl2/30 mM Tris, pH 7.4) at 21–25°C to facilitate binding and catalysis with the substrate usually tethered to a solid support to prevent loss of pool RNA caused by precipitation at elevated Mg2+ concentrations (1). Mg2+ was always added last. Round 1 was performed with the substrate RNA3 tethered to streptavidin agarose via a 5-fold excess of a biotinylated DNA oligonucleotide complementary to the sequence tag, and rounds 2, 4, and 5 were performed with biotinylated substrate directly attached to magnetic beads. Use of a solid support allowed specific retention of covalently ligated ribozymes, and selective PCR with the tag sequence as primer amplified only ligated sequences. Each round achieved >1000-fold enrichment of reacted RNA molecules. We introduced different guide sequences as well as DNA sequence tags on the substrate RNA in alternate rounds of selection to select for a general ligase (instead of one with restricted sequence requirements) and to increase the efficiency of the selection by preventing sequence dependency on the substrate.
In round 1, pool RNA and substrate RNA3 were annealed and reacted on a 7.5-ml streptavidin agarose column (Pierce), and unligated pool RNA was released from the solid support in 25–100 mM NaCl, followed by elution of any reacted (ligated) RNA in 0–5 mM NaCl. Radiolabeled RNA3 was used to monitor the presence of fractions containing covalently ligated RNA. These were pooled and ethanol-precipitated with glycogen as a carrier. Round 2 was performed similarly by using biotinylated RNA1 and 0.25 ml of M280 Streptavidin (Dynal, Great Neck, NY) magnetic beads. RNA collected from round 1 was affinity-purified on a second column (2.5 ml of streptavidin agarose) containing 2 nmol of a 20-base oligonucleotide complementary to the 3′ constant region to remove excess substrate (oligo 20.99 in ref. 1). Retained RNA was reverse-transcribed (SuperScript RNase H−, BRL) directly on the magnetic beads when biotinylated substrates were used.
In the first five rounds, 10–15 cycles of selective PCR (1) with the tag sequence as primer were followed by up to 33 cycles of nested PCR until a product was strongly visible on an ethidium bromide-stained agarose gel. Selective PCR (25–35 cycles) was used in later rounds when a product was readily visible, with only five cycles of nested PCR used to introduce the T7 promotor. Nonspecific RNA binding to the streptavidin-coated beads (Pierce, Dynal) was minimal (<0.01%) in most rounds. Progressively shorter incubation times, lower concentrations of Mg2+, and mutagenic PCR were introduced in later rounds to allow refinement of the catalytic pool.
Beginning with round 6, biotinylated RNA⋅cDNA hybrids were captured onto streptavidin-coated magnetic beads after reverse transcription (because rogue RNA molecules that bind the streptavidin support lose their affinity after reverse transcription and conversion to double-stranded molecules). These RNA⋅DNA hybrids were washed off the solid support while specifically retaining 5′-biotinylated duplexes in TE (10 mM Tris·HCl/1 mM EDTA) buffer. After DNA restriction-fragment and sequence analyses revealed the presence of a single class of closely related molecules in round 7, the addition of mutagenic PCR (7 mM MgCl2/0.2 mM each purine nucleotide/1 mM each pyrimidine nucleotide/0.5 mM MnCl2; see refs. 13 and 14) for mixtures of approximately 30, 60, and 90 doublings (1) with 200,000-fold dilutions between double sets of PCR introduced more variation. In each subsequent round, 30 cycles of nested PCR were performed with mutagenic conditions on 200,000-fold dilutions of DNA recovered after selective PCR. The MgCl2 concentration during ligation was reduced to 10 mM in rounds 8–11 and 5 mM in rounds 12–15. Incubation time was reduced from overnight to 3 hr in round 10, to 30 min in round 11, to 15 min in round 12, and to 2 min (after preincubation of RNA with substrate in the absence of MgCl2) in rounds 13 and 14. All reactions were quenched by adding EDTA. Selective PCR products were cloned into a UDG cloning vector (BRL).
Minimal Base-Pairing Requirements.
One round of selection was performed to determine the pairing requirements for ligation by ligating overnight 1 μM randomized hairpin RNA (Fig. 1d) to 3-fold excess RNA substrate containing the 10-nt sequence 5′-pppGACUCACACU fused to the 22-nt sequence tag from RNA2 in a 0.9-ml reaction volume (30 mM Tris⋅HCl, pH 8.5/0.4 M KCl/100 mM MgCl2). Product ligated to the 10-nt substrate (≈1.2%) was visualized with autoradiography and isolated on a 12% polyacrylamide gel containing 8.3 M urea, reverse transcribed, and amplified by rTth DNA polymerase (Perkin–Elmer) by using the 3′ sequence tag and a 5′ PCR primer containing the T7 promotor fused to the first eight nucleotides of the RNA 5′ end (GGUGUGGU).
Figure 1.

(a) Possible secondary structure of one of the ligase ribozymes selected from round 13, shown ligated to the substrate guide RNA (gRNA). The 5′ and 3′ constant regions of the pool are shaded (and sequences derived from the 5′ constant region are shown in blue throughout Fig. 1). The 3′-U6 tail (underlined and in green) and the 5′ biotin of the guide RNA substrate are marked. (CAT)4, a DNA sequence tag, and other sequences in red are not needed. The core region of the ribozyme is boxed in the center. The ∗ arrow indicates the 3′-terminal base needed for efficient ligation. Dashed lines and arrows indicate sites of allowed breakage for trans-ligation or truncation experiments. U–A pairs in magenta are weakly supported by sequence covariation. Substitutions in black increase activity, and mutations in light gray abolish activity, following a grayscale. The small arrow indicates attack of the 2′-hyrdoxyl of the 3′-U6 tail on the 5′-triphosphate of the G at the end of the RNA. (b) Very small cis-ligation with the same substrate as in a, kobs = 3.4 × 10−4 min−1. (c) Very small trans-ligation with a 10-nt 3′ substrate and a 29-nt ribozyme (boxed), kobs = 5.4 × 10−4 min−1 at 25°C and 30 × 10−4 min−1 at 45°C. (d and e) Two very simple catalytic motifs still ligate with up to 10% (d) and 4% (e) the efficiency of the ligation in b or c, demonstrating remarkable versatility of the small ligase motif. Randomized nucleotides (N) are not intended to be base paired.
RNA Labeling.
RNA molecules were either 32P-labeled at their 3′ termini with 3′-[α-32P]dATP (NEN) and poly(A) polymerase (BRL) or 5′-end labeled either with T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP or [γ-33P]ATP (NEN) or by T7 transcription in the presence of [α-32P]GTP and unlabeled ATP, CTP, and UTP (for RNase T2 digestion). T7 transcripts were treated with phosphatase (calf intestinal alkaline phosphatase, Boehringer Mannheim), phenol extracted, and precipitated before labeling with T4 kinase.
Ligation Reactions.
Reactions were performed in 5-μl volumes (20 μl for time course reactions) with 1 μM template (ribozyme), 1 μM 3′ substrate (for trans-ligations), and trace amounts of 5′-end-labeled substrate in 400 mM KCl, 30 mM buffer, 0.4 units/μl RNasin (Promega) at the indicated pH and with the indicated divalent cation. Tris buffer was used for pH 7.5–9.0 and 2-(N-cyclohexylamino)ethanesulfonic acid (Ches) buffer was used for pH 8.5–10.0 (both were obtained from Sigma). Metal salts [MgCl2 (99.995%), MnCl2 (99.99%), CaCl2 (99.99+%), SrCl2 (99.995%), BaCl2 (99.999%), and CdCl2 (99.99+%)] were purchased from Aldrich and of the highest purity available. Solutions were prepared in diethyl pyrocarbonate-treated H2O (Research Genetics, Huntsville, AL) and filtered through 0.22-μm Millipore filters before use. To avoid oxidation, 1 M stock solutions were stored at −20°C and mixed with buffer before use. All reactions were incubated at 22–25°C for between 0 hours and 7 days. Reactions were stopped by addition of an excess volume of EDTA in formamide loading buffer (95% deionized formamide/10 mM EDTA/0.02% xylene cyanol and bromphenol blue).
RNAse T2 Digestion.
RNase T2 reactions were carried to completion by incubation for 30 min at 37°C in 5 μl containing 50 mM sodium acetate (pH 4.5), 2 mM EDTA, and 10–20 units of RNase T2 (BRL). RNA for this experiment was labeled at the phosphate of the ligation junction by ligating a 10-base substrate RNA containing a 32P label at the α phosphate of the 5′-triphosphate to the 22-nt RNA (Fig. 1c) and gel-purifying the ligated product. Digestion products were analyzed on a 20% acrylamide, 8.3 M urea gel.
Manganese-Dependent, Site-Specific Cleavage.
RNA5 (15) was a gift from S. Kazakov (Somagenizo, Palo Alto, CA). Reaction volumes (10 μl) containing 10 mM MnCl2, 100 mM NaCl, and 50 mM Tris⋅acetate (pH 7.5) were incubated at 45°C for 1 hr. Reactions were stopped by addition of an equal volume of formamide loading buffer, incubated for 15 min at 37°C (to avoid nonspecific hydrolysis of RNA), and loaded directly onto a 20% acrylamide, 8.3 M urea gel.
RESULTS AND DISCUSSION
Selection for a General Ligase.
Inspired by the biological process of RNA editing, which uses multiple cycles of cleavage, U addition, and ligation to rewrite up to 90% of the coding-sequence information in kinetoplastid mitochondrial transcripts (16), we designed a substrate RNA and a pool 5′ constant region that contained a truncated NADH dehydrogenase subunit 7 (ND7) guide RNA from Leishmania tarentolae and 18 nucleotides of the 5′ editing domain in the ND7 mRNA, respectively (17). A side activity of RNA editing that has been observed both in vivo (18) and in vitro (17) is the covalent attachment of the guide RNA to the mRNA molecule. We sought to test whether this activity is an intrinsic property of the RNA molecules themselves. The one unifying feature among all guide RNA molecules is the presence of a 3′ oligo(U) tail (18); hence, we initially constructed all 5′ substrate molecules with a short (5–6 nt) oligo(U) tail. From a pool of 1.6 × 1015 unique sequences containing 100 nucleotides of random sequence flanked by constant regions, a single class of RNA molecules emerged after six cycles of selection for the ability to ligate each of three tagged substrate RNAs to their 5′ end. Covalent attachment to the substrate marks the pool RNA molecules with either biotin or a specific sequence tag (1), and the strategy of recovering biotin-tagged ribozymes as RNA⋅cDNA hybrids avoided recovery of single-stranded RNAs that bind only streptavidin.
All ribozymes that survived the selection contained a sequence within the random region that can anneal to the 5′ constant region of the pool molecules as well as to multiple substrate RNAs, forming a long imperfect stem, which positions the 3′ terminus of the oligo(U) tail of the substrate RNA near the 5′-triphosphate of the pool RNA (Fig. 1a). The 5′-end ligation catalyzed by this RNA involves nucleophilic attack by the small substrate RNA on the α phosphate of a 5′-triphosphate (1). We found that this ligation requires the presence of a 5′-triphosphate, or activation of the α phosphate by pyrophosphate, because activity is lost with phosphatase-treated or kinase-treated RNA (not shown) that contains either a 5′-hydroxyl or 5′-monophosphate, respectively.
Because of the difficulty of sampling such a large sequence space [fewer than 1016 molecules of a possible 4100 >1060], the ribozymes selected initially were expected to be suboptimal representatives of a class of catalytically active molecules (1). Seven more rounds of in vitro selection were performed by using heavy mutagenic PCR on the pool of ribozymes recovered after the seventh round of selection. This improved the reaction by reducing the required magnesium concentration and reaction time.
Alignment of 51 clones revealed a sequence consensus for this ribozyme with only a conserved 29-nt core region (Fig. 2). By assaying different clones, we verified that the extent of base pairing between ribozyme and substrate in the conserved region contributes to the rate of ligation. In addition, most sequence covariation allowing G·U pairs preserves pairing to the substrate in the core region.
Figure 2.

Alignment of 12 representative functional ribozyme clones from a total of 51 such clones. A dash indicates identity with the top sequence. A . indicates a deletion. The most conserved region is boxed. The constant regions are shaded in the reference sequence.
Remarkably, this ligase ribozyme has the fewest sequence requirements of any selected ribozyme, with only two nucleotides directly implicated in a nontemplate role. The only known smaller ribozyme promotes cleavage rather than ligation (15) and is derived from an RNA hairpin found in the Tetrahymena group I intron (19). The ligation catalyzed by this ribozyme thus differs from the ligations catalyzed by other RNA ligase ribozymes (10) by requiring a G–G juxtaposition at the site of ligation and very few selected nucleotides for activity (Fig. 1).
We constructed a minimal version of this RNA-ligase ribozyme. Deletion of sequences 3′ of the boxed conserved region (Fig. 1a) led to a 2-fold increase in activity; however, deletion of some sequences 5′ of the boxed region produced a slight decrease in activity, indicating that some of these bases may contribute modestly to catalysis. The smaller ribozyme contains only substrate nucleotides and a central 27-nt core derived from the originally selected ribozyme. This represents the smallest such ribozyme isolated by in vitro selection, with only two potentially unpaired nucleotides required for catalysis (Fig. 1b).
Ligation in Trans.
The ribozyme also functions efficiently in trans (Fig. 1c) but without turnover because of the slow dissociation of the enzyme–substrate complex (consisting of three annealed strands that together form the catalytic RNA system). The pseudo-first-order rate constant kobs for this reaction was 2.7 × 10−4 min−1 for the time course shown in Fig. 3. The 29-nt “ribozyme” is essentially as active as one containing the full 5′ end. To our surprise, we discovered that a general ligase ribozyme stripped of its template either upstream or downstream of the ligation site still ligates several sequences, albeit with approximately 4% the original efficiency (Fig. 1 d and e). An alignment of 36 successful ligase clones derived from a single round of selection (as in Fig. 1d) exhibited no marked sequence consensus, suggesting that the catalytic structures are versatile. In fact, the number of Watson–Crick or G·U base pairs found in the randomized region of these sequences was not significantly greater than expected for any pair of random hexamers (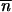 observed = 2–3 bp;
observed = 2–3 bp; 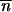 = 6(
= 6(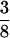 ) = 2.25 expected at random).
) = 2.25 expected at random).
Figure 3.

(Left) Time course of representative trans-ligation shown in Fig. 1c with 10-nt 3′ substrate; Lower band, labeled 22-nt guide RNA band; upper band, 32-nt ligated product band. kobs = 2.7 × 10−4 min−1. (Right) Time course of randomized cis-ligation shown in Fig. 1e. M, marker lane; lower band, labeled 41-nt partially randomized RNA; upper band, ligated 63-nt product.
Creation of 2′–5′ Linkages.
The regioselectivity of bond formation is exclusively 2′,5′-phosphodiester linkages as assayed by resistance of 2′,5′ linkages to ribonuclease T2 (Fig. 4). This is consistent with the absence of extended base pairing at the ligation junction, as a Watson–Crick duplex favors 3′,5′ linkages (20) but considerably destabilizes 2′,5′ linkages (20–22). Although nonbiological in extant systems, 2′,5′ linkages are favored in nonenzymatic oligoribonucleotide synthesis because of the increased nucleophilicity of the 2′-hydroxyl toward activated phosphate esters (23). However, recent experiments demonstrate that both 2′,5′ linkages and mixed linkages are active in template-directed oligomerization of mononucleotides (24) and oligonucleotides (25–26), and this has strengthened their possible role in primitive biochemistry.
Figure 4.

Ribonuclease T2 digestion to determine regiospecificity of bond formation. Lane 2 is the RNase T2 digestion product of the ligation in Fig. 1c (see Methods). Lane 1 is the RNase T2 digestion product of a ligated RNA of identical sequence, but with a labeled 3′,5′ linkage generated by T4 DNA ligase and a complementary DNA splint (42). Digestion products were analyzed on a 20% denaturing acrylamide gel. Lanes 3 and 4 are a time course of alkali hydrolysis at 5 and 10 min, respectively, of the ligated product digested in lane 1.
We examined the dependency of the ligation on time, Mg2+ concentration, and pH (Fig. 5). The reaction exhibits approximately linear dependence on Mg2+ concentration below 50 mM and a saturable Mg2+ binding site with half-maximal activity above 50 mM over a pH range of 7.0 to 9.0 (data not shown; ref. 27). The logarithm of kobs increases linearly with a slope of approximately 1 as a function of pH, indicating a single critical deprotonation step—probably at the 2′ hydroxyl. The pH plateau above pH 9.0 is consistent with an attacking 2′ hydroxyl (Fig. 5; pH >9.5 leads to degradation, deprotonation, and helix disruption and so could not be validated). Deletion analysis mapped the 3′ and 5′ terminal positions required for activity to a small conserved core, which provides mostly a template. Only two nucleotides are directly implicated in a nontemplate role: the G opposite the ligation site and a single bulged pyrimidine (Fig. 1).
Figure 5.

Biochemistry of ligation reaction. (Left) pH dependence and (Center and Right) effect of replacing Mg2+ with a series of divalent cations using ribozyme/substrate complex in Fig. 1c. (Center) kobs as a function of Mg2+ (⋄) and Mn2+ (□) at pH 7.0. (Right) kobs as a function of Mg2+ (□), Ca2+ (⋄), Cd2+ (○), Sr2+ (▵), and Ba2+ (crossed squares) at pH 9.0.
Mutational Analysis and Rate Enhancement.
Many single point mutations (summarized in Fig. 1a) abolish activity below our threshold of detection. For example, deletion of the single bulged U or introduction of any purine base opposite (and potentially paired with) this U results in complete loss of activity, as does mutation of the G opposite the cleavage site to any other base or mutation of the adjacent U·G base pair to a C-G base pair. On the other hand, conversion of this U·G pair to a U-A (Fig. 1a) leads to a 4- to 10-fold increase in both cis- and trans-ligation (kobs approx. 6 × 10−4⋅min−1).
We estimated the rate enhancement of our ribozyme-catalyzed ligation by comparing it against single and double point mutants that lack the bulged U and/or contain a C-G pair instead of a G–G juxtaposition at the ligation site. This gave an upper boundary of kobs for the background ligation of 3 × 10−8 min−1, a rate enhancement of the catalyzed reaction greater than 20,000-fold. Our ability to measure the background reaction containing the C-G pair may be hampered by the preferential hydrolysis of 2′,5′ linkages in a paired duplex (22); however, we estimated the same upper boundary for the single mutant lacking only the bulged U, which preserves the rest of the ligation site. This reaction also is 3,000-fold faster than a template-dependent uncatalyzed ligation with a similar 3′-terminal U-A base pair (kobs 8.5 × 10−8 min−1; ref. 20).
Ligation of Hexauridylate.
Because the guide RNA substrate in the original experiments contains an oligo(U) sequence at its 3′ end, we tested a 6-nt substrate, 5′,3′-(UUUUUU). Even this small RNA is a substrate for both cis- and trans-ligation (Fig. 6), albeit with 0.5% the efficiency of the 22-nt substrate shown in Fig. 1 c and e. As this sequence is small enough to be synthesized by mineral-catalyzed RNA condensation (28), this result suggests the possibility that we have uncovered an RNA ligation reaction with general prebiotic utility.
Figure 6.

Ligation of 5′,3′-(UUUUUU) (lower band) to a 10-nt substrate. Upper band is the 16-nt product. Lane 1 was incubated without substrate and lane M is a marker; lane 2, 3-day incubation; lane 3, 6-day incubation.
Manganese Dependency.
Surprisingly, the catalytic core built from random sequences actually contains the very small manganese-dependent cleavage motif formed during autocyclization of the Tetrahymena group I intron (refs. 15 and 19; Fig. 7). The trinucleotide UUU in a complex with GAAA (the smallest known catalytic RNA system) acts as a template catalyst (15, 29) to promote cleavage between the G and the A (15).
Figure 7.

Mn2+-dependent RNA cleavage. The complex of UUU and UGAAA undergoes site specific Mn-dependent cleavage at the arrowhead (15). The U is not required in the substrate but was present in the original sequence (19) and, surprisingly, the entire 8-nt motif shown here appears in our selected ligase, just opposite the ligation site (Figs. 1 a–c and e). The lines represent optional RNA sequences, and the circle represents at least one binding site for a divalent ion—Mn2+ or Cd2+ to promote cleavage—and presumably Mg2+ or other metals to promote ligation. Lanes M and 1 contain standards; lane 2 is unreacted 3′-end labeled 22-nt positive control synthetic RNA5 (15), and lane 3 is RNA5 digested to an 18-nt product. Lanes 4–6 represent Mn2+ cleavage of the 29-nt ribozyme strand shown in Fig. 1c to a 9-nt shorter product in the presence of Mn2+ and 0.05, 0.5, and 5 ng/μl poly(U).
Further experiments revealed that we had in fact selected a dual-catalytic RNA from random sequences. This RNA promotes both cleavage at one site (Fig. 7) and ligation at another (Fig. 1), with possibly two conformations surrounding a divalent-metal ion binding pocket (or pockets). We therefore tested the metal-ion dependence of the original ligation: at pH 7.0, the ligation proceeds ≈10-fold faster with Mn2+ than with Mg2+ (Fig. 5), but at higher pH the required concentration of Mn2+ leads to RNA degradation.
We subsequently confirmed that the selected ligase also undergoes self-cleavage in the presence of Mn2+ (Fig. 7) with kobs = 0.01 min−1 for self-cleavage of RNA5 (15), compared with 0.0036 min−1 for cleavage of GAAApCp (30) and kobs = 5–8 × 10−4 min−1 for cleavage of the 29-nt core ribozyme (template strand in Fig. 1c) in the presence of either 5–50 ng/μl poly(U), 0.5 nM–5 μM 5′,3′-UUUUUU, or 0.5 μM 22-nt substrate RNA shown in Figs. 1 c and e as template catalyst. In addition, the ligation shown in Fig. 1c proceeds remarkably well in optimal cleavage buffer conditions, with kobs = 0.003 min−1 at 45°C.
Other Divalent Metal Ions.
Cd2+ also supports G↓AAA cleavage (15) but supports ligation only weakly. Ca2+, Sr2+, and Ba2+ also catalyze the ligation. Overall the ligation rates (Mn2+ > Mg2+ > Ca2+ > Sr2+ = Ba2+) vary inversely with the pKa of their bound water molecules and follow the same order as hammerhead cleavage and nonenzymatic ligation (20), implicating a metal hydroxide or directly coordinated metal ion in the catalytic mechanism.
The Roles of RNA and the Metal.
The simplicity of the ligation depicted in Fig. 1 d and e suggests that this motif can be generalized even further. The GNRA tetraloops at the ends may act as terminal clamps, thus reducing the need for base pairing to juxtapose substrates along the ribozyme template. The role of divalent metal ions is particularly relevant, as they are generally thought to have been important prebiotic catalysts (31). Evoking the notion of a “Cheshire catalyst” (32), in which an RNA scaffold mainly positions a catalytic metal ion (9), we suggest that the core nucleotides—most notably the bulged pyrimidine (Fig. 1)—serve two roles: (i) to bring together the 2′ and 5′ termini in a configuration that resembles the transition state for ligation (33) and (ii) to properly position the metal ion(s) in a favorable geometry, whereas the conserved flanking nucleotides (or tetraloop) provide intrinsic binding energy that leads to the closed state required for catalysis (34).
A Case for RNA Preadaptation.
The self-cleaving GAAA/UUU motif arose in this case as an unintended consequence of selection for ligation at an independent site. In other words, it appears as a “spandrel” (35–36), an offshoot or structural byproduct of the design of an RNA ligase ribozyme from random sequences. A spandrel has been defined as “any geometric configuration of space inevitably left over as a consequence of other architectural decisions” (36). Because the ability to cleave within G↓AAA clearly did not emerge as an adaptation itself during the selection experiment—as it was neither selected for nor feasible in the magnesium-containing buffer—this additional activity presumably results from the need to construct a binding site for one or more divalent metal ions. In vitro evolution therefore provides an unambiguous example of preadaptation, in the sense that the product of evolution was ultimately suited for a catalytic function other than the one for which it was selected. It could thus be co-opted to specialize in its new role, such as cleavage, which would make this feature an exaptation (37). In a separate selection experiment, Faulhammer and Famulok (38) unveiled a DNA enzyme that was preadapted for calcium binding. The pool molecules had never been exposed to calcium during the selection; however, the optimal solution for magnesium-dependent cleavage of RNA coincidentally produced a good solution to the problem of calcium-dependent cleavage as well, as though the geometry of binding the metal hydroxide “anticipated” the best fit for binding calcium.
Conclusions.
The results presented here suggest that bifunctional catalytic RNA motifs can arise under fairly simple conditions and that multiple catalytic structures, including ligases, can evolve from basic preexisting building blocks. By extension, we predict that the combinatorial shuffling of catalytic modules (such as metal binding sites) into random sequence pools will greatly increase the density and representation of nucleic acid catalysts and that this will lead to a tremendous acceleration of in vitro evolution. Along similar lines, Burke and Willis (39) recently introduced recombination between RNA libraries to build aptamers (RNA ligands) that can bind two specific targets. Our finding that RNA structures as simple as a few unpaired nucleotides in a short helix may possess two catalytic activities suggests that similar metalloribozymes (i) arise surprisingly easily and would undoubtedly have been present in prebiotic evolution (as well as at high frequency in random pools) and (ii) could have contributed to the generation of RNA diversity by breaking apart and joining new sequence combinations.
Processes such as kinetoplastid RNA editing that may be primitive (40) require repeated cycles of both cleavage and ligation (41). These experiments demonstrate that a set of RNA molecules based initially on a Leishmania tarentolae guide RNA–mRNA combination are capable of performing both reactions. The full-length ND7 guide RNA is even a substrate for the ligase ribozyme (kobs = 1.7 × 10−5 min−1; however, kobs increases by a factor of 2 by extending the “anchor” region of pairing between ribozyme strand and guide RNA). The ribozyme-catalyzed ligation employs a different leaving group, however, than does kinetoplastid editing. These observations do hint at another possible role of the poly(U) tails on kinetoplastid guide RNAs: they could assist in the cleavage at frequent G↓AAA editing sites. This reaction might have played a more significant role early in the history of RNA editing.
Cleavage of RNA at specific sites has been suggested as a mechanism for generating abrupt translation termination signals (15); however, the presence of two metal-dependent activities in a single RNA molecule confers the ability to modulate biochemical activity in response to environmental cues. A single metal ion switch thus adds to the list of possible mechanisms that would have been available for control of gene expression in an RNA world.
Acknowledgments
We thank Sergei Kazakov for advice, Anne Torjussen, Caroline Fichtenberg, Monika Pfunder, and David Kutzler for assistance, Rob Knight, Drew Ronneberg, and two anonymous reviewers for comments, and Jack Szostak and members of his laboratory for suggestions and RNA oligonucleotides. This research was supported by National Science Foundation Grants MCB-9520253 and MCB-9604377 and a Burroughs Wellcome Fund New Investigator Award in Molecular Parasitology to L.F.L.
References
- 1.Bartel D P, Szostak J W. Science. 1993;261:1411–1418. doi: 10.1126/science.7690155. [DOI] [PubMed] [Google Scholar]
- 2.Green R, Ellington A D, Szostak J W. Nature (London) 1990;347:406–408. doi: 10.1038/347406a0. [DOI] [PubMed] [Google Scholar]
- 3.Beaudry A A, Joyce G F. Science. 1992;257:635–641. doi: 10.1126/science.1496376. [DOI] [PubMed] [Google Scholar]
- 4.Lehman N, Joyce J F. Nature (London) 1993;361:182–185. doi: 10.1038/361182a0. [DOI] [PubMed] [Google Scholar]
- 5.Piccirilli J A, McConnell T S, Zaug A J, Noller H F, Cech T R. Science. 1992;256:1420–1424. doi: 10.1126/science.1604316. [DOI] [PubMed] [Google Scholar]
- 6.Dai X, De Mesmaeker A, Joyce G F. Science. 1995;267:236–240. doi: 10.1126/science.7809628. [DOI] [PubMed] [Google Scholar]
- 7.Joyce G F, Dai X, De Mesmaeker A. Science. 1996;272:18–19. [PubMed] [Google Scholar]
- 8.Landweber L F, Simon P J, Wagner T A. Bioscience. 1998;48:94–103. [Google Scholar]
- 9.Pyle A M. Science. 1993;261:709–714. doi: 10.1126/science.7688142. [DOI] [PubMed] [Google Scholar]
- 10.Ekland E H, Szostak J W, Bartel D P. Science. 1995;269:364–370. doi: 10.1126/science.7618102. [DOI] [PubMed] [Google Scholar]
- 11.Zawadzki V, Gross H J. Nucleic Acids Res. 1991;19:1948. doi: 10.1093/nar/19.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milligan J F, Uhlenbeck O C. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 13.Caldwell R C, Joyce G F. PCR Meth Applic. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- 14.Fromant M, Blanquet S, Plateau P. Anal Biochem. 1995;224:347–353. doi: 10.1006/abio.1995.1050. [DOI] [PubMed] [Google Scholar]
- 15.Kazakov S, Altman S. Proc Natl Acad Sci USA. 1992;89:7939–7943. doi: 10.1073/pnas.89.17.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landweber L F, Gilbert W. Nature (London) 1993;363:179–182. doi: 10.1038/363179a0. [DOI] [PubMed] [Google Scholar]
- 17.Blum B, Simpson L. Proc Natl Acad Sci USA. 1992;89:11944–11948. doi: 10.1073/pnas.89.24.11944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blum B, Sturm N R, Simpson A M, Simpson L. Cell. 1991;65:543–550. doi: 10.1016/0092-8674(91)90087-f. [DOI] [PubMed] [Google Scholar]
- 19.Dange V, Van Atta R B, Hecht S M. Science. 1990;248:585–588. doi: 10.1126/science.2185542. [DOI] [PubMed] [Google Scholar]
- 20.Rohatgi R, Bartel D P, Szostak J W. J Am Chem Soc. 1996;118:3340–3344. doi: 10.1021/ja9537134. [DOI] [PubMed] [Google Scholar]
- 21.Usher D A. Nat New Biol. 1972;235:207–208. doi: 10.1038/newbio235207a0. [DOI] [PubMed] [Google Scholar]
- 22.Usher D A, McHale A H. Proc Natl Acad Sci USA. 1976;73:1149–1153. doi: 10.1073/pnas.73.4.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohrmann R, Orgel L E. Tetrahedron. 1978;34:853–855. [Google Scholar]
- 24.Prakash T P, Roberts C, Switzer C. Angew Chem Int Ed Engl. 1997;36:1522–1523. [Google Scholar]
- 25.Ertem G, Ferris J P. Nature (London) 1996;379:238–240. doi: 10.1038/379238a0. [DOI] [PubMed] [Google Scholar]
- 26.Sawai H, Totsuka S, Yamamoto K, Ozaki H. Nucleic Acids Res. 1998;26:2995–3000. doi: 10.1093/nar/26.12.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rohatgi R, Bartel D P, Szostak J W. J Am Chem Soc. 1996;118:3332–3339. doi: 10.1021/ja953712b. [DOI] [PubMed] [Google Scholar]
- 28.Ding P, Kawamura K, Ferris J P. Origins Life Evol Biosphere. 1996;26:151–171. doi: 10.1007/BF01809853. [DOI] [PubMed] [Google Scholar]
- 29.Orgel L E. J Theor Biol. 1986;123:127–149. doi: 10.1016/s0022-5193(86)80149-7. [DOI] [PubMed] [Google Scholar]
- 30.Bombard S, Kozelka J, Favre A, Chottard J-C. Eur J Biochem. 1998;252:25–35. doi: 10.1046/j.1432-1327.1998.2520025.x. [DOI] [PubMed] [Google Scholar]
- 31.Joyce G F, Orgel L E. In: The RNA World. Gesteland R F, Atkins J F, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. pp. 1–25. [Google Scholar]
- 32.Yarus M. FASEB J. 1993;7:31–39. doi: 10.1096/fasebj.7.1.8422972. [DOI] [PubMed] [Google Scholar]
- 33.Harada K, Orgel L E. Proc Natl Acad Sci USA. 1993;90:1576–1579. doi: 10.1073/pnas.90.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hertel K J, Peracchi A, Uhlenbeck O C, Herschlag D. Proc Natl Acad Sci USA. 1997;94:8497–8502. doi: 10.1073/pnas.94.16.8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gould S J, Lewontin R C. Proc R Soc London B. 1979;205:581–598. doi: 10.1098/rspb.1979.0086. [DOI] [PubMed] [Google Scholar]
- 36.Gould S J. Proc Natl Acad Sci USA. 1997;94:10750–10755. doi: 10.1073/pnas.94.20.10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gould S J, Vrba E. Paleobiology. 1982;8:4–15. [Google Scholar]
- 38.Faulhammer D, Famulok M. Angew Chem Int Ed Engl. 1996;35:2837–2841. [Google Scholar]
- 39.Burke D H, Willis J H. RNA. 1998;4:1165–1175. doi: 10.1017/s1355838298980542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landweber L F, Gilbert W. Proc Natl Acad Sci USA. 1994;91:918–921. doi: 10.1073/pnas.91.3.918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kable M L, Seiwert S D, Heidmann S, Stuart K. Science. 1996;273:1189–1195. doi: 10.1126/science.273.5279.1189. [DOI] [PubMed] [Google Scholar]
- 42.Moore M J, Sharp P A. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]