

Abstract
Different members of the Rel/NF-κB family may play different roles in immunity and inflammation. We report here that c-Rel–deficient mice are resistant to autoimmune encephalomyelitis and are defective in Th1, but not Th2 responses. The Th1 deficiency appears to be caused by selective blockade of IL-12 production by c-Rel–deficient antigen-presenting cells, as well as by a complete abrogation of IFN-γ expression in c-Rel–deficient T cells. Interestingly, c-Rel deficiency does not affect T-bet expression, suggesting that c-Rel may act downstream of T-bet during Th1 cell differentiation. Thus, unlike NF-κB1, which selectively regulates Th2 cell differentiation, c-Rel is essential for Th1 cell differentiation and Th1 cell–mediated autoimmune inflammation.
Introduction
Although it is well recognized that the Rel/NF-κB family of transcription factors is crucial for the development and function of the immune system, the precise roles of each Rel/NF-κB member in various immune cells are not well understood. In mammals, there are at least five Rel/NF-κB proteins: c-Rel, RelA (p65), RelB, NF-κB1 (p50/p105), and NF-κB2 (p52/p100) (1–4). Although all these proteins share a highly conserved 300–amino acid Rel homology domain at the N-terminal (which encompasses sequences required for DNA binding, protein dimerization, and nuclear localization), the C-terminal is less conserved, with only that of c-Rel, RelA, and RelB containing transcriptional transactivation domains (5). Additionally, although most members of the Rel/NF-κB family are constitutively expressed in many cell types, including lymphocytes, monocytes/macrophages, granulocytes, neurons, and glia, preferential expression in certain cell types has been reported (1–3, 6–12). Furthermore, in most cell types, the Rel/NF-κB proteins exist as inactive homo- or heterodimeric molecules in association with the inhibitory protein called IκB. There are at least five IκBs, which all act by masking the nuclear localization signal of Rel/NF-κB, preventing their nuclear translocation. A wide variety of stimuli, including cytokines, antigens, stress factors, and viral and bacterial products can activate Rel/NF-κB (1–4, 13). Activation of Rel/NF-κB involves phosphorylation and proteolytic degradation of the inhibitory protein IκB by specific IκB kinases. The free Rel/NF-κB in turn enters the nucleus and binds to the κB sites of gene promoters/enhancers. Many immune-related genes contain putative κB binding sites in their promoter regions, and may therefore be activated by Rel/NF-κB (1–4, 14–17). Thus the roles of individual Rel/NF-κB proteins can be regulated at many different levels.
Recent studies from several laboratories suggest that unique functions of individual Rel/NF-κB proteins may be dissected using gene knockout techniques. c-Rel knockout mice develop normally and acquire a structurally normal immune system (18–20). However, T cells derived from these mice produce reduced levels of IL-2, IL-3, IFN-γ, and GM-CSF, whereas B cells from these mice are more susceptible to apoptotic stimuli (19–21). Similarly, mice deficient in NF-κB1 p50/p105 do not suffer from any developmental defects but are more susceptible to intracellular and extracellular gram-positive bacterial infections, and are partially compromised in their B cell responses to LPS (3). Surprisingly, they are resistant to viral and gram-negative bacterial infections (3). In contrast, mice deficient in RelA die in utero, presumably due to enhanced hepatocyte apoptosis (2, 22). RelB-deficient mice develop normally, but suffer from severe disorders ranging from splenomegaly to chronic microbial infections (19, 23). Similarly, NF-κB2–deficient mice also suffer from severe immune disorders. Both their spleens and lymph nodes are bereft of B lymphocytes, undermining their capacity to form germinal centers (24, 25). These observations strongly suggest that members of the Rel/NF-κB family perform nonoverlapping functions, and that a loss-of-function mutation of a Rel/NF-κB gene may not be fully compensated for by other genes of this family.
Experimental autoimmune encephalomyelitis (EAE) is an inflammatory disease of the CNS that has long been used as an animal model for human multiple sclerosis. Development of EAE requires coordinated expression of a number of genes involved in the activation, differentiation, and effector functions of inflammatory cells. To investigate the roles of Rel/NF-κB in CNS inflammation, we studied EAE in mice deficient in NF-κB1 and c-Rel. We found that mice deficient in NF-κB1 are partially resistant to the development of EAE and are unable to support Th2 cell differentiation (26). This is consistent with a recent report by Das et al. that Gata3-mediated Th2 cell differentiation is blocked in NF-κB1–deficient mice, and that NF-κB1–deficient T cells are unable to differentiate into a Th2 lineage even under Th2-inducing conditions (27). We report here that c-Rel–deficient mice are completely defective in their Th1 responses, whereas their Th2 responses are enhanced. We will present evidence that c-Rel selectively regulates Th1 cell differentiation through IL-12 and IFN-γ, but not T-bet. T-bet is a newly described transcription factor specific for Th1 cells (28). We will then propose a new paradigm of Th cell differentiation in the context of Rel/NF-κB regulation of autoimmune inflammation.
Methods
Mice.
C57BL/6 mice that carry c-Rel gene mutation were generated by inserting the neor cassette into the fifth exon of the c-Rel gene as described previously (18). Normal C57BL/6, B6;129, and NF-κB1–deficient (NF-κB1–/–) B6;129 mice were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA). All mice were housed in the University of Pennsylvania Animal Care Facilities under pathogen-free conditions.
Bone marrow chimeric mice were generated by either irradiating c-Rel+/+ or c-Rel–/– C57BL/6 mice with two doses of 5 Gy spaced 3 hours apart, followed by intravenous injection of 107 bone marrow cells from CD45.1 congenic C57BL/6 mice; or by transferring c-Rel+/+ or c-Rel–/– bone marrow into irradiated CD45.1 congenic C57BL/6 mice. Repopulation of the immune system was monitored by flow cytometric analysis of the blood using anti–mouse CD45.1 Ab conjugated to phycoerythrin and anti–mouse CD45.2 Ab conjugated to FITC (BD Pharmingen, San Diego, California, USA). In the chimeric mice, approximately 90% of the T cells and more than 95% of the B cells and myeloid cells were derived from donor bone marrow. Mice were immunized 8–9 weeks after bone marrow transfer as described below.
Induction and clinical evaluation of EAE.
For induction of EAE, mice received a subcutaneous injection in the flank of 300 μg myelin oligodendrocyte glycoprotein (MOG) 38-50 peptide in 0.1 ml PBS emulsified in an equal volume of CFA containing 4 mg/ml of Mycobacterium tuberculosis H37RA (Difco Laboratories, St. Louis, Missouri, USA), and an intravenous injection of 200 ng pertussis toxin in 0.1 ml PBS. A second injection of pertussis toxin (200 ng per mouse) was given 48 hours later. Mice were examined daily for signs of EAE and scored as follows: 0, no disease; 1, tail paralysis; 2, hind limb weakness; 3, hind limb paralysis; 4, hind limb plus forelimb paralysis; 5, moribund or dead.
Antigens, Ab’s, recombinant cytokines, and ELISA.
Mouse MOG38-50 peptide was synthesized using Fmoc solid-phase methods, and purified by HPLC by Research Genetics (Huntsville, Alabama, USA). Pertussis toxin was purchased from List Biological Laboratories Inc. (Campbell, California, USA). The following reagents were purchased from BD Pharmingen: purified rat anti–mouse IL-2, IL-4, IL-10, IL-12 p40, and IFN-γ mAb’s; recombinant mouse IL-2, IL-4, IL-10, IL-12, and IFN-γ. Quantitative ELISA for IL-2, IL-4, IL-10, IL-12 p40, and IFN-γ was performed using paired mAb’s specific for corresponding cytokines per manufacturer recommendations.
Isolation of T cells, antigen-presenting cells, astrocytes, and microglia.
CD4+ T cells were isolated from spleen of naive C57BL/6 mice using the autoMACS magnetic cell sorter (Miltenyi Biotec, Auburn, California, USA) following sequential incubation of splenocytes with rat anti–mouse CD4 mAb (clone GK1-5; American Type Culture Collection, Rockville, Maryland, USA) and goat anti-rat IgG magnetic beads. More than 95% of the cells isolated in this manner are CD4+ as determined by flow cytometry. Splenic antigen-presenting cells (APCs) were prepared from the same cell suspension by depleting lymphocytes using mAb’s against mouse CD4, CD8 (clone 2.43; American Type Culture Collection), and CD19 (clone 1D3; BD Pharmingen) with the autoMACS cell sorter.
For IL-12 secretion experiments, a collagenase-digested splenocyte suspension was first incubated for 2 hours at 37°C. Cells in the suspension were discarded and the culture was allowed to continue for another 16 hours. Only dendritic cell–enriched (DC-enriched) APCs in the suspension were harvested and used for the experiment. To prepare APCs from bone marrow, femurs and tibiae were surgically removed and the marrow was flushed with complete RPMI medium. After removing erythrocytes, bone marrow cells were cultured at 106/ml in complete RPMI medium containing 3 ng/ml of murine GM-CSF and 5.5 ng/ml of murine IL-4 (R&D Systems Inc., Minneapolis, Minnesota, USA). Five days later, cells in suspension were harvested, washed, and used for IL-12 experiments. Microglia and astrocytes were prepared from newborn mice as described by Suzumura et al. with minor modifica-tions (29). Briefly, mixed glial cells were first cultured in complete DMEM at 106 cells/ml. After 8–10 days, microglia growing on the surface of adherent astrocytes were harvested by shaking for 1.5–2 hours at 260 rpm. The remaining cell monolayers were shaken at 100 rpm for another 24 hours to remove contaminating oligodendrocytes. Astrocytes were then detached and collected using 0.25% trypsin-EDTA.
Semiquantitative PCR analysis of p19, p35, and p40 mRNA in DCs.
DC-enriched APCs were cultured with or without 1–5 μg/ml of LPS (Sigma-Aldrich, St. Louis, Missouri, USA) for 5 hours. Total RNA was isolated using Trizol (Invitrogen Corporation, Carlsbad, California, USA), primed with random hexamers, and reverse transcribed using Moloney murine leukemia virus reverse transcriptase. Semiquantitative PCR of IL-12 p40, IL-12 p35, IL-23 p19, and β-actin were performed using the following oligonucleotides: IL-12 p40, 5′-AAGTTCTCTCCTCTTCCCTGTCGC-3′ and 5′-TCTTGTGGAGCAGCAGATGTGAG-3′ (which result in a 423-bp product); IL-12 p35, 5′-CAATCACGCTACCTCCTCTTTTTG-3′ and 5′-CTCCCTCTTGTTGTGGAGAAGT-C-3′ (which result in a 308-bp product); IL-23 p19, 5′-ACCCACAAGGACTCAAGGACAAC-3′ and 5′-TGCCCTTCACGCAAAACAAAAC-3′ (which result in a 476-bp product); β-actin, 5′-GTGGGCCGCTCTAGGCA-CCAA-3′ and 5′-CTCTTTGATGTCACGCACGATTTC-3′ (which result in a 540-bp product). PCR products were analyzed by agarose gel electrophoresis.
Western blot.
Nuclear lysates of T cells were fractionated by 10% PAGE and transferred onto a nitrocellulose membrane. After blocking with 5% nonfat milk, 10 mM Tris, 100 mM NaCl, and 0.1% Tween 20, the membrane was incubated sequentially with polyclonal goat anti–T-bet Ab’s and horseradish peroxidase–labeled anti-goat IgG (Santa Cruz Biotechnology Inc., Santa Cruz, California, USA). Color was developed using SuperSignal chemiluminescent substrates (Pierce Chemical Co., Rockford, Illinois, USA).
Statistical analysis.
Disease severity, day of onset, and cytokine concentrations were analyzed by ANOVA. Disease scores were analyzed by Mann-Whitney test.
Results
c-Rel–deficient mice are resistant to EAE.
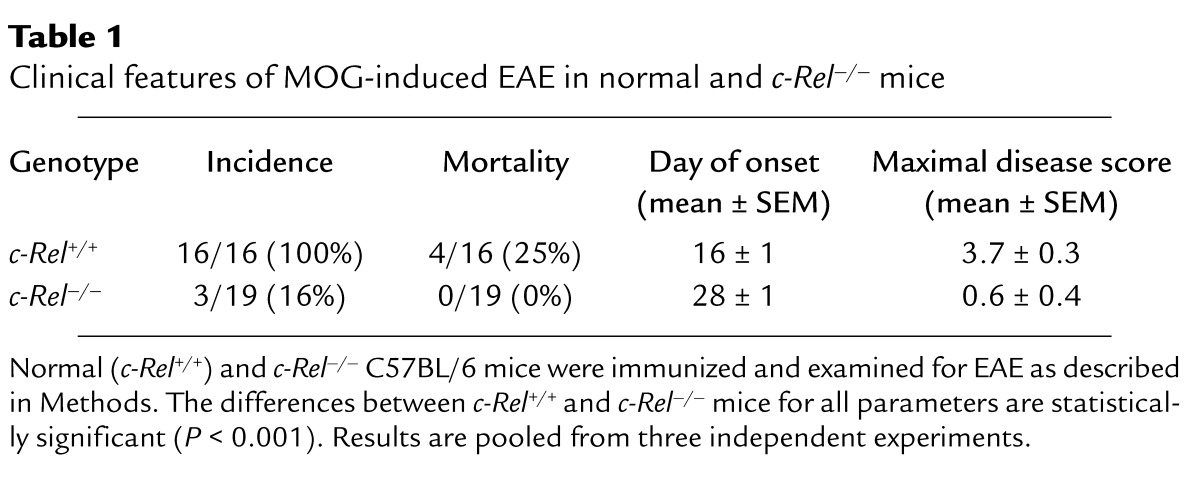
To study the roles of c-Rel in the development of EAE, we immunized normal and c-Rel–deficient mice (18) with MOG38-50 peptide and monitored the disease by both physical examination and histochemistry. Figure 1a illustrates typical disease courses in control and c-Rel–deficient C57BL/6 mice. EAE developed in all control mice, starting approximately 13 days after immunization and reaching a maximal clinical score of 3.7 three weeks later. The disease took a chronic progressive course with a mortality rate of approximately 25%. By contrast, in the c-Rel–deficient group, only 16% of the mice developed symptoms of EAE. Disease severity was also dramatically reduced in this group, and the onset of disease was delayed by approximately 12 days (Figure 1a, Table 1).
Figure 1.

c-Rel expressed by the immune system but not the CNS is required for the development of EAE. Groups of C57BL/6 mice were immunized to induce EAE as described in Methods. Data presented are mean ± SEM of the disease scores. (a) Normal c-Rel+/+ mice (filled squares, n = 8) and c-Rel–/– mice (open circles, n = 8). (b) Irradiated c-Rel+/+ mice that were injected with c-Rel+/+ bone marrow cells (filled squares, n = 15) and irradiated c-Rel+/+ mice that were injected with c-Rel–/– bone marrow cells (open circles, n = 15). (c) Irradiated c-Rel–/– mice that were injected with c-Rel+/+ bone marrow cells (filled squares, n = 9) and irradiated c-Rel+/+ mice that were injected with c-Rel+/+ bone marrow cells (open circles, n = 8). The differences between the two groups in a and b are statistically significant (P < 0.002) as determined by Mann-Whitney test. Results are representative of two independent experiments.
Table 1.
Clinical features of MOG-induced EAE in normal and c-Rel–/– mice
Consistent with these clinical findings, histological examination of the CNS revealed dramatic differences between the two groups. In the control group, multiple inflammatory foci were observed in the cerebrum, cerebellum, brain stem, and spinal cord, with severe demyelination in the white matter (Figure 2b). By contrast, no lesions or demyelination were detected in the CNS of most c-Rel–deficient mice that showed no symptoms of EAE (Figure 2a). In those c-Rel–deficient mice that did develop EAE, we observed inflammatory infiltrates and demyelination similar to those found in the control mice.
Figure 2.
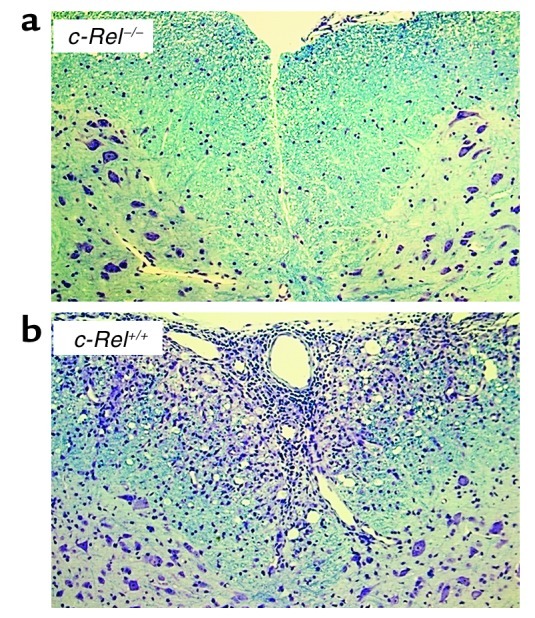
Histological profiles of spinal cords. Mice were treated as described in Figure 1a and perfused with PBS and phosphate-buffered formalin at the end of the experiment. Spinal cords were harvested, fixed in 10% formalin, and embedded in paraffin. Five-micrometer-thick paraffin sections were stained with both Luxol fast blue and cresyl violet. (a) Spinal cord of a c-Rel–/– mouse with no signs of EAE. (b) Spinal cord of a c-Rel+/+ mouse with a disease score of 4.
Since c-Rel is expressed not only by cells of the immune system but also by cells of the CNS, we next investigated whether c-Rel expressed by different organ systems plays different roles in EAE. Bone marrow chimeric mice were generated either by injecting c-Rel–/– or c-Rel+/+ bone marrow cells into irradiated c-Rel+/+ recipients (Figure 1b) or by injecting c-Rel+/+ bone marrow cells into irradiated c-Rel–/– and c-Rel+/+ recipients (Figure 1c). Mice were then immunized with MOG peptide to induce EAE as for Figure 1a. Remarkably, c-Rel deficiency in bone marrow–derived cells alone was sufficient to inhibit EAE (Figure 1b), whereas c-Rel deficiency in cells not derived from bone marrow had no effect on EAE (Figure 1c). Taken together, these results indicate that c-Rel expressed by the immune system plays an indispensable role in the development of autoimmune inflammation in the CNS.
c-Rel and NF-κB1 regulate different types of T cell responses in vivo.
Resistance to EAE in c-Rel–deficient mice can be due either to the inability of myelin-specific T cells to differentiate into effector T cells in the periphery or to the inability of differentiated effector T cells to induce demyelinating inflammation in the CNS, or both. To address this issue, we examined whether activation and differentiation of myelin-specific T cells were normal in c-Rel–deficient animals. Splenocytes were collected from both control and c-Rel–deficient mice preimmunized with MOG peptide and tested in vitro for their cytokine production and proliferation in response to the specific peptide. As shown in Figure 3, splenocytes of control animals proliferated vigorously in response to MOG peptide and produced both Th1 (IL-2 and IFN-γ) and Th2 (IL-4) cytokines. By contrast, splenocytes from c-Rel–deficient animals produced no detectable IFN-γ regardless of the MOG concentration in the culture. IL-2 secretion and cell proliferation were also reduced in the c-Rel–/– cultures (Figure 3). Unexpectedly, IL-4 production was dramatically increased in these cultures. Since IFN-γ can potentially inhibit the differentiation of Th2 cells, it is possible that the increase in IL-4 production is caused by the decrease in IFN-γ secretion.
Figure 3.

Complete blockade of Th1 IFN-γ response in c-Rel–deficient mice. Normal (black bars) and c-Rel–/– (white bars) mice were treated as in Figure 1a and sacrificed 36 days after immunization. Splenocytes (7.5 × 105/ml) were cultured in complete DMEM with or without 5–25 μg/ml of MOG38-50 peptide. Culture supernatants were collected 40 hours later and tested for cytokines by ELISA. For proliferation assays, cells were pulsed with 3H-thymidine at 48 hours and radioactivity was determined 16 hours later. Results are shown as mean + SD from a total of 12 mice (6 mice per group). The differences between the two groups are statistically significant for all parameters (P < 0.01). The experiments were repeated twice with similar results.
These results strongly suggest that Th1, but not Th2 pathway of T cell differentiation is blocked in c-Rel–/– mice. This is in startling contrast to the Th2 deficiency observed in mice deficient in NF-κB1, another member of the Rel/NF-κB family (26, 27). As shown in Figure 4, NF-κB1–/– mice are defective primarily in their IL-4 secretion, whereas their IFN-γ production is relatively normal. Das et al. recently demonstrated that this deficiency in IL-4 production is caused by selective blockade of Gata3-mediated Th2 cell differentiation in NF-κB1–/– mice; Th1 cell differentiation is not affected by NF-κB1 deficiency (27). Also shown in Figure 4 are IFN-γ and IL-4 responses of control C57BL/6 and B6;129 mice. It is to be noted that although the genetic backgrounds of these mice are not identical, they mounted similar Th1 and Th2 responses following immunization with MOG peptide.
Figure 4.

c-Rel and NF-κB1 regulate different types of T cell responses. C57BL/6 or B6;129 mice were treated and tested as for Figure 3. Data presented are IFN-γ and IL-4 concentrations of splenocyte cultures activated with 25 μg/ml of MOG peptide. Control C57BL/6 culture (open circle), control B6;129 culture (open square), c-Rel–/– C57BL/6 culture (filled circle), and NF-κB1–/– B6;129 culture (filled square).
Taken together, these results indicate that c-Rel and NF-κB1 preferentially regulate Th1 and Th2 cell responses, respectively.
c-Rel–deficient cells are defective in IL-12 production.
Activation and differentiation of T cells require antigen presentation and costimulation by APCs. c-Rel may regulate T cell activation and differentiation directly through T cells or indirectly through APCs. To investigate the latter possibility, we examined whether the expression of APC molecules required for T cell activation and differentiation are normal in c-Rel–deficient mice. To our initial surprise, we found no significant differences between normal and c-Rel–deficient APCs in their expression of B7-1, B7-2, class I, and class II MHC molecules (as determined by flow cytometry), with or without LPS stimulation (our unpublished observations). Importantly, we found that c-Rel–deficient cells produced significantly smaller amounts of IL-12 p40, a cytokine essential for Th1 cell differentiation. Figure 5 is representative of such experiments. APCs derived from normal spleen or bone marrow, and microglia and astrocytes derived from CNS all produced significant amounts of IL-12 p40 upon stimulation with LPS. By contrast, much-reduced amounts of IL-12 p40 were produced by c-Rel–deficient cells of all these lineages. Similar reductions in IL-12 p40 production were also observed in splenocyte cultures stimulated with MOG, although the total amounts of IL-12 in the latter cultures were significantly lower than in LPS-treated cultures (our unpublished observations). Since IL-12 p40 can pair with either p35 (to form IL-12) or p19 (to form IL-23), we examined the gene expression of p35 and p19 as well as p40 by PCR. As shown in Figure 6a, the expression of each of these three genes was markedly reduced in c-Rel–/– APCs. As shown in Figure 6b, T-bet is expressed by both Th0 and Th1 cells.
Figure 5.

IL-12 p40 production by normal and c-Rel–deficient cells. DC-enriched APCs from spleen (a) and bone marrow (b); and microglia (c) and astrocytes (d) from brain and spinal cord were prepared as described in Methods. All cells were cultured at 106/ml with or without 1 μg/ml (for spleen and bone marrow APCs) or 5 μg/ml (for microglia and astrocytes) LPS. Culture supernatants were collected at 40 hours and IL-12 concentrations were determined by ELISA. Results are representative of two experiments.
Figure 6.
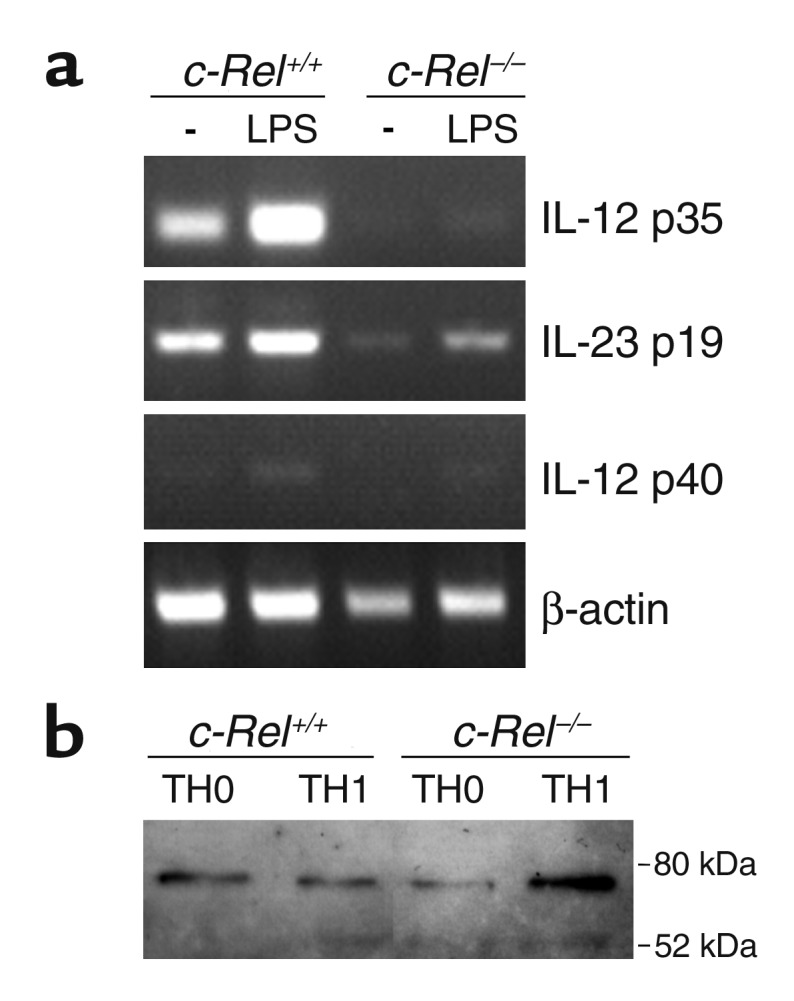
Regulation of gene expression by c-Rel. (a) p35, p40, and p19 expression in APCs. DC-enriched APCs from normal and c-Rel–deficient C57BL/6 mice were cultured with or without (–) LPS for 5 hours as described in Figure 5 legend. Total RNA was extracted, and gene expression was determined by RT-PCR as described in Methods. (b) T-bet expression in T cells. CD4+ T cells were isolated and treated as described in Figure 9, and their nuclear extracts were prepared 48 hours after the second stimulation. T-bet expression was determined by Western blot as described in Methods.
These results indicate that c-Rel may regulate IL-12 and IL-23 gene expression in various cell types. Consistent with this view, Grumonta et al. (30) and Sanjabi et al. (31) recently reported that unlike other members of the Rel/NF-κB family, c-Rel is required for the activation of the IL-12 p35 and p40 promoters in DCs and macrophages, respectively. Thus, c-Rel may be a selective transcriptional regulator of the IL-12 and IL-23 genes.
c-Rel–deficient APCs support Th2, but not Th1 responses in vitro.
Results described above suggest that the Th1 deficiency in c-Rel–/– mice may result from reduced IL-12 production by c-Rel–/– APCs. To test this theory, we examined whether c-Rel–deficient APCs are able to support Th1 and Th2 responses in vitro. CD4+ T cells and APCs were isolated from C57BL/6 mice and stimulated in vitro with anti-CD3 mAb for 5 days (Figure 7). Cells were then collected and restimulated, and their cytokine profiles were determined. As shown in Figure 7, c-Rel+/+ APCs were able to support both Th1 and Th2 responses of normal (c-Rel+/+) T cells. By contrast, c-Rel–/– APCs could support IL-2, IL-4, and IL-10, but not IFN-γ responses of c-Rel+/+ T cells. On the other hand, normal APCs were not able to support Th1 cytokine responses of c-Rel–/– T cells, suggesting that APCs may not be solely responsible for the Th1 deficiency in c-Rel–deficient mice. Again, Th2 cytokine responses were not significantly affected by c-Rel deficiency in either T cell or APC compartments, indicating that c-Rel is not required for the development of Th2 responses.
Figure 7.

c-Rel expressed both by T cells and by APCs is required for the development of Th1 responses. Purified splenic APCs and naive CD4+ T cells were cultured at a ratio of 1:5 with 106 cells/ml in the presence of plate-bound anti-CD3 mAb (2 μg/ml). Each culture contained one type of APC (c-Rel+/+ or c-Rel–/–) and one type of T cell (c-Rel+/+ or c-Rel–/–). Five days later, cells were washed and restimulated with plate-bound anti-CD3 mAb (2 μg/ml) and anti-CD28 mAb (2 μg/ml) for 48 hours. Supernatants were collected and cytokine concentrations were determined by ELISA. Results are representative of two experiments.
c-Rel–deficient T cells are completely defective in their IFN-γ expression.
Results presented above suggest that reduced production of IL-12 by APCs may be only partially responsible for the Th1 deficiency in c-Rel–deficient mice. To test whether c-Rel gene mutation directly affects T cell activation and differentiation, we purified both c-Rel+/+ and c-Rel–/– T cells and compared their responses to anti-CD3 mAb and anti-CD28 mAb. As shown in Figure 8, anti-CD3 mAb alone induced significant secretion of both Th1 and Th2 cytokines by c-Rel+/+ T cells, but only Th2 cytokine secretion (bottom row) by c-Rel–/– T cells. Importantly, anti-CD28 mAb completely abrogated the IL-2 deficiency but had no effect on IFN-γ production by c-Rel–/– T cells. Unexpectedly, anti-CD28 mAb enhanced IL-4 and IL-10 responses of c-Rel–/–, but not c-Rel+/+, T cells (Figure 8).
Figure 8.

Selective blockade of IFN-γ production in c-Rel–deficient T cells. Normal and c-Rel–deficient CD4+ T cells were purified and cultured at 106 cells/ml with or without plate-bound anti-CD3 mAb (2 μg/ml) and anti-CD28 mAb (2 μg/ml). Culture supernatants were collected 48 hours later and cytokine concentrations were determined by ELISA. Results are representative of two experiments.
These results suggest that c-Rel–deficient T cells are able to respond to both T cell receptor (anti-CD3 mAb) and costimulatory (anti-CD28 mAb) signals and are not intrinsically defective in their activation program. In the presence of TCR and CD28 signals, c-Rel–deficient T cells can become activated and mount significant Th2 and IL-2 responses as normal T cells do (Figure 8). However, c-Rel–deficient T cells do suffer from a severe functional defect: they are unable to produce IFN-γ even in the presence of CD28 stimulation, suggesting that c-Rel may selectively regulate the maturation of IFN-γ–producing T cells.
c-Rel–/– T cells fail to differentiate into Th1 cells even under Th1-inducing conditions.
To directly test whether c-Rel–/– T cells are intrinsically defective in their Th1 differentiation program, we studied Th1 cell differentiation in vitro. Naive CD4+ T cells were purified and cultured with anti-CD3 mAb, anti-CD28 mAb, and IL-2, in the absence or presence of IL-12 and anti–IL-4 mAb (Th1-inducing condition). The presence of anti-CD28 mAb and IL-2 in the culture ensures that the activation requirements for both c-Rel+/+ and c-Rel–/– T cells are met and that c-Rel deficiency does not block Th cell differentiation indirectly by hindering T cell activation (see Figure 8). Addition of IL-12 to the culture eliminates the requirement for endogenous IL-12 (which is reduced in c-Rel–deficient mice), whereas addition of anti–IL-4 mAb eliminates the effect of IL-4 on Th1 cell differentiation. Cultures without IL-12 and anti–IL-4 mAb are considered to be Th0 cultures.
As shown in Figure 9, c-Rel+/+ T cells can differentiate into either Th0 or Th1 cells depending on the culture conditions. By contrast, no Th1 cells were generated from c-Rel–/– naive T cells under Th1 conditions. This Th1 blockade was complete, since neither IFN-γ– nor IL-2–producing cells were detected. Interestingly, c-Rel–deficient T cells produced significant amounts of IL-2 and IL-4 under Th0-inducing conditions, although the kinetics of the cytokine secretion was slightly delayed. The presence of these IL-2+/IFN-γ– Th1-like cells may explain why IL-2 was detected in c-Rel–/– mice (Figure 3) and cultures (Figure 6 and Figure 7) even in the absence of IFN-γ. Taken together, these results suggest that c-Rel–deficient T cells are intrinsically defective in their Th1 differentiation program, which cannot be rescued by IL-12.
Figure 9.

CD4+ T cell differentiation in vitro. Purified CD4+ T cells were cultured at 106/ml in the presence of IL-2 (50 U/ml), plate-bound anti-CD28 mAb (2 μg/ml), and anti-CD3 mAb (2 μg/ml) without (a, Th0, left column) or with (b, Th1, right column) IL-12 (10 ng/ml) and anti-IL4 mAb (20 ng/ml). Five days later, cells were washed and restimulated with plate-bound anti-CD3 mAb and anti-CD28 mAb for up to 72 hours. Culture supernatants were collected at 24, 48, and 72 hours, and cytokine concentrations were determined by ELISA. Results are representative of two experiments. Only those bars for SEM that are wider than the symbol are shown.
c-Rel may act downstream of T-bet.
To explore the potential relationship between c-Rel and T-bet in Th1 cell differentiation, we investigated whether T-bet expression is affected by c-Rel deficiency. Deficiency in c-Rel had little effect on T-bet expression. This indicates that c-Rel may act downstream of T-bet in the Th1 differentiation pathway.
Discussion
Upon encountering specific antigens, naive CD4+ T cells can differentiate into Th0, Th1, and Th2 cells (32). These effector cells secrete different patterns of cytokines and perform different functions (32). For instance, myelin-specific Th1 cells secreting IL-2, IFN-γ, and TNF-α are highly pathogenic and can initiate EAE in healthy animals; by contrast, myelin-specific Th2 cells secreting IL-4 and IL-10 are not encephalitogenic in immune-competent animals and may suppress Th1 cell–mediated EAE (33). Several transcriptional factors have been implicated in regulating the commitment of CD4+ T cells to different Th lineages. These include STAT-4, STAT-6, Gata3, T-bet, c-Maf, NF-AT, AP-1, and NF-κB1 (34–36). Results reported here indicate that c-Rel is another transcription factor that is crucial for Th cell differentiation. However, unlike NF-κB1, which is required for Th2 cell differentiation, c-Rel appears to be required for Th1, but not Th2, cell differentiation. Thus, although NF-κB1 and c-Rel share sequence homology and mechanisms of action, they play radically different roles in Th cell differentiation, with c-Rel being involved in the Th1 pathway and NF-κB1 in the Th2 pathway. These unexpected findings provide new insights into the mechanisms of regulation of Th cell differentiation. Since c-Rel deficiency does not significantly affect T-bet expression, it is likely that c-Rel acts downstream of T-bet in Th1 cell differentiation. This puts c-Rel in the same category as STAT-4, which according to a recent report may also act downstream of T-bet during Th1 cell differentiation (37). By Western blot analysis, we found that c-Rel deficiency did not affect STAT-4 expression or phosphorylation in cells cultured as described in Figure 9 legend (our unpublished observations). Thus, c-Rel, STAT-4, and T-bet may act in concert to regulate the Th1 differentiation pathway.
The mechanisms of c-Rel regulation of Th1 cell differentiation in vivo are not fully understood. c-Rel may indirectly regulate Th1 cell differentiation through modulating the expression of molecules that affect Th1 cell differentiation. Our observation that c-Rel–deficient APCs produce reduced amounts of IL-12, which is crucial for Th1 cell differentiation, does support this view. However, since adding IL-12 to c-Rel–/– T cell cultures does not rescue Th1 cell differentiation (see Figure 7), additional mechanisms must be involved in c-Rel–mediated regulation of Th1 cell differentiation. We are in the process of identifying downstream c-Rel target genes that may be involved in regulating Th1 cell differentiation.
The inductive phase of EAE primarily involves activation and differentiation of myelin-specific lymphocytes. In the present study, this was achieved by immunizing mice with MOG peptide together with CFA. Although it is clear that MOG and adjuvant activate specific T cells by generating both the T cell receptor (the first) and the costimulatory (the second) signals, the transcriptional elements involved in this process are poorly understood. Data presented here suggest that c-Rel may be one of the transcriptional regulators that are important for this process. In the absence of c-Rel, autoreactive T cells may not be fully activated and may not effectively differentiate into Th1-type cells; therefore, severe autoimmune encephalomyelitis may not develop even with active immunization with myelin antigen and adjuvant.
It is to be noted that experiments reported here do not directly address the question of whether c-Rel plays a role in the effector stage of EAE. Because differentiation of autoreactive Th1 cells is significantly compromised in c-Rel–deficient mice, it is difficult to examine the functions of c-Rel–deficient effector cells in EAE. It is also to be noted that although c-Rel is expressed by various neural cells (6–9) and its deficiency significantly affects IL-12 production by astrocytes and microglia (see Figure 5), c-Rel expressed by the CNS may not affect the progression of autoimmune inflammation (Figure 1c). Consistent with this view, a recent report by Campbell et al. suggests that c-Rel may not be involved in the later stages of synovial inflammation in an animal model of rheumatoid arthritis (38). Thus, the roles of c-Rel in autoimmune inflammation may be mediated primarily through its effect on immune cells.
Acknowledgments
This work was supported by NIH grants AI-50059, NS-40188, NS-40447, and AR-44914.
Footnotes
See the related Commentary beginning on page 741.
Conflict of interest: No conflict of interest had been declared.
Nonstandard abbreviations used: Experimental autoimmune encephalomyelitis (EAE); myelin oligodendrocyte glycoprotein (MOG); antigen-presenting cell (APC); dendritic cell (DC).
References
- 1.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 2.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 3.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80:321–330. doi: 10.1016/0092-8674(95)90415-8. [DOI] [PubMed] [Google Scholar]
- 4.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- 5.Bull P, Morley KL, Hoekstra MF, Hunter T, Verma IM. The mouse c-rel protein has an N-terminal regulatory domain and a C-terminal transcriptional transactivation domain. Mol Cell Biol. 1990;10:5473–5485. doi: 10.1128/mcb.10.10.5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakalkin G, Yakovleva T, Terenius L. NF-kappa B-like factors in the murine brain. Developmentally-regulated and tissue-specific expression. Brain Res Mol Brain Res. 1993;20:137–146. doi: 10.1016/0169-328x(93)90119-a. [DOI] [PubMed] [Google Scholar]
- 7.Grumont RJ, Gerondakis S. Murine c-rel transcription is rapidly induced in T-cells and fibroblasts by mitogenic agents and the phorbol ester 12-O-tetradecanoylphorbol-13-acetate. Cell Growth Differ. 1990;1:345–350. [PubMed] [Google Scholar]
- 8.Grumont RJ, Gerondakis S. The murine c-rel proto-oncogene encodes two mRNAs the expression of which is modulated by lymphoid stimuli. Oncogene Res. 1990;5:245–254. [PubMed] [Google Scholar]
- 9.Brownell E, et al. Detection of c-rel-related transcripts in mouse hematopoietic tissues, fractionated lymphocyte populations, and cell lines. Mol Cell Biol. 1987;7:1304–1309. doi: 10.1128/mcb.7.3.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:2642–2647. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaltschmidt C, et al. Transcription factor NF-kappa B is activated in microglia during experimental autoimmune encephalomyelitis. J Neuroimmunol. 1994;55:99–106. doi: 10.1016/0165-5728(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 12.Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-kappa B activity in neurons. Mol Cell Biol. 1994;14:3981–3992. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas LH, Friedland JS, Sharland M, Becker S. Respiratory syncytial virus-induced RANTES production from human bronchial epithelial cells is dependent on nuclear factor-kappa B nuclear binding and is inhibited by adenovirus-mediated expression of inhibitor of kappa B alpha. J Immunol. 1998;161:1007–1016. [PubMed] [Google Scholar]
- 14.Ting AT, Pimentel-Muinos FX, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15:6189–6196. [PMC free article] [PubMed] [Google Scholar]
- 15.Ishikawa H, et al. Chronic inflammation and susceptibility to bacterial infections in mice lacking the polypeptide (p)105 precursor (NF-kappaB1) but expressing p50. J Exp Med. 1998;187:985–996. doi: 10.1084/jem.187.7.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu SF, Ye X, Malik AB. In vivo inhibition of nuclear factor-kappa B activation prevents inducible nitric oxide synthase expression and systemic hypotension in a rat model of septic shock. J Immunol. 1997;159:3976–3983. [PubMed] [Google Scholar]
- 17.Wingren AG, et al. T cell activation pathways: B7, LFA-3, and ICAM-1 shape unique T cell profiles. Crit Rev Immunol. 1995;15:235–253. doi: 10.1615/critrevimmunol.v15.i3-4.30. [DOI] [PubMed] [Google Scholar]
- 18.Liou HC, et al. c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int Immunol. 1999;11:361–371. doi: 10.1093/intimm/11.3.361. [DOI] [PubMed] [Google Scholar]
- 19.Gerondakis S, et al. Rel-deficient T cells exhibit defects in production of interleukin 3 and granulocyte-macrophage colony-stimulating factor. Proc Natl Acad Sci USA. 1996;93:3405–3409. doi: 10.1073/pnas.93.8.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tumang JR, et al. c-Rel is essential for B lymphocyte survival and cell cycle progression. Eur J Immunol. 1998;28:4299–4312. doi: 10.1002/(SICI)1521-4141(199812)28:12<4299::AID-IMMU4299>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 21.Grumont RJ, Rourke IJ, Gerondakis S. Rel-dependent induction of A1 transcription is required to protect B cells from antigen receptor ligation-induced apoptosis. Genes Dev. 1999;13:400–411. doi: 10.1101/gad.13.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y. NF-kappa B RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med. 1997;185:953–961. doi: 10.1084/jem.185.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weih F, et al. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80:331–340. doi: 10.1016/0092-8674(95)90416-6. [DOI] [PubMed] [Google Scholar]
- 24.Caamano JH, et al. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med. 1998;187:185–196. doi: 10.1084/jem.187.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franzoso G, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187:147–159. doi: 10.1084/jem.187.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hilliard B, Samoilova EB, Liu TT, Rostami AM, Chen Y. Experimental autoimmune encephalomyelitis in nuclear factor-kB-deficient mice: roles of nuclear factor-kB in the activation and differentiation of autoreactive T cells. J Immunol. 1999;163:2937–2943. [PubMed] [Google Scholar]
- 27.Das J, et al. A critical role for NF-kappa B in GATA3 expression and Th2 differentiation in allergic airway inflammation. Nat Immunol. 2001;2:45–50. doi: 10.1038/83158. [DOI] [PubMed] [Google Scholar]
- 28.Szabo SJ, et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 29.Suzumura A, Mezitis SG, Gonatas NK, Silberberg DH. MHC antigen expression on bulk isolated macrophage-microglia from newborn mouse brain: induction of Ia antigen expression by gamma-interferon. J Neuroimmunol. 1987;15:263–278. doi: 10.1016/0165-5728(87)90121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grumonta R, et al. c-Rel regulates interleukin 12 p70 expression in CD8+ dendritic cells by specifically inducing p35 gene transcription. J Exp Med. 2001;194:1021–1032. doi: 10.1084/jem.194.8.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci USA. 2000;97:12705–12710. doi: 10.1073/pnas.230436397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- 33.Kuchroo VK, et al. B7-1 and B7-2 costimulatory molecules differentially activate the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 34.Ho IC, Kim JI, Szabo SJ, Glimcher LH. Tissue-specific regulation of cytokine gene expression. Cold Spring Harb Symp Quant Biol. 1999;64:573–584. doi: 10.1101/sqb.1999.64.573. [DOI] [PubMed] [Google Scholar]
- 35.O’Garra A. Immunology. Commit ye helpers. Nature. 2000;404:719–720. doi: 10.1038/35008191. [DOI] [PubMed] [Google Scholar]
- 36.O’Garra A, Murphy K. Role of cytokines in development of Th1 and Th2 cells. Chem Immunol. 1996;63:1–13. doi: 10.1159/000319475. [DOI] [PubMed] [Google Scholar]
- 37.Mullen AC, et al. Role of T-bet in commitment of Th1 cells before IL-12-dependent selection. Science. 2001;292:1907–1910. doi: 10.1126/science.1059835. [DOI] [PubMed] [Google Scholar]
- 38.Campbell IK, Gerondakis S, O’Donnell K, Wicks IP. Distinct roles for the NF-κB1 (p50) and c-Rel transcription factors in inflammatory arthritis. J Clin Invest. 2000;105:1799–1806. doi: 10.1172/JCI8298. [DOI] [PMC free article] [PubMed] [Google Scholar]