

Abstract
In type 2 diabetes, chronic hyperglycemia is suggested to be detrimental to pancreatic β cells, causing impaired insulin secretion. IL-1β is a proinflammatory cytokine acting during the autoimmune process of type 1 diabetes. IL-1β inhibits β cell function and promotes Fas-triggered apoptosis in part by activating the transcription factor NF-κB. Recently, we have shown that increased glucose concentrations also induce Fas expression and β cell apoptosis in human islets. The aim of the present study was to test the hypothesis that IL-1β may mediate the deleterious effects of high glucose on human β cells. In vitro exposure of islets from nondiabetic organ donors to high glucose levels resulted in increased production and release of IL-1β, followed by NF-κB activation, Fas upregulation, DNA fragmentation, and impaired β cell function. The IL-1 receptor antagonist protected cultured human islets from these deleterious effects. β cells themselves were identified as the islet cellular source of glucose-induced IL-1β. In vivo, IL-1β–producing β cells were observed in pancreatic sections of type 2 diabetic patients but not in nondiabetic control subjects. Similarly, IL-1β was induced in β cells of the gerbil Psammomys obesus during development of diabetes. Treatment of the animals with phlorizin normalized plasma glucose and prevented β cell expression of IL-1β. These findings implicate an inflammatory process in the pathogenesis of glucotoxicity in type 2 diabetes and identify the IL-1β/NF-κB pathway as a target to preserve β cell mass and function in this condition.
Introduction
Type 2 diabetes mellitus results from an inadequate adaptation of the functional pancreatic β cell mass in the face of insulin resistance. In turn, hyperglycemia by itself has secondary adverse effects on β cells. Indeed, several studies have shown that chronic elevation of blood glucose concentration impairs β cell function, leading to the concept of glucotoxicity (1–7). Moreover, elevated glucose concentrations induce β cell apoptosis in cultured islets from diabetes-prone Psammomys obesus (8) and from humans (9, 10); somewhat higher concentrations of glucose are required to induce β cell apoptosis in rodent islets (8, 11, 12). Various molecular mechanisms have been proposed to underlie glucose-induced β cell dysfunction, including formation of advanced glycation end products (13), direct impairment of insulin gene transcription and proinsulin biosynthesis (14, 15), and reduced binding activity of pancreatic duodenal homeobox 1 (PDX-1) (7). Recently, we proposed a mechanism underlying glucose-induced β cell apoptosis in human islets that involves upregulation of Fas receptors by elevated glucose levels (9). However, the mediator of glucose-induced Fas expression and its role in glucotoxicity remains unknown.
IL-1β has been proposed to mediate both impaired function and destruction of pancreatic β cells during the development of autoimmune type 1 diabetes (16). In keeping with this, treatment of rodent islets with IL-1β results in a potent inhibition of insulin secretion followed by islet destruction (17–23). In human islets, IL-1β has been shown to impair insulin release and to induce Fas expression, enabling Fas-triggered apoptosis (9, 24–28). Finally, activation of the nuclear transcription factor NF-κB is required for IL-1β–induced Fas expression (29–31). Part of these IL-1β effects are reminiscent of the toxic effects of elevated glucose concentrations. Together, the above findings led us to postulate that glucose may induce IL-1β secretion from β cells in the absence of an autoimmune process. We now identify β cells as the cellular source of glucose-induced IL-1β in cultured human islets and confirm this using tissue sections from the pancreas of type 2 diabetic patients and of Psammomys obesus. The role of such endogenously produced IL-1β in β cell glucotoxicity was also explored.
Methods
Islet isolation and culture.
Islets were isolated from pancreata of 11 organ donors at the Department of Surgery, University of Geneva Medical Center, as described (32–34). Islet purity was greater than 95% as judged by dithizone staining (if this degree of purity was not achieved by routine isolation, islets were hand picked). The donors, aged 40–70 years, were heart-beating cadaver organ donors, and none had a previous history of diabetes or metabolic disorders. For long-term in vitro studies, the islets were cultured on extracellular matrix–coated plates derived from bovine corneal endothelial cells (Novamed Ltd., Jerusalem, Israel), allowing the cells to attach to the dishes and spread, preserving their functional integrity (7, 35). Islets were cultured in CMRL 1066 medium (hereafter referred to as culture medium) containing 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FCS (Invitrogen Ltd., Carlsbad, California, USA). Two days after plating, when most islets were attached and began to flatten, the medium was changed to culture medium containing 5.5, 11.1, or 33.3 mM glucose. In some experiments, islets were additionally cultured with 2 ng/ml recombinant human IL-1β, 1,000 U/ml recombinant human IFN-γ (ReproTech EC Ltd., London, United Kingdom), 500 ng/ml IL-1 receptor antagonist (IL-1Ra; R&D Systems Inc., Minneapolis, Minnesota, USA), 1 ng/ml membrane-bound Fas ligand (FasL; Upstate Biotechnology Inc., Lake Placid, New York, USA) (36), or with 100 μM pyrrolidinedithiocarbamate (PDTC) for 2 hours for every 2 days in culture (Sigma-Aldrich, St. Louis, Missouri, USA).
Animals.
Psammomys obesus of both sexes (age 2.0–3.5 months) from the diabetes-prone and diabetes-resistant lines of the Hebrew University colonies were originally obtained from Harlan Laboratories Ltd. (Jerusalem, Israel). After weaning, diabetes-prone Psammomys obesus were maintained on a low-energy diet containing 2.38 kcal/g (Koffolk Ltd., Petach Tikva, Israel) until the start of the experiments, whereas diabetes-resistant Psammomys obesus were maintained on a high-energy diet containing 2.93 kcal/g (Weizmann Institute of Science, Rehovot, Israel) to identify animals that develop diabetes and exclude them from the study (∼30–40% of the animals in the diabetes-resistant colony). All nonfasted animals with random blood glucose concentrations below 7.8 mmol/l (tested with the Glucometer Elite from Bayer Corp., Elkhart, Indiana, USA) were considered nondiabetic. Diabetes-prone Psammomys obesus switched to a high-energy diet received an injection of 0.4 g/kg phlorizin (Sigma-Aldrich) or solvent (40% propylene glycol) every 12 hours and were killed after 8 days. Psammomys obesus were anesthetized with ketamine (Ketalar; Parke-Davis & Co., Gwent, United Kingdom) and exsanguinated by cardiac puncture. The pancreas was rapidly removed and immersion-fixed in 10% phosphate-buffered formalin. The animal studies were approved by the Institutional Animal Care and Use Committee of Hebrew University and the Hadassah Medical Organization.
Detection of IL-1β–expressing β cells.
Pancreata from routine necropsies and from Psammomys obesus were immersion-fixed in formalin, followed by paraffin embedding. Sections were deparaffinized and rehydrated, and endogenous peroxidase was blocked by submersion in 0.3% H2O2 for 15 minutes. Sections were then incubated in methanol for 4 minutes. After washing with PBS, cultured islets and isolated β cells were fixed in 4% paraformaldehyde (30 minutes at room temperature) followed by permeabilization with 0.5% Triton X-100 (4 minutes at room temperature). Both tissue sections and cultured cells were double-labeled for IL-1β and insulin by 1 hour of exposure to 10% BSA followed by incubation for 1 hour at 37°C with mouse anti–IL-1β antibody (1:30 dilution; R&D Systems Inc.). Detection was performed using donkey anti-mouse Cy3-conjugated antibody (1:100 dilution; Jackson ImmunoResearch Laboratories, West Grove, Pennsylvania, USA). Subsequently, specimens were incubated for 30 minutes at 37°C with guinea pig anti-insulin antibody diluted 1:50 (DAKO Corp., Carpinteria, California, USA), followed by a 30-minute incubation with a 1:20 dilution of fluorescein-conjugated rabbit anti–guinea pig antibody (DAKO Corp.). For positive control of IL-1β staining, human mononuclear cells were isolated as described previously (37) and exposed for 2 hours at 37°C to 1 μg/ml LPS (Becton, Dickinson and Co., Franklin Lakes, New Jersey, USA). Coverslips were air-dried and mounted onto slides, then fixed and permeabilized for 5 minutes at room temperature in 1:1 acetone/methanol and stained for IL-1β as described.
For mRNA in situ hybridization of IL-1β, DNA templates were generated by PCR with incorporation of a T3 or a T7 promoter into the antisense or sense primer. The following primers were used: T3, 5′-AAGCGCGCAATTAACCCTCACTAAAGGGTCAGCACCTCTCAAGCAGAA-3′ and T7, 5′-GGCCAGTAATTGTAATACGACTCACTATAGGGAGGCGGCCCTGAAAGGAGAGAGCTGA-3′. Purification of PCR product was performed with Nucleospin Extract 2 in 1 (Macherey-Nagel GmbH, Düren, Germany) according to the manufacturer’s instructions. After phenol/chloroform purification, digoxigenin-labeled RNA probes were prepared using RNA T7- and T3-polymerase and RNA digoxigenin labeling mix (Roche Diagnostics GmbH, Mannheim, Germany). Tissue sections were treated with 20 μg/ml proteinase K (Roche Diagnostics GmbH) and prehybridized for 2 hours at 55°C in hybridization buffer containing 50% formamide, 5× sodium chloride–sodium citrate, 50 μg/ml salmon sperm (Sigma-Aldrich), 1× Denhardt solution, and 250 μg/ml RNA type IV from calf liver (Sigma-Aldrich). Hybridization was performed overnight at 52°C in 100 μl hybridization buffer containing 30 ng of digoxigenin-labeled RNA probe. Sections were then blocked with 5% milk powder at room temperature and incubated for 1 hour at 37°C with anti-digoxigenin–rhodamine Fab fragment (20 μg/ml; Roche Diagnostics GmbH), followed by insulin immunostaining as described above.
After staining, samples were embedded in Kaiser’s glycerol gelatin (Merck KGaA, Darmstadt, Germany) and analyzed by light and fluorescence microscopy (Axiolab; Carl Zeiss Jena GmbH, Jena, Germany).
Western blot analysis.
Islets were maintained in culture medium in nonadherent plastic dishes. One day after isolation, medium was changed and groups of 200 islets were incubated for 44 hours in culture medium containing 5.5 or 33.3 mM glucose without or with 2 ng/ml IL-1β or 500 ng/ml IL-1Ra. At the end of the incubations, islets were washed in PBS, suspended in 50 μl sample buffer containing 125 mM Tris-HCl (pH 6.8), 4% SDS, 10% glycerol, 0.3% bromophenol blue, and 1.8% β-mercaptoethanol and boiled for 5 minutes. Equivalent amounts of each treatment group were run on 15% SDS polyacrylamide gels. Proteins were electrically transferred to nitrocellulose filters and incubated with rabbit anti-Fas (C20; Santa Cruz Biotechnology Inc., Santa Cruz, California, USA), mouse anti–IL-1β (R&D Systems Inc.), rabbit anti–IL-1β (recognizing precursor and cleaved forms of human IL-1β; New England BioLabs Inc., Beverly, Massachusetts, USA), mouse anti–NF-κB (p65) (Active Motif LLC, Carlsbad, California, USA), mouse anti–NOS-2 antibody C-11 (Santa Cruz Biotechnology Inc.) recognizing mouse, rat, and human origin of inducible nitric oxide synthase (iNOS), or mouse anti-actin antibody (C-2; Santa Cruz Biotechnology Inc.), followed by incubation with horseradish peroxidase–linked anti-mouse or anti-rabbit IgG’s (Santa Cruz Biotechnology Inc.). The emitted light was captured on x-ray film after adding LumiGLO reagent (Phototope-HRP Western blot detection kit; New England BioLabs Inc.). As a marker, biotinylated protein molecular weight standard (New England BioLabs Inc.) was run in parallel. Between incubations, nitrocellulose membranes were stripped for 30 minutes at 50°C in 40 ml of a solution containing 280 μl β-mercaptoethanol, 5 ml 0.5 M Tris-HCl (pH 6.8), and 10% SDS, and then washed for 1 hour in Tris-buffered saline containing 0.1% Tween-20. Intensity of bands was analyzed using MultiAnalyst (Bio-Rad Laboratories Inc., Hercules, California, USA).
NF-κB activation.
Islets were cultured in suspension as described above and washed with PBS. Activation of NF-κB complex was quantified with an ELISA-based kit (Trans-AM NF-κB; Active Motif LLC) using attached oligonucleotides binding to an NF-κB consensus site and detected by an anti-p65 or p50 subunit antibody according to the manufacturer’s instructions. In parallel, islets were fixed in Bouin solution for 15 minutes and resuspended in 40 μl of 2% melted agarose in PBS (40°C), followed by rapid centrifugation and paraffin embedding. Sections were deparaffinized and rehydrated, endogenous peroxidase blocked by submersion in 0.3% H2O2 for 15 minutes, and incubated in methanol for 4 minutes. Sections were incubated with a 1:50 dilution of either mouse anti–NF-κB (p65) or rabbit anti–inhibitory κB (I-κB) (C-21; Santa Cruz Biotechnology Inc.) antibodies, detected by donkey anti-mouse or anti-rabbit Cy3-conjugated antibodies, and double stained for insulin as described above.
RNA extraction, RT-PCR, and sequencing of RT-PCR product.
Islets were cultured in suspension as described above. Total RNA was extracted using an RNeasy mini kit (Qiagen Inc., Basel, Switzerland), and RT-PCR was performed using the Superscript II RNase H– reverse transcriptase kit and oligo(dT) (24) (Life Technologies Inc., Gaithersburg, Maryland, USA) according to the instructions from the manufacturers. The primers were: human IL-1β, 5′-AAGCTGATGGCCCTAAACAG-3′ and 5′-AGGTGCATCGTGCACATAAG-3′; human Fas, 5′-GCATCTGGACCCTCCTACCT-3′ and 5′-CAGTCTGGTTCATCCCCATT-3′; and human iNOS, 5′-ACGTGCGTTACTCCACCAACA-3′ and 5′-CATAGCGGATGAGCTGAGCATT-3′.
PCR conditions for IL-1β and Fas were: denaturation for 30 seconds at 94°C, annealing for 30 seconds at 60°C, and elongation for 30 seconds at 72°C, followed for real-time PCR by quantification 5 seconds at 80°C; repetition for 45 cycles. Saturation of PCR product occurred between 22 and 28 cycles. The size of the amplification product was 250 bp. The purified PCR products were sequenced to confirm amplification of the correct gene. The conditions for PCR amplification of iNOS were: denaturation for 30 seconds at 94°C, annealing for 40 seconds at 55°C, and elongation for 30 seconds at 72°C; repetition for 35 cycles. For quantitative analysis, we used the LightCycler quantitative PCR system (Roche Diagnostics GmbH) and performed quantitative PCR with a commercial kit (LightCycler–DNA Master SYBR Green I; Roche Diagnostics GmbH). The amounts of Fas and IL-1β mRNA were standardized against GAPDH (5′-AACAGCGACACCCACTCCTC-3′ and 5′-GGAGGGGAGATTCAGTGTGGT-3′).
β cell apoptosis.
The free 3′-OH strand breaks resulting from DNA degradation were detected using the TUNEL technique (38). After washing with PBS, cultured islets were fixed in 4% paraformaldehyde for 30 minutes at room temperature, followed by permeabilization with 0.5% Triton X-100 for 4 minutes at room temperature. The TUNEL assay was performed according to the manufacturer’s instructions (In Situ Cell Death Detection Kit, AP; Roche Molecular Biochemicals, Mannheim, Germany). The preparations were then rinsed with Tris-buffered saline and incubated for 10 minutes at room temperature with the 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (BCIP/NBT) liquid substrate system (Sigma-Aldrich). Thereafter, islets were incubated with a guinea pig anti-insulin antibody as above, followed by detection using the streptavidin-biotin-horseradish peroxidase complex (Zymed Laboratories Inc., South San Francisco, California, USA). In parallel to the TUNEL reaction, we used the DNA-binding dye propidium iodide (Sigma-Aldrich) to assess the effects of glucose on necrosis. Cultured islets were washed with PBS (without paraformaldehyde fixation), incubated for 10 minutes on ice with 10 μg/ml propidium iodide in PBS, washed with PBS, and embedded in fluorescent mounting medium (DAKO Corp.). The samples were immediately evaluated by fluorescence microscopy for positively stained necrotic nuclei.
Cytokine release.
Cytokine release was evaluated in the culture medium collected before the termination of each experiment. The following ELISA kits were used: human IL-1α, IL-1β, and IL-12 (R&D Systems Inc.), human TNF-α (Endogen Inc., Boston, Massachusetts, USA), and human IFN-γ (Invitrogen Ltd.).
Insulin release and content.
To determine acute insulin release in response to glucose stimulation, islets were washed in RPMI 1640 medium with 10% FCS and containing 3.3 mM glucose, and preincubated for 1 hour in the same medium. The medium was then discarded and replaced with fresh medium containing 3.3 mM glucose for 1 hour for basal secretion, followed by an additional 1 hour incubation in medium containing 16.7 mM glucose. Supernatants were collected and frozen for insulin assays. Thereafter islets were washed with PBS and extracted with HCl (0.18 N) in 70% ethanol for 24 hours at 4°C. The acid-ethanol extracts were collected for determination of insulin content. Insulin was determined using a human insulin RIA kit (CIS Bio International, Gif-sur-Yvette, France).
Evaluation and statistical analysis.
Samples were evaluated in a randomized manner by a single investigator (K. Maedler) who was blinded to the treatment conditions. Care was taken to score islets of similar size. Some larger islets did not completely spread and were several cells thick. Such islets were excluded because a monolayer is a prerequisite for single-cell evaluation. The mean surface area of the evaluated islet monolayers was 0.031 ± 0.012 mm2 and 0.029 ± 0.011 mm2 in islets cultured at 5.5 and 33.3 mM glucose, respectively (no significant difference). Thus, the exclusion of larger islets occurred to a similar extent in each dish regardless of the treatment. Saisam software (Microvision Instruments, Evry, France) was used to measure area. Data were analyzed by Student t test or by ANOVA with a Bonferroni correction for multiple group comparisons.
Results
Glucose induces IL-1β production and release in human islets.
Human islets were exposed to elevated glucose concentrations for 4 days. Measurement of IL-1β released in the culture medium revealed a 2.2-fold increase in islets cultured at 33.3 mM compared with islets in 5.5 mM glucose (Figure 1a). To exclude a nonspecific effect of this high concentration of D-glucose, osmolarity was corrected with 27.8 mM L-glucose together with 5.5 mM D-glucose, resulting in a similar release of IL-1β to that observed with 5.5 mM D-glucose alone. No IL-1β was detectable in unused culture medium. The specificity of IL-1β release was assessed by comparison with the release of other cytokines. Limited amounts of IL-1α and TNF-α were found, but neither was regulated by glucose and no significant amount of IFN-γ or IL-12 was detectable (Table 1). The time-course effect of 33.3 mM glucose on IL-1β secretion became significant only after 20 hours of exposure to high glucose, and persisted after 44 hours (Figure 1b). Western blot analysis and quantitative RT-PCR measurement of IL-1β production in human islets revealed that elevated glucose concentration induces not only IL-1β release but also IL-1β protein and RNA synthesis (Figure 1, c and d). However, Western blot analysis gave quite variable results in terms of increased IL-1β in response to glucose. This may be due to varying numbers of non-endocrine cells (including most notably macrophages) in the different islet preparations. IL-1β production from such accompanying cells is not stimulated by glucose. Therefore, the higher the contribution in total IL-1β production, the lower the expected glucose effect.
Figure 1.

Glucose induces IL-1β expression and release in human islets. (a) Secretion of IL-1β from human islets cultured on extracellular matrix–coated dishes for 4 days in 5.5, 11.1, or 33.3 mM D-glucose or in 5.5 mM D-glucose plus 27.8 mM L-glucose. Each bar represents the mean ± SEM of eight experiments from eight separate donors. *P < 0.01 compared with islets cultured in 5.5 mM glucose alone. (b) Secretion of IL-1β from human islets during 44 hours of culture in suspension with 5.5 or 33.3 mM D-glucose. Data were collected from four tubes per treatment in two separate experiments from two donors. Data are represented as mean ± SEM. *P < 0.01 compared with islets cultured in 5.5 mM glucose. (c) Immunoblotting of pro–IL-1β, IL-1β, and actin. Human islets cultured in suspension at 5.5 or 33.3 mM glucose were analyzed after 44 hours of incubation. One experiment of eleven (from eleven donors) is shown. In seven experiments, glucose induced IL-1β. In three experiments, IL-1β remained unchanged, and in one it was decreased. (d) RT-PCR detection and quantification of IL-1β mRNA expression. Total RNA was isolated from human islets cultured for 44 hours in medium containing 5.5 or 33.3 mM glucose. In the LightCycler quantitative PCR system, the level of IL-1β expression was normalized against GAPDH and the results were expressed as mRNA levels relative to control incubations at 5.5 mM. Results are presented as mean ± SEM for six independent experiments from six donors. *P < 0.05 relative to islets cultured in 5.5 mM glucose.
Table 1.
Cytokines released by human islets cultured at low and high glucose concentrations

Identification of the islet cellular source of glucose-dependent IL-1β production.
We next identified the islet cells producing IL-1β. Exposure of cultured human islets to 33.3 mM glucose for 4 days induced IL-1β expression in clusters of β cells, as determined by double immunostaining of islets plated on extracellular matrix–coated dishes with anti–IL-1β and anti-insulin antibodies. Figure 2, a–d, shows representative images from one experiment of six from six separate donors. In each experiment, IL-1β–positive β cells were observed in islets cultured at 33.3 mM glucose. To exclude false-positive results due to IL-1β secreted by other cells binding to β cell membranes, islets were treated with IL-1Ra. Under these conditions, IL-1Ra should antagonize the interaction of IL-1β with its surface receptors on β cells. Coincubation with IL-1Ra displayed similar results, confirming β cell production of IL-1β (not shown).
Figure 2.

Expression of IL-1β by human β cells exposed to a diabetic milieu. Double immunostaining for IL-1β appears in red (b and d) and insulin in green (a and c) in human islets cultured on extracellular matrix–coated dishes and exposed for 4 days to media containing 5.5 mM glucose (a and b) or 33.3 mM glucose (c and d). Double immunostaining for IL-1β in red (f and h) and insulin in green (e and g) in tissue sections of pancreata from a nondiabetic patient (e and f) and from a patient with type 2 diabetes (g and h). In situ hybridization for IL-1β mRNA in red (j, l, and n) double immunostained for insulin in green (i, k, and m) in tissue sections of pancreata from a patient with type 2 diabetes (k, l, m, and n) with anti-sense probe (l) and with sense probe (negative control) (n), and from a nondiabetic patient (i and j) using anti-sense probe (j). Immunostaining for IL-1β in LPS-treated macrophages (positive control) (o). Magnification: ×250.
IL-1β is not expressed in normal human pancreatic islets (28). However, based on the in vitro studies, it was anticipated that it might be expressed in islets of patients with type 2 diabetes, as a result of hyperglycemia. Expression of IL-1β was therefore studied in sections of pancreata from five poorly controlled type 2 diabetic patients, all with documented fasting blood glucose higher than 8 mM. Double immunostaining of the pancreatic sections for IL-1β and insulin revealed localization of IL-1β in clusters of β cells in all pancreata. Clusters of IL-1β–expressing β cells were exhibited in 22.5% ± 3.4% of the islets in each pancreas (representative images are sown in Figure 2, g–h). The presence of IL-1β mRNA transcripts was verified by in situ hybridization in β cells of diabetic patients (Figure 2, k and l). IL-1β expression could not be detected in β cells of nondiabetic controls (Figure 2, e, f, i, and j) or in the exocrine pancreas. A digoxigenin-labeled sense probe was used as control and gave no signal (Figure 2n).
β cell expression of IL-1β during development of diabetes in Psammomys obesus is glucose-dependent.
To examine whether induction of IL-1β in vivo is also regulated by glucose, three diabetes-resistant and eight diabetes-prone Psammomys obesus fed a low- or high-energy diet were studied. The hyperglycemic animals were further treated with phlorizin, which corrects hyperglycemia by inhibiting renal tubular reabsorption of glucose. No IL-1β–expressing β cells were observed in islets of diabetes-resistant (not shown) or in fasted diabetes-prone Psammomys obesus (Figure 3, a and b). After 8 days on a high-energy diet, islets of severely hyperglycemic diabetes-prone Psammomys obesus exhibited IL-1β expression in most β cells, which barely expressed insulin (Figure 3, c and d). Normalization of blood glucose by injection of phlorizin in animals fed a high-energy diet restored insulin stores and prevented IL-1β expression (Figure 3, e and f).
Figure 3.
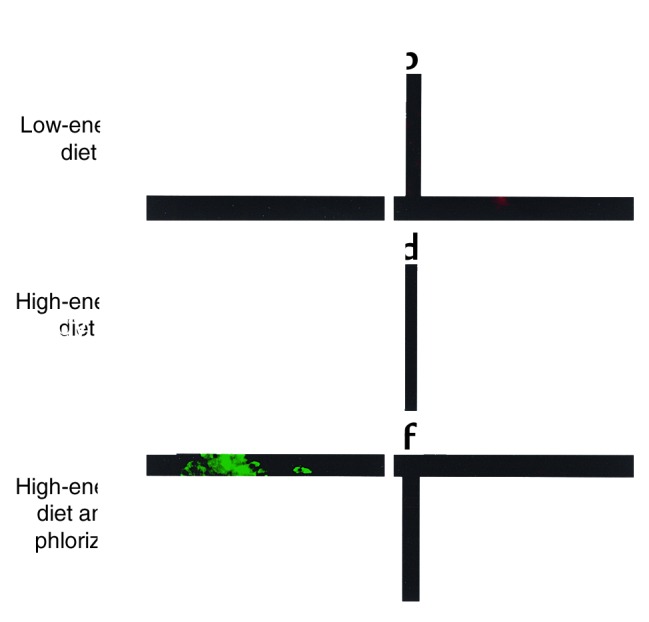
β cell expression of IL-1β during development of diabetes in Psammomys obesus. Double immunostaining for IL-1β in red (b, d, and f) and insulin in green (a, c, and e) in tissue sections of pancreata from a fasted Psammomys obesus on a low-energy diet (blood glucose 4 mM) (a and b), from an animal on a high-energy diet for 8 days without injection (blood glucose 13.6 mM) (c and d), and with injections of the glucopenic drug phlorizin (blood glucose 5.6 mM) (e and f). Magnification: ×250.
IL-1β mediates glucose-induced NF-κB activation, Fas expression, and β cell apoptosis.
The functional role of glucose-induced IL-1β was tested using IL-1Ra as an inhibitor. In human islets, elevated glucose concentrations induced a 1.9-fold increase in NF-κB activity (Figure 4a). This was prevented by IL-1Ra. Moreover, high glucose induced the expression of the p65 subunit (Figure 4b). NF-κB is bound in the cytoplasm to I-κB proteins (39). Exposure of human islets to 33 mM glucose decreased I-κB expression (Figure 4c), leading to apparition of NF-κB (p65) in β cell nuclei (Figure 4d). Glucose-dependent induction of Fas protein and mRNA were also hindered by IL-1Ra (Figure 4, b, e, and f).
Figure 4.

Glucose decreases β cell I-κB expression and induces IL-1β–meditated NF-κB activation, Fas expression, and DNA fragmentation. (a) Relative NF-κB activity. Human islets were cultured in suspension for 44 hours in 5.5 or 33.3 mM glucose alone or in the presence of IL-1β, IL-1Ra, or both. HeLa cells stimulated with 5 ng/ml IL-1α were used as positive control. Each bar represents the mean ± SEM of three experiments from three separate donors. *P < 0.05 relative to islets cultured in 5.5 mM glucose alone. (b) Immunoblotting of NF-κB (p65), Fas, and actin. Human islets cultured in suspension at 5.5 or 33.3 mM glucose with and without IL-1β or IL-1Ra were analyzed after 44 hours of incubation. The antibodies were blotted on the same membrane after stripping. One of three experiments from three donors is shown. Each experiment displayed similar results. (c) Double immunostaining for I-κB in red (boxes 1 and 3) and insulin in green (boxes 2 and 4) in sections of cultured human islets exposed for 44 hours to media containing 5.5 mM (boxes 1 and 2) or 33.3 mM glucose (boxes 3 and 4). (d) Double immunostaining for NF-κB (p65) in red (boxes 1 and 3) and insulin in green (boxes 2 and 4) in human islets exposed for 44 hours to media containing 5.5 mM (boxes 1 and 2) or 33.3 mM glucose (boxes 3 and 4). The arrows mark β cell nuclei stained positive for NF-κB. Magnification: ×750. (e) RT-PCR detection and quantification of Fas mRNA expression. Total RNA was isolated from human islets cultured for 44 hours in medium containing 5.5 or 33.3 mM glucose alone or in the presence of IL-1Ra. In the LightCycler quantitative PCR system, the level of Fas expression was normalized against GAPDH, and the results were expressed as mRNA levels relative to control incubations at 5.5 mM. Results are shown as mean ± SEM of six independent experiments from six donors. *P < 0.05 compared with islets cultured in 5.5 mM glucose alone. **P < 0.05 compared with islets cultured in 33.3 mM glucose. (f) Double immunostaining for Fas in red (boxes 1, 3, 5, 7, 9, and 11) and insulin in green (boxes 2, 4, 6, 8, 10, and 12) in human islets exposed for 4 days to media containing 5.5 mM glucose without IL-1β (boxes 1 and 2), with IL-1β alone (boxes 3 and 4), or with IL-1Ra (boxes 5 and 6) or 33.3 mM glucose without (boxes 7 and 8) and with IL-1Ra (boxes 9 and 10) or IL-1β (boxes 11 and 12). Magnification: ×250. (g) Human islets were cultured for 4 days in 5.5 and 33.3 mM glucose alone or in the presence of IL-1β and/or IL-1Ra, or (h) with and without PDTC. Results are mean ± SEM of the percentage of TUNEL-positive β cells. The mean number of islets scored from each donor was 49 (range 35–63) for each treatment condition. Islets were isolated from five organ donors. *P < 0.01 relative to islets cultured in 5.5 mM glucose. **P < 0.01 relative to islets cultured in 33.3 mM glucose. +P < 0.01 relative to islets cultured in 5.5 mM glucose plus IL-1β.
Next, the ability of IL-1Ra to protect against glucose-induced β cell apoptosis was evaluated. Exposure of human islets cultured on extracellular matrix–coated dishes to elevated glucose concentrations increased the number of nuclei displaying DNA fragmentation (TUNEL-positive nuclei) (Figure 4, g and h). Exposure of islet cultures for 4 days to increasing glucose concentrations (from 5.5 to 33.3 mM) did not, however, lead to propidium iodide uptake, thereby excluding necrosis (not shown). IL-1Ra did not significantly change baseline apoptosis at 5.5 mM glucose. However, IL-1Ra protected the β cells from apoptosis induced by 33.3 mM glucose (Figure 4g). Similarly, the NF-κB inhibitor PDTC inhibited glucose-induced DNA fragmentation (Figure 4h). Addition of exogenous FasL did not lead to a significant increase of β cell death (fold increase of 2.8 ± 0.4 of TUNEL-positive β cells in 33.3 mM glucose alone, and 3.0 ± 0.4 in 33 mM glucose plus exogenous FasL, compared with control in 5.5 mM glucose). Finally, we examined whether high glucose induces iNOS expression. Neither 33 mM glucose nor IL-1β alone induced iNOS mRNA (Figure 5) or protein expression (not shown).
Figure 5.
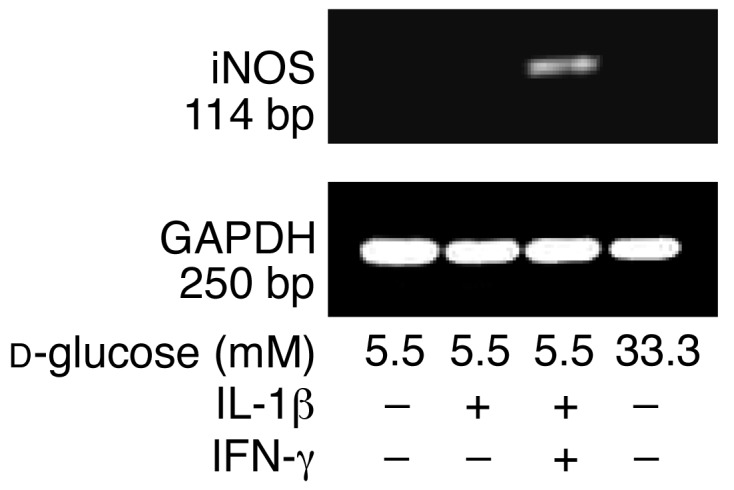
Failure of glucose and IL-1β to induce iNOS mRNA expression in human islets. RT-PCR analysis of iNOS expression by islets cultured for 44 hours in 5.5 and 33.3 mM glucose or with IL-1β alone or in combination with IFN-γ (positive control). GAPDH was used as control. One of three experiments from three donors is shown.
IL-1Ra and PDTC improve impaired β cell function due to IL-1β–mediated glucotoxicity.
Chronic exposure of human islets to 33.3 mM glucose or IL-1β for 4 days abolished acute glucose-stimulated insulin release (Figure 6, a and c). Coincubation with IL-1Ra or PDTC partially restored the glucose stimulation. Insulin content of islets cultured in high glucose was decreased compared with control (5.5 mM glucose) and remained unaffected by IL-1Ra (Figure 6b).
Figure 6.

IL-1Ra and PDTC restore glucose-stimulated insulin secretion in human islets exposed to high glucose. Islets were cultured on extracellular matrix–coated dishes for 4 days in 5.5 and 33.3 mM glucose (control) or with IL-1Ra, PDTC, and IL-1β in combination or individually. (a and c) Basal and stimulated insulin secretion during successive 1-hour incubations at 3.3 (basal) and 16.7 (stimulated) mM glucose following the 4-day culture period. (b) Insulin content. Data are represented as mean ± SEM of three experiments from three separate donors. In each experiment, the data were collected from three plates per treatment. *P < 0.01 compared with islets cultured in 5.5 mM glucose alone. **P < 0.01 compared with islets cultured in 33.3 mM glucose alone. ***P < 0.01 compared with islets cultured in 5.5 mM glucose plus IL-1β.
Discussion
In advanced stages of type 2 diabetes, β cell function often degenerates to such a degree that insulin therapy becomes necessary. The islet demise in humans with late type 2 diabetes is probably due to a combination of genetic and environmental components as well as secondary events including hyperglycemia-induced impairment of β cell function and apoptosis. The hallmark of type 1 diabetes is a specific and massive destruction of the β cells, mostly by apoptosis. Although both diseases have fundamental etiological differences, increasing evidence links both types of diabetes, and a significant fraction of individuals originally diagnosed with type 2 diabetes are cryptic type 1 diabetics or evolve with time to a type 1 state and exhibit anti–β cell autoimmunity (40–43). Moreover, apoptotic cells can provoke an immune response under the appropriate conditions, for example, when present in high enough numbers or exposed to cytokines such as IL-1β (44, 45). Thus, induction of IL-1β by elevated glucose concentrations as shown in the present study or as a part of an autoimmune response may connect type 2 and type 1 diabetes.
Resident islet macrophages are fundamental in the development of autoimmune diabetes (46, 47). Activation of resident macrophages results in the expression and release of IL-1β (48). Recently, it has been shown that viral replicative intermediate double-stranded RNA stimulates rat islet β cell production of IL-1, a mechanism by which viral infection may mediate β cell damage in autoimmune diabetes (49). Interestingly, this same study revealed that, following such stimulation, rat β cells also produce IL-1β–converting enzyme, the enzyme required for activation of IL-1β. The results presented in this study indicate that human β cells themselves are capable of producing IL-1β independently of any viral infection or immune-mediated process, in response to glucose. The fact that IL-1β–producing β cells were also detected in tissue sections of type 2 diabetic patients and in hyperglycemic but not euglycemic Psammomys obesus fully supports the in vitro observations and the central hypothesis of this study.
Diabetes-prone Psammomys obesus fed a high-energy diet develop diabetes. As in some humans with type 2 diabetes, initial hyperinsulinemia in this animal model of diabetes progresses to hypoinsulinemia at later stages of the disease due to insufficient insulin secretion and reduced pancreatic insulin reserves (50). In a longitudinal study, we analyzed β cell turnover during nutrition-induced diabetes (8). During the development of hyperglycemia, an initial and transient increase in β cell replication occurs, followed by a prolonged increase in the number of apoptotic β cells. We now extend this study by the observation of IL-1β–expressing β cells during insulin depletion in islets of hyperglycemic Psammomys obesus. Normalization of plasma glucose by treatment of the animals with phlorizin prevented IL-1β expression and restored insulin expression in the islets, suggesting that in addition to its role as a mediator of glucose-induced β cell apoptosis, IL-1β may participate in control of pancreatic insulin reserves.
To the best of our knowledge, iNOS cannot be induced by IL-1β alone in human islets (51, 52). Moreover, cytokine-induced Fas expression is NO-independent (53, 54). In line with these reports, in the present study glucose did not induce iNOS. Therefore, the IL-1β–mediated deleterious effects are probably independent of NO. However, we can not exclude that NO is produced by β cells in an iNOS-independent way, since IL-1β–induced nitrite production has been demonstrated in the past (24, 25, 55), although this has not been observed by others (56).
IL-1β stimulates I-κB degradation and NF-κB nuclear localization in a time-dependent manner that is maximal following 20–30 minutes of exposure (31). Therefore, it is not clear how NF-κB activation remains detectable following a 44-hour incubation in high glucose. The prolonged period of activation may be related to the interplay of all factors required for NF-κB activation. Indeed, we show that high glucose induced p65 expression and decreased I-κB. Moreover, other modulators of IL-1β–induced NF-κB activation, such as endogenous IL-1Ra, may interfere. All these factors may influence the time course of glucose-induced NF-κB activation. Regardless, at least 20 hours of exposure to high glucose is required to induce IL-1β, leading to NF-κB activation, Fas expression, and β cell death. This is in line with glucotoxic effects, which appear only after prolonged exposure to high glucose.
Inhibition of NF-κB activation by an adenoviral vector encoding the repressor I-κB protects human islets from Fas-triggered apoptosis and results in normal insulin response in the presence of IL-1β (55). Similarly, in purified rat β cells, inhibition of cytokine-induced NF-κB activation prevents β cell apoptosis (57). Thus, the present finding that glucose decreases I-κB expression and induces NF-κB activation via IL-1β opens the path to prevent glucotoxic effects by inhibition of NF-κB activation.
So far, IL-1β production and release by islets has been considered to be limited to type 1 diabetes. Here we demonstrate that high concentrations of glucose induce IL-1β production and secretion in human β cells, leading to Fas receptor upregulation, NF-κB activation, β cell apoptosis, and dysfunction. Moreover, we observed IL-1β–producing β cells in diabetic patients and diabetic Psammomys obesus. The pathway by which hyperglycemia causes impairment and loss of insulin-producing cells thus shares features with immune-mediated processes. It follows that the proinflammatory cytokine IL-1β may be a crucial factor contributing to β cell glucotoxicity in the pathogenesis of type 2 diabetes.
Acknowledgments
This work was supported by Swiss National Science Foundation grants 3200-067049.01 (M.Y. Donath) and 3200-061776.00 (P.A. Halban), and by Juvenile Diabetes Research Foundation grants 4-1999-844 (P.A. Halban) and I-1998-9 (N. Kaiser). M.Y. Donath is supported by the Max Cloetta Foundation. We thank G. Schoedon for helpful suggestions, P.U. Heitz, P. Komminoth, and P. Saremaslani for providing the sections of human pancreas, and G. Siegfried-Kellenberger and C.S. Manzano for technical assistance.
Footnotes
Pavel Sergeev and Frédéric Ris contributed equally to this work.
Conflict of interest: No conflict of interest has been declared.
Nonstandard abbreviations used: IL-1 receptor antagonist (IL-1Ra); Fas ligand (FasL); pyrrolidinedithiocarbamate (PDTC); inducible nitric oxide synthase (iNOS); inhibitory κB (I-κB).
References
- 1.Unger RH, Grundy S. Hyperglycaemia as an inducer as well as a consequence of impaired islet cell function and insulin resistance: implications for the management of diabetes. Diabetologia. 1985;28:119–121. doi: 10.1007/BF00273856. [DOI] [PubMed] [Google Scholar]
- 2.Kaiser N, Corcos AP, Sarel I, Cerasi E. Monolayer culture of adult rat pancreatic islets on extracellular matrix: modulation of B-cell function by chronic exposure to high glucose. Endocrinology. 1991;129:2067–2076. doi: 10.1210/endo-129-4-2067. [DOI] [PubMed] [Google Scholar]
- 3.Leahy JL, Cooper HE, Deal DA, Weir GC. Chronic hyperglycemia is associated with impaired glucose influence on insulin secretion. A study in normal rats using chronic in vivo glucose infusions. J Clin Invest. 1986;77:908–915. doi: 10.1172/JCI112389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson RP. Type II diabetes, glucose “non-sense,” and islet desensitization. Diabetes. 1989;38:1501–1505. doi: 10.2337/diab.38.12.1501. [DOI] [PubMed] [Google Scholar]
- 5.Rossetti L, Giaccari A, DeFronzo RA. Glucose toxicity. Diabetes Care. 1990;13:610–630. doi: 10.2337/diacare.13.6.610. [DOI] [PubMed] [Google Scholar]
- 6.Eizirik DL, Korbutt GS, Hellerstrom C. Prolonged exposure of human pancreatic islets to high glucose concentrations in vitro impairs the beta-cell function. J Clin Invest. 1992;90:1263–1268. doi: 10.1172/JCI115989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marshak S, et al. Impaired beta-cell functions induced by chronic exposure of cultured human pancreatic islets to high glucose. Diabetes. 1999;48:1230–1236. doi: 10.2337/diabetes.48.6.1230. [DOI] [PubMed] [Google Scholar]
- 8.Donath MY, Gross DJ, Cerasi E, Kaiser N. Hyperglycemia-induced beta-cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes. 1999;48:738–744. doi: 10.2337/diabetes.48.4.738. [DOI] [PubMed] [Google Scholar]
- 9.Maedler K, et al. Glucose induces b-cell apoptosis via upregulation of the Fas-receptor in human islets. Diabetes. 2001;50:1683–1690. doi: 10.2337/diabetes.50.8.1683. [DOI] [PubMed] [Google Scholar]
- 10.Federici M, et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: a potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes. 2001;50:1290–1301. doi: 10.2337/diabetes.50.6.1290. [DOI] [PubMed] [Google Scholar]
- 11.Efanova IB, et al. Glucose and tolbutamide induce apoptosis in pancreatic beta-cells. A process dependent on intracellular Ca2+ concentration. J Biol Chem. 1998;273:33501–33507. doi: 10.1074/jbc.273.50.33501. [DOI] [PubMed] [Google Scholar]
- 12.Maedler K, et al. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes. 2001;50:69–76. doi: 10.2337/diabetes.50.1.69. [DOI] [PubMed] [Google Scholar]
- 13.Tajiri Y, Moller C, Grill V. Long-term effects of aminoguanidine on insulin release and biosynthesis: evidence that the formation of advanced glycosylation end products inhibits B cell function. Endocrinology. 1997;138:273–280. doi: 10.1210/endo.138.1.4851. [DOI] [PubMed] [Google Scholar]
- 14.Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest. 1992;90:320–325. doi: 10.1172/JCI115865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robertson RP, Olson LK, Zhang HJ. Differentiating glucose toxicity from glucose desensitization: a new message from the insulin gene. Diabetes. 1994;43:1085–1089. doi: 10.2337/diab.43.9.1085. [DOI] [PubMed] [Google Scholar]
- 16.Mandrup-Poulsen T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia. 1996;39:1005–1029. doi: 10.1007/BF00400649. [DOI] [PubMed] [Google Scholar]
- 17.Mandrup-Poulsen T, Bendtzen K, Nielsen JH, Bendixen G, Nerup J. Cytokines cause functional and structural damage to isolated islets of Langerhans. Allergy. 1985;40:424–429. doi: 10.1111/j.1398-9995.1985.tb02681.x. [DOI] [PubMed] [Google Scholar]
- 18.Bendtzen K, et al. Cytotoxicity of human pI 7 interleukin-1 for pancreatic islets of Langerhans. Science. 1986;232:1545–1547. doi: 10.1126/science.3086977. [DOI] [PubMed] [Google Scholar]
- 19.Spinas GA, et al. Interleukin 1 dose-dependently affects the biosynthesis of (pro)insulin in isolated rat islets of Langerhans. Diabetologia. 1987;30:474–480. doi: 10.1007/BF00279615. [DOI] [PubMed] [Google Scholar]
- 20.Mandrup-Poulsen T, et al. Affinity-purified human interleukin I is cytotoxic to isolated islets of Langerhans. Diabetologia. 1986;29:63–67. doi: 10.1007/BF02427283. [DOI] [PubMed] [Google Scholar]
- 21.Spinas GA, et al. Low concentrations of interleukin-1 stimulate and high concentrations inhibit insulin release from isolated rat islets of Langerhans. Acta Endocrinol (Copenh) 1986;113:551–558. doi: 10.1530/acta.0.1130551. [DOI] [PubMed] [Google Scholar]
- 22.Yamada K, et al. Mouse islet cell lysis mediated by interleukin-1-induced Fas. Diabetologia. 1996;39:1306–1312. doi: 10.1007/s001250050574. [DOI] [PubMed] [Google Scholar]
- 23.Corbett JA, Lancaster JR, Jr, Sweetland MA, McDaniel ML. Interleukin-1 beta-induced formation of EPR-detectable iron-nitrosyl complexes in islets of Langerhans. Role of nitric oxide in interleukin-1 beta-induced inhibition of insulin secretion. J Biol Chem. 1991;266:21351–21354. [PubMed] [Google Scholar]
- 24.Giannoukakis N, et al. Adenoviral gene transfer of the interleukin-1 receptor antagonist protein to human islets prevents IL-1beta-induced beta-cell impairment and activation of islet cell apoptosis in vitro. Diabetes. 1999;48:1730–1736. doi: 10.2337/diabetes.48.9.1730. [DOI] [PubMed] [Google Scholar]
- 25.Giannoukakis N, et al. Prevention of beta cell dysfunction and apoptosis activation in human islets by adenoviral gene transfer of the insulin-like growth factor I. Gene Ther. 2000;7:2015–2022. doi: 10.1038/sj.gt.3301333. [DOI] [PubMed] [Google Scholar]
- 26.Loweth AC, Williams GT, James RF, Scarpello JH, Morgan NG. Human islets of Langerhans express Fas ligand and undergo apoptosis in response to interleukin-1beta and Fas ligation. Diabetes. 1998;47:727–732. doi: 10.2337/diabetes.47.5.727. [DOI] [PubMed] [Google Scholar]
- 27.Rabinovitch A, Sumoski W, Rajotte RV, Warnock GL. Cytotoxic effects of cytokines on human pancreatic islet cells in monolayer culture. J Clin Endocrinol Metab. 1990;71:152–156. doi: 10.1210/jcem-71-1-152. [DOI] [PubMed] [Google Scholar]
- 28.Stassi G, et al. Nitric oxide primes pancreatic beta cells for Fas-mediated destruction in insulin-dependent diabetes mellitus. J Exp Med. 1997;186:1193–1200. doi: 10.1084/jem.186.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon G, Corbett JA, Rodi CP, Sullivan P, McDaniel ML. Interleukin-1 beta-induced nitric oxide synthase expression by rat pancreatic beta-cells: evidence for the involvement of nuclear factor kappa B in the signaling mechanism. Endocrinology. 1995;136:4790–4795. doi: 10.1210/endo.136.11.7588208. [DOI] [PubMed] [Google Scholar]
- 30.Darville MI, Eizirik DL. Cytokine induction of Fas gene expression in insulin-producing cells requires the transcription factors NF-kappaB and C/EBP. Diabetes. 2001;50:1741–1748. doi: 10.2337/diabetes.50.8.1741. [DOI] [PubMed] [Google Scholar]
- 31.Flodstrom M, Welsh N, Eizirik DL. Cytokines activate the nuclear factor kappa B (NF-kappa B) and induce nitric oxide production in human pancreatic islets. FEBS Lett. 1996;385:4–6. doi: 10.1016/0014-5793(96)00337-7. [DOI] [PubMed] [Google Scholar]
- 32.Linetsky E, et al. Improved human islet isolation using a new enzyme blend, liberase. Diabetes. 1997;46:1120–1123. doi: 10.2337/diab.46.7.1120. [DOI] [PubMed] [Google Scholar]
- 33.Oberholzer J, et al. Human islet transplantation: lessons from 13 autologous and 13 allogeneic transplantations. Transplantation. 2000;69:1115–1123. doi: 10.1097/00007890-200003270-00016. [DOI] [PubMed] [Google Scholar]
- 34.Ricordi C, Lacy PE, Finke EH, Olack BJ, Scharp DW. Automated method for isolation of human pancreatic islets. Diabetes. 1988;37:413–420. doi: 10.2337/diab.37.4.413. [DOI] [PubMed] [Google Scholar]
- 35.Kaiser N, Corcos AP, Sarel I, Cerasi E. Monolayer culture of adult rat pancreatic islets on extracellular matrix: modulation of B-cell function by chronic exposure to high glucose. Endocrinology. 1991;129:2067–2076. doi: 10.1210/endo-129-4-2067. [DOI] [PubMed] [Google Scholar]
- 36.Jodo S, et al. Apoptosis-inducing membrane vesicles. A novel agent with unique properties. J Biol Chem. 2001;276:39938–39944. doi: 10.1074/jbc.M107005200. [DOI] [PubMed] [Google Scholar]
- 37.Schneemann M, et al. Nitric oxide synthase is not a constituent of the antimicrobial armature of human mononuclear phagocytes. J Infect Dis. 1993;167:1358–1363. doi: 10.1093/infdis/167.6.1358. [DOI] [PubMed] [Google Scholar]
- 38.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–6874. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- 40.Mathis D, Vence L, Benoist C. beta-Cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 41.Pietropaolo M, Barinas-Mitchell E, Pietropaolo SL, Kuller LH, Trucco M. Evidence of islet cell autoimmunity in elderly patients with type 2 diabetes. Diabetes. 2000;49:32–38. doi: 10.2337/diabetes.49.1.32. [DOI] [PubMed] [Google Scholar]
- 42.Rowley MJ, Mackay IR, Chen QY, Knowles WJ, Zimmet PZ. Antibodies to glutamic acid decarboxylase discriminate major types of diabetes mellitus. Diabetes. 1992;41:548–551. doi: 10.2337/diab.41.4.548. [DOI] [PubMed] [Google Scholar]
- 43.Wilkin TJ. The accelerator hypothesis: weight gain as the missing link between Type I and Type II diabetes. Diabetologia. 2001;44:914–922. doi: 10.1007/s001250100548. [DOI] [PubMed] [Google Scholar]
- 44.Bellone M, et al. Processing of engulfed apoptotic bodies yields T cell epitopes. J Immunol. 1997;159:5391–5399. [PubMed] [Google Scholar]
- 45.Trudeau JD, et al. Neonatal beta-cell apoptosis: a trigger for autoimmune diabetes? Diabetes. 2000;49:1–7. doi: 10.2337/diabetes.49.1.1. [DOI] [PubMed] [Google Scholar]
- 46.Lacy PE, Finke EH. Activation of intraislet lymphoid cells causes destruction of islet cells. Am J Pathol. 1991;138:1183–1190. [PMC free article] [PubMed] [Google Scholar]
- 47.Lacy PE. The intraislet macrophage and type I diabetes. Mt Sinai J Med. 1994;61:170–174. [PubMed] [Google Scholar]
- 48.Arnush M, Scarim AL, Heitmeier MR, Kelly CB, Corbett JA. Potential role of resident islet macrophage activation in the initiation of autoimmune diabetes. J Immunol. 1998;160:2684–2691. [PubMed] [Google Scholar]
- 49.Heitmeier MR, Arnush M, Scarim AL, Corbett JA. Pancreatic beta-cell damage mediated by beta-cell production of IL-1: a novel mechanism for virus-induced diabetes. J Biol Chem. 2001;276:11151–11158. doi: 10.1074/jbc.M009159200. [DOI] [PubMed] [Google Scholar]
- 50.Gadot M, et al. Hyperproinsulinemia and insulin deficiency in the diabetic Psammomys obesus. Endocrinology. 1994;135:610–616. doi: 10.1210/endo.135.2.8033810. [DOI] [PubMed] [Google Scholar]
- 51.Chen MC, Proost P, Gysemans C, Mathieu C, Eizirik DL. Monocyte chemoattractant protein-1 is expressed in pancreatic islets from prediabetic NOD mice and in interleukin-1 beta-exposed human and rat islet cells. Diabetologia. 2001;44:325–332. doi: 10.1007/s001250051622. [DOI] [PubMed] [Google Scholar]
- 52.Eizirik DL, Darville MI. beta-cell apoptosis and defense mechanisms: lessons from type 1 diabetes. Diabetes. 2001;50:S64–S69. doi: 10.2337/diabetes.50.2007.s64. [DOI] [PubMed] [Google Scholar]
- 53.Liu D, et al. Cytokines induce apoptosis in beta-cells isolated from mice lacking the inducible isoform of nitric oxide synthase (iNOS–/–) Diabetes. 2000;49:1116–1122. doi: 10.2337/diabetes.49.7.1116. [DOI] [PubMed] [Google Scholar]
- 54.Zumsteg U, Frigerio S, Hollander GA. Nitric oxide production and Fas surface expression mediate two independent pathways of cytokine-induced murine beta-cell damage. Diabetes. 2000;49:39–47. doi: 10.2337/diabetes.49.1.39. [DOI] [PubMed] [Google Scholar]
- 55.Giannoukakis N, Rudert WA, Trucco M, Robbins PD. Protection of human islets from the effects of interleukin-1beta by adenoviral gene transfer of an Ikappa B repressor. J Biol Chem. 2000;275:36509–36513. doi: 10.1074/jbc.M005943200. [DOI] [PubMed] [Google Scholar]
- 56.Corbett JA, Sweetland MA, Wang JL, Lancaster JR, Jr, McDaniel ML. Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of Langerhans. Proc Natl Acad Sci USA. 1993;90:1731–1735. doi: 10.1073/pnas.90.5.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heimberg H, et al. Inhibition of cytokine-induced NF-kappaB activation by adenovirus-mediated expression of a NF-kappaB super-repressor prevents beta-cell apoptosis. Diabetes. 2001;50:2219–2224. doi: 10.2337/diabetes.50.10.2219. [DOI] [PubMed] [Google Scholar]