

Cholesterol has long been known to function as both a structural lipid and a precursor molecule for bile acid and steroid hormone synthesis. In addition, this ubiquitous lipid is now known to contribute fundamentally to the development and function of the CNS and the bones, and, as detailed in other articles in this Perspective series, it plays major roles in signal transduction, sperm development, and embryonic morphogenesis. Over the past decade, the identification of multiple congenital anomaly/mental retardation syndromes due to inborn errors of cholesterol synthesis has underscored the importance of cholesterol synthesis in normal development.
The prototypical example of a human malformation syndrome that results from a defect in cholesterol synthesis is the RSH/Smith-Lemli-Opitz syndrome (SLOS). To date, five additional human syndromes resulting from impaired cholesterol synthesis have been described. These include desmosterolosis, X-linked dominant chondrodysplasia punctata type 2 (CDPX2), CHILD syndrome (congenital hemidysplasia with ichthyosiform erythroderma/nevus and limb defects), Greenberg dysplasia, and, most recently, Antley-Bixler syndrome. Natural mouse mutations corresponding to CDPX2 (tattered) and CHILD syndrome (bare patches and striated) have been identified, and mouse models corresponding to SLOS and lathosterolosis have been produced by gene disruption. Identification of the biochemical defects present in these disorders has given insight into the role that cholesterol plays in normal embryonic development, has provided the initial step in understanding the pathophysiological processes underlying these malformation syndromes, and has given rise to treatment protocols for patients with SLOS.
Cholesterol is synthesized from lanosterol, the first sterol in the cholesterol synthesis pathway, via a series of enzymatic reactions shown in Figure 1. These include the demethylation at C4α, C4β, and C14, which converts the C30 molecule lanosterol to C27 cholesterol; isomerization of the Δ8(9) double bond to a Δ7 double bond; desaturation to form a Δ5 double bond; and finally, reduction of Δ14, Δ24, and Δ7 double bonds (Figure 1). Analyses of human and murine syndromes resulting from cholesterol synthetic defects have helped illuminate both the normal functions of cholesterol and a range of normal and teratogenic functions of the various precursor sterols that accumulate in these disorders. Here, I consider the clinical, molecular, biochemical, and developmental aspects of these disorders, focusing on the five malformation syndromes presented in Table 1.
Figure 1.

Cholesterol synthesis pathway and human malformation syndromes. The sterol ring structure and carbon position numbering are shown at the top of the figure. Human malformation syndromes are in boldface type. Cholesterol is synthesized from squalene in a series of enzymatic reactions denoted by numbers: 1, squalene monooxygenase and squalene cyclase; 2, lanosterol 14-α-demethylase; 3, 3β-hydroxysterol Δ14-reductase; 4, C4 demethylation complex (C4-sterol methyloxidase, C4-sterol decarboxylase [NSDHL], and 3-ketoreductase); 5, 3β-hydroxysterol Δ8,Δ7-isomerase; 6, lathosterol 5-desaturase; 7, 3β-hydroxysterol Δ7-reduct-ase; 8, 3β-hydroxysterol Δ24-reductase.
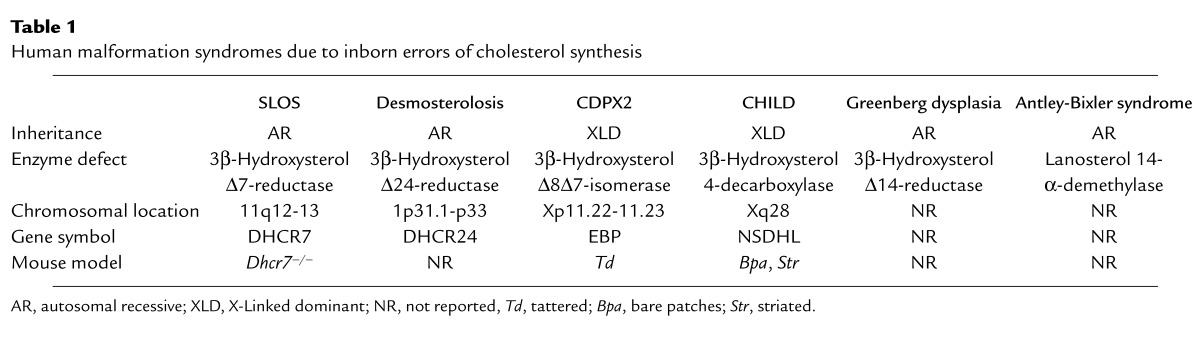
Table 1.
Human malformation syndromes due to inborn errors of cholesterol synthesis
Inborn errors of cholesterol metabolism
Smith-Lemli-Opitz syndrome.
The Smith-Lemli-Opitz syndrome (SLOS; MIM no. 270400; see refs. 1–3 for detailed reviews), the prototypical example of a human malformation syndrome resulting from an inborn error of cholesterol synthesis, was first described in 1964. Smith, Lemli, and Opitz initially described three male patients with similar facial features, mental retardation, microcephaly, developmental delay, and hypospadias, and they designated the novel disorder as the RSH syndrome, referring to the names of the first three patients. In various populations, the clinical incidence of this condition has been reported to range from 1 in 10,000 to 1 in 60,000, although SLOS is thought to be more common in individuals of Northern European descent.
SLOS, along with a severe variant (type II SLOS, or Rutledge lethal multiple congenital anomaly syndrome; MIM no. 268670), is now known to represent a single, clinically heterogeneous genetic disorder. Infants at the severe end of the SLOS phenotypic spectrum often die due to multiple major congenital anomalies. Conversely, individuals at the mild end of the spectrum show only minor physical stigmata, coupled with behavioral problems that can include aspects of autism (4) and self-injurious behavior. Although near-normal intelligence has been reported, moderate to severe mental retardation is typical. The classical SLOS face is distinctive (Figure 2, a–c): Typical craniofacial features include microcephaly, metopic prominence, ptosis, a small upturned nose, cleft palate, broad alveolar ridges, and micrognathia, with cataracts developing pre- or postnatally. Limb abnormalities are frequent in SLOS, and syndactyly of the second and third toes is the most frequent single physical finding (Figure 2e). Because the SLOS phenotypic spectrum is so broad, the presence of second-third toe syndactyly in a child with significant mental or behavioral problems should prompt consideration of SLOS. Other limb anomalies include short and proximally placed thumbs, single palmar creases, and postaxial polydactyly of either the upper or lower extremities (Figure 2d). Congenital heart defects are common. Slow growth and poor weight gain are typical. Most SLOS infants are poor feeders, and gastrostomy tube placement is required in many cases. Gastrointestinal anomalies include colonic aganglionosis, pyloric stenosis, and malrotation. Genital malformations are common in male patients. Hypospadius is a typical finding, and more severely affected patients may have ambiguous genitalia (Figure 2f).
Figure 2.
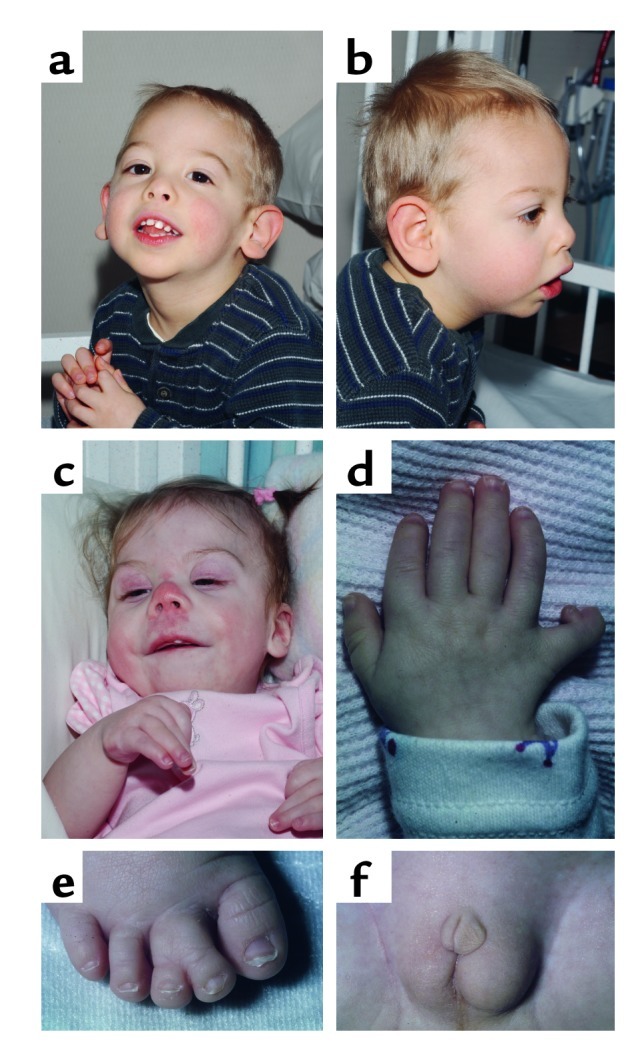
Phenotypic features of SLOS. (a–c) Facial appearance including microcephaly, ptosis, a small upturned nose, and micrognathia. (d) Hand malformations including postaxial polydactyly and a short thumb. (e) Second-third toe syndactyly. (f) Ambiguous genitalia in a 46, XY patient.
In 1993, Irons and coworkers (5) reported decreased serum levels of cholesterol and elevated levels of 7-dehydrocholesterol (7-DHC) in two SLOS patients. The conversion of 7-DHC to cholesterol, the last enzymatic reaction in the Kandutsch-Russel cholesterol synthetic pathway, is catalyzed by the 3β-hydroxysterol Δ7-reductase (Figure 1). In 1998, three groups, including ours (6), independently cloned the human 3β-hydroxysterol Δ7-reductase gene (DHCR7), which encodes this enzyme, and identified DHCR7 mutations in SLOS patients. The DHCR7 protein is predicted to be a 475–amino acid polypeptide integral membrane protein with up to nine transmembrane domains. To date, 79 SLOS disease alleles have been identified in DHCR7, the most frequent of which is IVS8-1G→C (32%). This splice acceptor mutation, which results in the inclusion of 134 bp of intronic sequence in the DHCR7 mRNA, is a null allele, and IVS8-1G→C homozygotes typically have a severe phenotype. Other relatively common alleles include T93M (9%), W151X (7%), V326L (6%), R404C (5%), and R352W (3%). Establishing a genotype/phenotype correlation for SLOS has been confounded by the large number of different alleles and the fact that most patients are compound heterozygotes. The observation that patients with the same DHCR7 genotype may have markedly different phenotypic severity suggests that other genetic, developmental, or maternal factors, perhaps affecting cholesterol biosynthesis or homeostasis, significantly influence a given patient’s phenotype.
The IVS8-1G→C allele has been reported to have a carrier frequency of 1.06% (16 in 1,503) in an Oregon population (7). A 1% carrier frequency for the IVS8-1G→C mutation predicts an IVS8-1G→C homozygosity incidence of 1 in 40,000. Since the IVS8-1G→C mutation accounts for a third of the ascertained mutant DHCR7 alleles, this finding has been used to predict carrier frequencies of 1 in 30 and disease incidences on the order of 1 in 1,590 to 1 in 13,500 (7). This predicted disease incidence is much higher than clinical data would indicate, suggesting that a substantial number of severely affected fetuses with two severe alleles may be lost prenatally. An alternative explanation, that not all SLOS patients are currently being ascertained, could be addressed in a newborn screening trial. Indeed, given the availability of a therapeutic intervention, such a screen should be considered.
The high carrier frequency of the IVS8-1G→C mutation has led to speculation that heterozygotes are at a competitive advantage. Decreased cardiac or thromboembolic disease has been proposed as a possible advantage but seems unlikely, since these disorders usually affect individuals after their reproductive years. However, since many viruses rely on a membrane fusion event to infect cells, it is plausible that the presence of low levels of 7-DHC in the plasma membrane could protect the host from some viral diseases, a possibility that has yet to be tested in mouse models or human cell lines. In addition, since 7-DHC in the skin is the precursor for vitamin D synthesis, increased 7-DHC levels in heterozygotes could be adaptive in a Northern European population at risk of vitamin D–deficient rickets.
Dietary cholesterol supplementation has been attempted in SLOS, but the efficacy of this approach is probably limited by inefficient cholesterol transport across the blood-brain barrier and by the inability to reverse prior developmental effects of cholesterol deprivation. Biochemically, cholesterol supplementation leads to an improved plasma cholesterol/total sterol ratio, and prolonged therapy can even decrease plasma 7-DHC levels. Observational studies have reported multiple benefits of dietary cholesterol supplementation, including improved nutrition and growth, improved muscle tone and strength, decreased photosensitivity, decreased irritability, decreased tactile defensiveness, increased sociability and alertness, decreased self-injurious behavior, and decreased aggressiveness. To date, a controlled, blinded trial of dietary cholesterol supplementation confirming these observations has not been published. In a preliminary statement on one blinded trial, Kelley (3) reported that he found no difference between supplementation with 50 mg/kg/d and with 150 mg/kg/d. Since 50 mg/kg/d exceeds daily cholesterol needs in children, the clinical effect may already be maximal at this dose. Although dietary cholesterol supplementation has no effect on fixed developmental malformations, it does appear to improve the overall health status of these patients and to lessen the behavioral problems associated with this disorder.
Other treatment modalities have been considered as well. Bile acid supplementation has not shown a clear benefit. Fresh frozen plasma, a source of lipoproteins containing cholesterol, has been used for prenatal therapy and appears to be efficacious in the acute management of ill SLOS patients. Inhibition of HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis, has been suggested as a means to reduce any toxic effects of 7-DHC. However, in a teratogenic rat model of SLOS, one inhibitor of this enzyme decreased cholesterol levels without decreasing 7-DHC levels (8). This animal study, combined with prior evidence that these drugs can precipitate an acute metabolic crisis in mevalonic aciduria patients and with anecdotal reports of poor outcomes in SLOS patients treated with HMG-CoA reductase inhibitors, led to the concern that inhibition of endogenous cholesterol synthesis in SLOS could be detrimental.
Nevertheless, one small trial of simvastatin therapy in SLOS has been reported. Jira et al. (9) treated two SLOS patients with simvastatin, and they reported decreased dehydrocholesterol levels, improved serum dehydrocholesterol/cholesterol ratios in serum and cerebral spinal fluid (CSF), and improved growth. Curiously, they also observed a paradoxical increase in plasma cholesterol levels. This surprising finding might be explained by upregulation of the mutant DHCR7. Because statins induce transcription of HMG-CoA reductase and many genes involved in cholesterol synthesis are coordinately regulated, simvastatin treatment may well have induced the synthesis of the 3β-hydroxysterol Δ7-reductase protein in these patients. If a mutant allele encodes a protein with residual enzymatic function, such an increase in protein levels could result in increased conversion of 7-DHC to cholesterol. Conversely, simvastatin upregulation of a null allele would clearly not increase the conversion of 7-DHC to cholesterol, but it could inhibit the endogenous sterol synthesis. Since 7-DHC appears to substitute for cholesterol in some cellular functions, the net effect of this situation may be detrimental.
Defining the mechanism of action of the HMG-CoA reductase inhibitors in SLOS will be important for safely designing a clinical trial. If upregulation of a partially functioning enzyme is the mechanism of the observed effect, initial trials should be limited to patients who can be demonstrated to have significant residual enzymatic function. Since simvastatin crosses the blood-brain barrier, it may directly affect the biochemical defect in the CNS and thus, perhaps, the behavioral phenotype of the disease.
Several pharmacological inhibitors of 3β-hydroxysterol Δ7-reductase have been used in studies of SLOS teratogenesis (reviewed in ref. 10). However, these studies are confounded by the effects of these drugs on maternal cholesterol synthesis and on other enzymes involved in cholesterol biosynthesis. Wassif et al. (10) produced a genetic mouse model of SLOS by disruption of Dhcr7. As found in human patients, Dhcr7–/– pups have markedly reduced tissue cholesterol levels and increased 7-DHC levels. Phenotypic overlap between this mouse model and the human syndrome includes intrauterine growth retardation; variable craniofacial malformations, including cleft palate; poor feeding with an abnormal suck; and neurological abnormalities, including apparent hypotonia, apparent weakness, and decreased movement.
Desmosterolosis.
As shown in Figure 1, two major pathways of cholesterol synthesis operate in parallel. The first uses 7-DHC as the immediate precursor of cholesterol. In the second, the enzyme 3β-hydroxysterol Δ24-reductase reduces the Δ24 double bond in desmosterol to yield cholesterol. Desmosterolosis (MIM no. 602398; see refs. 3, 11 for extensive reviews), an autosomal recessive malformation syndrome resulting from a deficiency of this enzyme, was the second human malformation syndrome shown to arise from an inborn error of cholesterol synthesis.
Since only two cases have been identified, the desmosterolosis phenotype has yet to be fully delineated. The initial patient, identified by Clayton et al. (12), was an infant who died soon after birth. This patient had macrocephaly, craniofacial malformations including cleft palate and a thick alveolar ridge, a congenital heart malformation, hypoplastic lungs, renal hypoplasia, ambiguous genitalia, short limbs, and osteosclerosis. The second patient was a 4-year-old male with marked developmental delay, microcephaly, dysmorphic facial features, and limb malformations (13). Sterol analysis by liquid gas chromatography-mass spectrometry (LGC-MS) showed marked elevations of desmosterol in serum and tissues from both of these patients.
The human 3β-hydroxysterol Δ24-reductase gene (DHCR24), identified and cloned by Waterham et al. (13), encodes a 516–amino acid polypeptide with one predicted transmembrane domain. The first patient had one missense mutation, Y471S, on one allele, and two missense mutations, N294T and K306N, on the second allele. The second patient was homozygous for an E191K mutation. All four missense mutations independently decreased DHCR24 activity, and the combination of N294T with K306N was more severe than either mutation by itself.
X-linked dominant chondrodysplasia punctata type 2.
Chondrodysplasia punctata is a defect of endochondral bone formation characterized by abnormal foci of calcification in cartilaginous elements. In x-rays, chondrodysplasia punctata appears as epiphyseal stippling. X-linked dominant chondroplasia punctata type 2 (CDPX2, also known as Conradi-Hünermann-Happle syndrome or Happle syndrome; MIM no. 302960) was first delineated by Happle (14) in 1979. This X-linked dominant disorder is typically lethal in hemizygous males. Heterozygous females present at birth with generalized congenital ichthyosiform erythroderma, a scaly hyperkeratotic rash in linear and blotchy patterns following the lines of Blaschko, consistent with the effects of X-inactivation. Older patients suffer from ichthyosis, atrophoderma, layered splitting of the nails, trichorrhexis nodosa, and patchy alopecia. Skeletal abnormalities include chondrodysplasia punctata, asymmetrical rhizomelic limb shortening, scoliosis, and hexadactyly. Craniofacial anomalies include an asymmetrical head shape, due to unilateral hypoplasia, and a flat nasal bridge. Asymmetrical and sectorial cataracts are found in CDPX2. Like the distribution of the dermatological findings, the asymmetrical nature of the craniofacial malformations and the cataracts is likely due to differential X-inactivation.
Because chondrodysplasia punctata has been described in SLOS, Kelley et al. (15) investigated the possibility that abnormal sterol synthesis could cause other genetic disorders with chondrodysplasia punctata. This group identified five CDPX2 patients with increased tissue 8-dehydrocholesterol and cholesta-8(9)-en-3β-ol, consistent with a deficiency of 3β-hydroxysterol Δ8,Δ7-isomerase activity (Figure 1). This enzyme was first identified as a high-affinity emopamil-binding protein (EBP) (16), a putative four–transmembrane domain integral membrane protein of 230 amino acids. Mutations of the EBP gene in CDPX2 patients, first reported by Derry et al. (17) and Braverman et al. (18), include a preponderance of presumed null alleles, suggesting that mild missense alleles either are less penetrant or yield a different phenotypic presentation.
In the tattered mouse, a missense mutation (G107R) of Ebp confers an X-linked male-lethality trait (17). Heterozygous females are growth-retarded and have a “tattered” appearance due to hyperkeratotic patches of alopecia. The hemizygous male phenotype includes intrauterine growth retardation, micrognathia, short limbs, retarded ossification, and intestinal agenesis.
CHILD syndrome.
CHILD syndrome (MIM # 308050), an X-linked dominant disorder which consists of congenital hemidysplasia with ichthyosiform erythroderma/nevus and limb defects, overlaps both phenotypically and genetically with CDPX2. This disorder is distinguished by the striking unilateral presentation of ichthyosiform and inflammatory nevus in heterozygous females, although contralateral skin involvement can be present. In 1980, Happle et al. (19) reviewed the CHILD phenotype in 20 cases and found that ipsilateral limb defects range from phalangeal hypoplasia to limb truncation, and ipsilateral skeletal defects may include hypoplasia of the calvaria, mandible, scapula, vertebrae, or ribs. Chondrodysplasia punctata was noted in five of these 20 cases. Congenital heart disease has also been described, as well as ipsilateral renal agenesis and lung hypoplasia. Unlike CDPX2, cataracts are not reported in CHILD syndrome, and the patient’s right side is more often affected than the left. Defects in cholesterol synthesis in patients with CHILD syndrome were simultaneously reported by two groups in 2000. König et al. (20) reported mutations in the NADPH sterol dehydrogenase–like protein gene (NSDHL) in six patients, and Grange et al. (21) reported mutation of the EBP gene in a single patient. The NSDHL protein likely functions in concert with a C4-sterol methyloxidase and a 3-ketoreductase to remove the C4 methyl groups of 4,4-dimethylcholesta-8(9)-en-3β-ol or 4,4-dimethylcholesta-8,24-dien-3β-ol to yield cholesta-8(9)-en-3β-ol or zymosterol, respectively (22). This enzymatic reaction immediately precedes the reaction catalyzed by EBP (Figure 1).
Although the diagnosis of the patient with the EBP mutation has been debated, CHILD syndrome as a clinical diagnosis is distinguished by its predominantly unilateral presentation. This presentation may reflect a disturbance of laterality determination induced by the presence of abnormal sterol metabolites. The function of Hedgehog proteins is affected by abnormal sterol metabolites, and both Sonic hedgehog (Shh) and Indian hedgehog (Ihh) are involved in left-right axis determination (see ref. 23). Although an epigenetic effect of random X-inactivation could lead to a unilateral presentation, this mechanism would not explain why all patients with NSDHL mutations appear to present with CHILD syndrome, whereas only some patients with EBP mutations present with a CHILD phenotype. However, because of the sequential nature of these two inborn errors of cholesterol synthesis, the elevated 4,4-dimethylcholesta-8-en-3β-ol and 4,4-dimethylcholesta-8,24-dien-3β-ol levels that are characteristic of NSDHL mutations could also occur as a result of mutations in EBP. A sterol-induced effect on left-right axis determination, perhaps specific to one of these metabolites, needs to be considered. Accumulation of different precursor sterols in patients with NSDHL or EBP mutations may explain the differences in the dermatological findings between these two syndromes and the development of cataracts in CDPX2 but not in CHILD syndrome.
Mutations of the Nsdhl are found in the bare patches (Bpa) and striated (Str) mice. Bpa and Str are allelic phenotypes with a hypomorphic presentation in Str. Heterozygous Bpa females demonstrate growth retardation and have patchy hyperkeratotic skin lesions, short limbs, chondrodysplasia punctata, and asymmetrical cataracts. The Bpa mutation is a preimplantation lethal for hemizygous male embryos. Liu et al. (24) showed that Bpa and Str mice carry mutations of Nsdhl and sterol profiles consistent with impaired Nsdhl function. The striking unilateral presentation of CHILD syndrome is not observed in Bpa, Str, or tattered (Td) mice (22).
Greenberg dysplasia.
Greenberg dysplasia (MIM no. 215140), also called hydrops–ectopic calcification–moth-eaten skeletal dysplasia (HEM dysplasia), is a rare autosomal recessive skeletal dysplasia first described by Greenberg et al. (25). The phenotypic findings of this lethal disorder can include dysmorphic facial features, hydrops fetalis, cystic hygroma, incomplete lung lobation, pulmonary hypoplasia, extramedullary hematopoiesis, intestinal malrotation, polydactyly, and very short limbs. Radiological findings include a distinctive “moth-eaten” appearance of the long bones, platyspondyly with abnormal ossification centers, ectopic ossification of both the ribs and the pelvis, and deficient ossification of the skull. Histological characterization showed marked disorganization of cartilage and bone, with absence of cartilage column formation, nodular calcifications in cartilage, and islands of cartilage surrounded by bone.
Based on the observation that the ossification abnormalities found in HEM dysplasia are similar to the ossification abnormalities found in CDPX2, Kelley (3) analyzed sterols from cartilage obtained from four fetuses with HEM dysplasia. He found increased levels of cholesta-8,14-dien-3β-ol and cholesta-8,14,24-trien-3β-ol, consistent with a deficiency of 3β-hydroxysterol Δ14-reductase activity. To date, the genetic cause of HEM dysplasia has not been identified.
Antley-Bixler syndrome.
Antley-Bixler syndrome (MIM no. 207410) is one of a large number of craniosynostosis syndromes. Reviewing phenotypic findings in 22 cases, Bottero et al. (26) noted a variety of other morphogenetic defects, including brachycephaly, proptosis, midface hypoplasia with choanal atresia or stenosis, dysplastic ears, radiohumeral synostosis, femoral bowing, multiple joint contractures and long bone fractures, as well as genital anomalies in most subjects, and imperforate anus and congenital heart defects in occasional cases.
Antley-Bixler syndrome appears to affect a heterogenous group of patients with multiple genetic and teratogenic etiologies. Mutations of the FGF receptor 2 gene (FGFR2) have been reported in a number of patients with an Antley-Bixler–like phenotype (27). Mutations of FGF receptor gene family members (FGFR1, FGFR2, and FGFR3) are common in craniosynostosis syndromes. Reardon et al. (27) identified FGFR2 mutations in seven of sixteen Antley-Bixler patients and described various abnormalities of steroid biogenesis in a different subset of these patients. Although only one of the patients exhibited both an abnormality in steroid biogenesis and an FGFR2 mutation, this group has proposed that some cases of Antley-Bixler syndrome might be digenic in origin, with abnormal steroid biogenesis or an alteration of the steroid environment potentiating the effects of a hypomorphic FGFR2 mutation.
Reardon et al. (27) also reviewed the role of fluconazole in the genesis of teratogenic cases of Antley-Bixler syndrome. This synthetic triazole antifungal agent appears to be a teratogen, causing a malformation syndrome with a phenotype similar to that of Antley-Bixler syndrome. Fluconazole inhibits the cytochrome P450 enzyme lanosterol 14-α-demethylase, which demethylates lanosterol. Based on this association with lanosterol 14-α-demethylase inhibition, Kelley et al. (28) hypothesized that autosomal recessive cases of Antley-Bixler syndrome might be due to a deficiency CYP51, which encodes this enzyme, and they showed that lymphoblasts from one Antley-Bixler patient who did not have a mutation of FGFR2 exhibited markedly increased levels of lanosterol and dihydrolanosterol. While the subject studied appeared to be free of pathogenic mutations in CYP51, these data support the idea that loss of lanosterol 14-α-demethylase activity during embryonic development underlies at least some cases of autosomal recessive Antley-Bixler syndrome.
Sterols in development
The identification of human and murine malformation syndromes due to inborn errors of cholesterol synthesis has led to a better appreciation of the multiple functions of sterols during development. To understand the pathophysiological processes underlying the various malformations and clinical problems found in this group of malformation syndromes, one needs to consider the consequences of both the loss of cholesterol or its products and the teratogenic effects of accumulating precursor sterols.
Because some cholesterol precursors are biologically active, abnormally low or high concentrations of specific sterols may have functional consequences. For instance, 7-DHC is the precursor for vitamin D synthesis. The B-ring of 7-DHC is photolyzed by ultraviolet light to form cholecalciferol (vitamin D3). Since serum vitamin D levels are elevated in patients with SLOS (our unpublished data), calcium homeostasis may be altered. Likewise, although functions for these metabolites have yet to be been determined, both 7-DHC and 8-DHC are naturally present at high concentrations in the rat epididymis (29). In addition, 4,4-dimethyl-5α-cholesta-8,24-dien-3β-ol and 4,4-dimethyl-5α-cholesta-8,14,24-trien-3β-ol are meiosis-activating sterols that have been shown to activate the LXRα nuclear receptor (30). Altered gene regulation by these nuclear receptor transcription factors could clearly have profound consequences on development. Another metabolite, desmosterol, is synthesized in the CNS and accumulates just prior to the onset of myelination (31). It is not known whether this accumulation of desmosterol is necessary for the normal myelination process or merely reflects limited DHCR24 activity in this tissue. Desmosterol is also a major sterol in the testes and spermatozoa, and its concentration in the flagella has led to the hypothesis that it increases membrane fluidity to allow tail movement in sperm motility (32). Thus, in addition to being a precursor for cholesterol, desmosterol likely has independent functions. It is likely that the accumulation of bioactive cholesterol precursors contributes to the developmental malformations found in the inborn errors of cholesterol synthesis.
Low levels of cholesterol or the substitution of other sterols for cholesterol may also have detrimental effects during development. In particular, the accumulation of 7- and 8-DHC in SLOS can give rise to aberrant bile acids (33) and steroid hormones (34) whose biological effects have not been defined. 7-DHC itself may impair the function of proteins with sterol-sensing domains, such as HMG-CoA reductase, the Niemann-Pick type C protein (NPC1; see Maxfield and Wüstner, this Perspective series, ref. 35), the sterol regulatory element binding protein cleavage–activating protein (SCAP; ref. 36), and two proteins, Patched and Dispatched, that participate in Hedgehog signaling during development (see Jeong and McMahon, this series, ref. 37). Fitzky et al. (38) have shown that 7-DHC inhibits cholesterol synthesis by accelerating the degradation of HMG-CoA reductase protein, and Wassif et al. (39) have shown that 7-DHC impairs LDL cholesterol metabolism in a manner similar to that seen in Neimann-Pick type C.
In addition to its role as a precursor molecule, cholesterol is a structural lipid in cellular membranes. With their elevated 7-DHC levels, SLOS fibroblasts are reported to have increased membrane fluidity (40). Altered 7-DHC/cholesterol ratios may also affect cellular interactions and morphology during development (41), perhaps by perturbing signal transduction in lipid rafts, ordered lipid microenvironments in the plasma membrane consisting of cholesterol and sphingolipids (see Simons and Ehehalt, this Perspective series, ref. 42; and ref. 43). Indeed, incorporation of 7-DHC changes the structural properties of lipid rafts (44) and may affect local protein structures or interactions. The effects of other cholesterol synthetic intermediates on the structural properties of lipid rafts have not yet been thoroughly investigated in vitro or in the various mouse models of cholesterol biosynthesis disorders.
The requirement for cholesterol in the maturation of the Hedgehog family morphogens has now been studied extensively in vertebrate and invertebrate systems (see Jeong and McMahon, this Perspective series, ref. 37). There are three vertebrate Hedgehog family members. Shh functions in the patterning of the neural tube and the limbs, Ihh is important in endochondral bone formation, and Desert hedgehog (Dhh) is necessary for normal testicular development (23). In the course of autocatalytic cleavage of the Hedgehog protein, a molecule of cholesterol is added covalently to the protein’s amino-terminal signaling domain (45), thus attaching these proteins to the plasma membrane and altering the secretion and movement of these signaling proteins during development (23). Disturbance of Hedgehog signaling likely underlies some of the developmental abnormalities found in the cholesterol synthetic disorders. Shh signaling appears to be impaired in SLOS, as well as in model systems treated with inhibitors of postsqualene cholesterol biosynthesis. Indeed, pharmacologic inhibition of 3β-hydroxysterol Δ7-reductase activity is teratogenic, causing holoprosencephaly, a phenotype seen in Shh–/– mouse embryos and in individuals with either SHH mutations (46) or severe SLOS (1, 41). The inhibitor AY9944 has been shown to perturb the normal expression of Shh, as well as of the developmentally important transcription factors Otx2 and Pax6 in the ventral neural tube (47). Because Hedgehog and the Hedgehog receptor complex of Patched and Smoothened localize to lipid raft domains (48, 49), altered physiochemical properties of lipid rafts incorporating sterols other than cholesterol (44) may represent a mechanism by which Hedgehog signaling could be impaired in the cholesterol synthetic defects.
Common clinical findings in the various malformation syndromes that occur in inborn errors of cholesterol synthesis include abnormalities of bone formation and limb patterning (11). Thus, rhizomesomelic limbs have been reported in SLOS, desmosterolosis, Greenberg dysplasia, and CDPX2. Postaxial polydactyly is found in SLOS, CDPX2, and Greenberg dysplasia. Chondrodysplasia punctata has been reported in SLOS, CDPX2, and CHILD syndrome. Other abnormalities, such as osteosclerosis in desmosterolosis, limb aplasia in CHILD syndrome, and the dramatic “moth-eaten” appearance of the skeleton in Greenberg dysplasia, are unique to a given syndrome. Disturbance of SHH, which plays a major role in both proximal-distal and preaxial-postaxial patterning of the limb, and of IHH, which is involved in the normal progression of cartilage to bone during endochondral bone formation (23), likely underlies some of the shared developmental abnormalities. However, the unique aspects of some of the skeletal defects argue for specific teratogenic effects of the various precursor sterols during skeletal development.
Sterols in CNS function and disease
Sterols and sterol synthetic enzymes appear to play a major role in CNS function. The mental retardation and behavioral phenotype found in SLOS may be due to developmental abnormalities, or to disturbance of CNS function arising from a deficit of cholesterol in this tissue, as suggested by the apparently beneficial effect of dietary cholesterol supplementation on behavioral aspects of this disorder. Understanding the pathophysiological processes underlying the clinical problems found in the inborn errors of cholesterol synthesis may offer insights into pathophysiological processes in more common disorders of the CNS.
Epidemiological studies have suggested an association between decreased serum cholesterol levels and problems such as depression, suicide, and increased impulsivity and aggression. It has been postulated that decreased cholesterol levels contribute to the serotonergic abnormalities found in these disorders (50). Indeed, older children and adults with SLOS frequently have clinical depression (11), and both impulsivity and aggression are part of the SLOS behavioral phenotype (4), consistent with impaired serotonergic function. Alternatively, the Hedgehog signaling pathway is involved in the generation of serotonergic neurons in the developing brain, so an abnormal sterol environment during development could disturb normal development of this neuronal population. Further study of existing mouse models of SLOS and the development of viable hypomorphic mouse models will allow some of these questions to be addressed.
Dietary cholesterol supplementation may also help prevent the development of autistic behavior in SLOS. In a retrospective study, Tierney et al. (4) found that the age of initiation of dietary cholesterol supplementation appeared to influence the development of autistic behavioral characteristics. Of nine SLOS patients who were started on dietary cholesterol supplementation prior to 5 years of age, two (22%) met the Autism Diagnostic Interview-Revised (ADI-R) criteria for autism. In contrast, seven of eight (88%) SLOS patients started on dietary cholesterol supplementation after 5 years of age met these criteria. In a pilot study by Tierney and Kelley (reported in ref. 3), cholesterol therapy reduced the mean ADI-R social domain score of five 4- to 5-year-old SLOS children from 18.2 ± 2.8 to 6.8 ± 1.8, where a normal score is 0 and scores above 10 are judged to be consistent with autism. Although SLOS patients likely represent a small subset of patients with autism, the characterization of this aspect of the SLOS behavioral phenotype may provide broader insights into the roles of sterols in the biology of autism.
The effect of dietary cholesterol supplementation suggests that the behavioral phenotype of SLOS is, at least in part, due to a biochemical disturbance, rather than to irreversible developmental problems arising during embryogenesis. Because the blood-brain barrier excludes significant transfer of dietary cholesterol to the CNS, it may be that the beneficial effects are mediated by neurosteroids, molecules that appear to modulate the activity of a number of neurotransmitter receptors, including NMDA glutamate, GABAA, and cholinergic receptors (51, 52). Analogs of pregnenolone, pregnanetriol, DHEA, and androstenediol derived from 7-DHC have been identified (34, 53), but it is not yet known whether any of these compounds can either substitute for or antagonize the effects of their normal, 7-hydroxy analogs.
The ς-receptors are a class of receptors thought to mediate immunosuppressive, antipsychotic, and neuroprotective effects of a large number of drugs and neurosteroids (51, 52, 54). Three ς-receptor family members have been described, and at least two of these may have role in cholesterol synthesis (54). At the amino acid level, the guinea pig ς1-receptor is 30% identical and 69% similar to the yeast ERG2 protein, a sterol Δ8,Δ7-isomerase involved in ergosterol synthesis. ERG2 has a predicted secondary structure similar to that of the ς1-receptor and binds numerous ς1 ligands. Although the human gene product fails to complement ERG2 mutant yeast, the ς1-receptor may function as a sterol isomerase in mammalian neurons, as suggested by Gibbs and Farb (51). Remarkably, the mammalian 3β-hydroxysterol Δ8,Δ7-isomerase protein (first identified as a high-affinity EBP, as noted above) is also a member of the ς-receptor family, although it is structurally distinct from the ς1-receptor. This parallel is very intriguing and suggests that sterol isomerase function and ς-receptor function may be somehow linked. A third member of the ς-receptor family, ς2, has yet to be purified or cloned. It will be interesting to determine whether this protein is structurally related to a sterol isomerase or to other enzymes in cholesterol synthesis.
Poor feeding, a major clinical problem encountered in SLOS infants, is also observed in Dhcr7–/– pups (10). These pups die during the first day of life due to an abnormal suck and failure to feed. Neurophysiological testing shows a decreased NMDA receptor response of cortical neurons to glutamate stimulation. Given both that pharmacological blockade of NMDA receptor function in newborn mice impairs suckling and that disruption of the NMDAε2 subunit gene results in an uncoordinated suck similar to what we observe in Dhcr7–/– newborns, we speculate that impaired NMDA receptor function underlies the poor feeding and abnormal suck observed in Dhcr7–/– pups. Impaired NMDA function in the Dhcr7–/– mice could have several possible explanations. Decreased cholesterol synthesis by glial cells could disrupt normal synaptogenesis (55). Alternatively, the substitution of 7-DHC and 7-dehydrodesmosterol for cholesterol and desmosterol, respectively, could directly inhibit the NMDA receptor or impair translocation of the receptor subunits to the synaptosomal membrane. NMDA function could also be impaired indirectly by an effect on neurosteroid function, since NMDA glutamate receptor function is increased by neurosteroids, and decreased cholesterol levels might reduce the production of neurosteroids. Increased 7-DHC levels might also inhibit neurosteroid formation or lead to synthesis of an inhibitory analog. The finding of decreased NMDA glutamate receptor function in Dhcr7–/– cortical neurons may provide insight into some of the neurological and feeding problems found in SLOS infants. Further characterization of neurotransmitter function in these model systems should help clarify the pathophysiological basis of the behavioral and mental disturbances found in this human syndrome and may suggest therapies to ameliorate these problems.
One other link that has been proposed between cholesterol and neurological dysfunction concerns Alzheimer disease (AD). This link was suggested in part by epidemiological data showing that elevated serum cholesterol levels are a risk factor for later development of AD. In addition, animal studies indicate that dietary cholesterol supplementation causes β-amyloid accumulation, that diet-induced hypercholesterolemia can accelerate the development of AD-like pathology, and that cholesterol levels may directly affect the activity of β- and γ-secretases, processing enzymes that act on the amyloid precursor protein (reviewed in ref. 56; see also Simons, this Perspective series, ref. 42). Further evidence comes from genetic associations of APOE, which encodes a lipoprotein involved in cholesterol transport in the brain, since carriers of the ε4 allele of this gene are at markedly increased risk of developing AD (57). Also consistent with either a primary or a secondary disturbance of CNS cholesterol homeostasis in these patients, levels of 24S-hydroxycholesterol, which plays a major role in CNS cholesterol homeostasis, are elevated in the cerebrospinal fluid of AD patients (58). Recent epidemiological studies have shown that the prevalence of AD is decreased in patients on HMG-CoA inhibitors (59). Fassbender et al. (60), likewise, have recently reported that that simvastatin reduces β-amyloid peptide accumulation in vitro and in vivo.
A number of observations link AD to inborn errors of cholesterol synthesis. First, the 3β-hydroxysterol Δ7-reductase inhibitor BM15.766, which has been used to produce teratogenic SLOS rodent models, reduces plaque formation in a transgenic mouse model of AD (61). Since the number of adult patients with SLOS is small, and no pathological analysis has been reported from older patients, it is not known whether SLOS patients or DHCR7 heterozygotes are at increased or decreased risk for AD. Second, DHCR24 is identical to a previously identified gene named seladin-1 (selective Alzheimer disease indicator 1), whose expression is decreased in neurons from patients with AD. When overexpressed in cell culture, this gene product appears to have a protective effect against β-amyloid toxicity (62). Sterol profiles, including that of desmosterol, have not been well characterized in AD, but the recognition that seladin-1 is an enzyme involved in cholesterol synthesis suggests that desmosterol or other Δ24-sterols contribute to the pathogenesis of AD.
Summary
The inborn errors of cholesterol synthesis are an interesting group of human malformation syndromes for both basic and clinical science. Cholesterol and precursor sterols are essential for many normal cellular and developmental processes. The importance of sterols in normal embryonic development has been underscored by the discovery that inborn errors of cholesterol synthesis cause human and murine malformation syndromes. Defining the clinical problems found in this group of disorders and studying the developmental perturbations caused by inborn errors of cholesterol synthesis continues to yield insights into the function of cholesterol and its precursors during normal development and to suggest potential therapeutic interventions.
Acknowledgments
I would like to thank Richard Kelley with sharing a copy of his Antley-Bixler manuscript with me prior to its publication. I would like to acknowledge the assistance of Lina Correa-Cerro, Diana Cozma, Brooke Wright, and Christopher Wassif in the preparation of this review. Finally, I would like to express my appreciation to the families of my patients.
Footnotes
Conflict of interest: No conflict of interest has been declared.
Nonstandard abbreviations used: Smith-Lemli-Opitz syndrome (SLOS); chondrodysplasia punctata type 2 (CDPX2); congenital hemidysplasia with ichthyosiform erythroderma/nevus and limb defects syndrome (CHILD syndrome); emopamil-binding protein (EBP); hydrops–ectopic calcification–moth-eaten skeletal dysplasia (HEM dysplasia); Sonic hedgehog (Shh); Indian hedgehog (Ihh); Desert hedgehog (Dhh); Alzheimer disease (AD); selective Alzheimer disease indicator 1 (seladin-1); cerebral spinal fluid (CSF); liquid gas chromatography-mass spectrometry (LGC-MS); Autism Diagnostic Interview-Revised (ADI-R).
References
- 1.Kelley RI, Hennekam RC. The Smith-Lemli-Opitz syndrome. J Med Genet. 2000;37:321–335. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Porter FD. RSH/Smith-Lemli-Opitz syndrome: a multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis. Mol Genet Metab. 2000;71:163–174. doi: 10.1006/mgme.2000.3069. [DOI] [PubMed] [Google Scholar]
- 3.Kelley RI. Inborn errors of cholesterol biosynthesis. Adv Pediatr. 2000;47:1–53. [PubMed] [Google Scholar]
- 4.Tierney E, et al. Behavior phenotype in the RSH/Smith-Lemli-Opitz syndrome. Am J Med Genet. 2001;98:191–200. doi: 10.1002/1096-8628(20010115)98:2<191::aid-ajmg1030>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 5.Irons M, Elias ER, Salen G, Tint GS, Batta AK. Defective cholesterol biosynthesis in Smith-Lemli-Opitz syndrome. Lancet. 1993;341:1414. doi: 10.1016/0140-6736(93)90983-n. [DOI] [PubMed] [Google Scholar]
- 6.Wassif CA, et al. Mutations in the human sterol delta7-reductase gene at 11q12-13 cause Smith-Lemli-Opitz syndrome. Am J Hum Genet. 1998;63:55–62. doi: 10.1086/301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Battaile KP, Battaile BC, Merkens LS, Maslen CL, Steiner RD. Carrier frequency of the common mutation IVS8-1G>C in DHCR7 and estimate of the expected incidence of Smith-Lemli-Opitz syndrome. Mol Genet Metab. 2001;72:67–71. doi: 10.1006/mgme.2000.3103. [DOI] [PubMed] [Google Scholar]
- 8.Xu G, et al. Treatment of the cholesterol biosynthetic defect in Smith-Lemli-Opitz syndrome reproduced in rats by BM 15.766. Gastroenterology. 1995;109:1301–1307. doi: 10.1016/0016-5085(95)90592-8. [DOI] [PubMed] [Google Scholar]
- 9.Jira PE, et al. Simvastatin. A new therapeutic approach for Smith-Lemli-Opitz syndrome. J Lipid Res. 2000;41:1339–1346. [PubMed] [Google Scholar]
- 10.Wassif CA, et al. Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith-Lemli-Opitz syndrome. Hum Mol Genet. 2001;10:555–564. doi: 10.1093/hmg/10.6.555. [DOI] [PubMed] [Google Scholar]
- 11.Nwokoro NA, Wassif CA, Porter FD. Genetic disorders of cholesterol biosynthesis in mice and humans. Mol Genet Metab. 2001;74:105–119. doi: 10.1006/mgme.2001.3226. [DOI] [PubMed] [Google Scholar]
- 12.Clayton P, Mills K, Keeling J, FitzPatrick D. Desmosterolosis: a new inborn error of cholesterol biosynthesis. Lancet. 1996;348:404. doi: 10.1016/s0140-6736(05)65020-9. [DOI] [PubMed] [Google Scholar]
- 13.Waterham HR, et al. Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet. 2001;69:685–694. doi: 10.1086/323473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Happle R. X-linked dominant chondrodysplasia punctata. Review of literature and report of a case. Hum Genet. 1979;53:65–73. doi: 10.1007/BF00289453. [DOI] [PubMed] [Google Scholar]
- 15.Kelley RI, et al. Abnormal sterol metabolism in patients with Conradi-Hunermann-Happle syndrome and sporadic lethal chondrodysplasia punctata. Am J Med Genet. 1999;83:213–219. doi: 10.1002/(sici)1096-8628(19990319)83:3<213::aid-ajmg15>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 16.Moebius FF, et al. Purification and amino-terminal sequencing of the high affinity phenylalkylamine Ca2+ antagonist binding protein from guinea pig liver endoplasmic reticulum. J Biol Chem. 1994;269:29314–29320. [PubMed] [Google Scholar]
- 17.Derry JM, et al. Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. Nat Genet. 1999;22:286–290. doi: 10.1038/10350. [DOI] [PubMed] [Google Scholar]
- 18.Braverman N, et al. Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hunermann syndrome. Nat Genet. 1999;22:291–294. doi: 10.1038/10357. [DOI] [PubMed] [Google Scholar]
- 19.Happle R, Koch H, Lenz W. The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur J Pediatr. 1980;134:27–33. doi: 10.1007/BF00442399. [DOI] [PubMed] [Google Scholar]
- 20.König A, Happle R, Bornholdt D, Engel H, Grzeschik KH. Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet. 2000;90:339–346. [PubMed] [Google Scholar]
- 21.Grange DK, Kratz LE, Braverman NE, Kelley RI. CHILD syndrome caused by deficiency of 3beta-hydroxysteroid-delta8, delta7-isomerase. Am J Med Genet. 2000;90:328–335. doi: 10.1002/(sici)1096-8628(20000214)90:4<328::aid-ajmg13>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 22.Herman GE. X-Linked dominant disorders of cholesterol biosynthesis in man and mouse. Biochim Biophys Acta. 2000; 1529:357–373. doi: 10.1016/s1388-1981(00)00160-8. [DOI] [PubMed] [Google Scholar]
- 23.Ingham PW. Hedgehog signaling: a tale of two lipids. Science. 2001;294:1879–1881. doi: 10.1126/science.1064115. [DOI] [PubMed] [Google Scholar]
- 24.Liu XY, et al. The gene mutated in bare patches and striated mice encodes a novel 3beta-hydroxysteroid dehydrogenase. Nat Genet. 1999;22:182–187. doi: 10.1038/9700. [DOI] [PubMed] [Google Scholar]
- 25.Greenberg CR, et al. A new autosomal recessive lethal chondrodystrophy with congenital hydrops. Am J Med Genet. 1988;29:623–632. doi: 10.1002/ajmg.1320290321. [DOI] [PubMed] [Google Scholar]
- 26.Bottero L, Cinalli G, Labrune P, Lajeunie E, Renier D. Antley-Bixler syndrome. Description of two new cases and a review of the literature. Childs Nerv Syst. 1997;13:275–280. doi: 10.1007/s003810050082. [DOI] [PubMed] [Google Scholar]
- 27.Reardon W, et al. Evidence for digenic inheritance in some cases of Antley-Bixler syndrome? J Med Genet. 2000;37:26–32. doi: 10.1136/jmg.37.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelley RI, et al. Abnormal sterol metabolism in a patient with Antley-Bixler syndrome and ambiguous genitalia. Am J Med Genet. 2002;110:95–102. doi: 10.1002/ajmg.10510. [DOI] [PubMed] [Google Scholar]
- 29.Lindenthal B, et al. Neutral sterols of rat epididymis. High concentrations of dehydrocholesterols in rat caput epididymidis. J Lipid Res. 2001;42:1089–1095. [PubMed] [Google Scholar]
- 30.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 31.Hinse CH, Shah SN. The desmosterol reductase activity of rat brain during development. J Neurochem. 1971;18:1989–1998. doi: 10.1111/j.1471-4159.1971.tb09604.x. [DOI] [PubMed] [Google Scholar]
- 32.Connor WE, Lin DS, Wolf DP, Alexander M. Uneven distribution of desmosterol and docosahexaenoic acid in the heads and tails of monkey sperm. J Lipid Res. 1998;39:1404–1411. [PubMed] [Google Scholar]
- 33.Honda A, et al. Bile acid synthesis in the Smith-Lemli-Opitz syndrome: effects of dehydrocholesterols on cholesterol 7alpha-hydroxylase and 27-hydroxylase activities in rat liver. J Lipid Res. 1999;40:1520–1528. [PubMed] [Google Scholar]
- 34.Shackleton CH, Roitman E, Kelley R. Neonatal urinary steroids in Smith-Lemli-Opitz syndrome associated with 7-dehydrocholesterol reductase deficiency. Steroids. 1999;64:481–490. doi: 10.1016/s0039-128x(99)00022-7. [DOI] [PubMed] [Google Scholar]
- 35.Maxfield, F.R., and Wüstner, D. 2002. Intracellular cholesterol transport. J. Clin. Invest. In press. doi:10.1172/JCI200216500. [DOI] [PMC free article] [PubMed]
- 36.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi:10.1172/JCI200215593. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeong J, McMahon AP. Cholesterol modification of Hedgehog family proteins. J Clin Invest. 2002;110:591–596. doi:10.1172/JCI200216506. doi: 10.1172/JCI16506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fitzky BU, et al. 7-Dehydrocholesterol–dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome. J Clin Invest. 2001;108:905–915. doi:10.1172/JCI200212103. doi: 10.1172/JCI12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wassif CA, et al. Cholesterol storage defect in RSH/Smith-Lemli-Opitz syndrome fibroblasts. Mol Genet Metab. 2002;75:325–334. doi: 10.1016/S1096-7192(02)00010-0. [DOI] [PubMed] [Google Scholar]
- 40.Tulenko TN, LaBelle E, Boesze-Battaglia K, Mason RP, Tint GS. A membrane bilayer defect in the Smith-Lemli-Opitz syndrome. FASEB J. 1998;12:A827. (Abstr.) [Google Scholar]
- 41.Dehart DB, Lanoue L, Tint GS, Sulik KK. Pathogenesis of malformations in a rodent model for Smith-Lemli-Opitz syndrome. Am J Med Genet. 1997;68:328–337. doi: 10.1002/(sici)1096-8628(19970131)68:3<328::aid-ajmg15>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 42.Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi:10.1172/JCI200216390. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. doi: 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- 44.Xu X, et al. Effect of the structure of natural sterols and sphingolipids on the formation of ordered sphingolipid/sterol domains (rafts). Comparison of cholesterol to plant, fungal, and disease-associated sterols and comparison of sphingomyelin, cerebrosides, and ceramide. J Biol Chem. 2001;276:33540–33546. doi: 10.1074/jbc.M104776200. [DOI] [PubMed] [Google Scholar]
- 45.Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 46.Roessler E, et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- 47.Gofflot F, Kolf-Clauw M, Clotman F, Roux C, Picard JJ. Absence of ventral cell populations in the developing brain in a rat model of the Smith-Lemli-Opitz syndrome. Am J Med Genet. 1999;87:207–216. doi: 10.1002/(sici)1096-8628(19991126)87:3<207::aid-ajmg3>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 48.Rietveld A, Neutz S, Simons K, Eaton S. Association of sterol- and glycosylphosphatidylinositol-linked proteins with Drosophila raft lipid microdomains. J Biol Chem. 1999;274:12049–12054. doi: 10.1074/jbc.274.17.12049. [DOI] [PubMed] [Google Scholar]
- 49.Karpen HE, et al. The sonic hedgehog receptor patched associates with caveolin-1 in cholesterol-rich microdomains of the plasma membrane. J Biol Chem. 2001;276:19503–19511. doi: 10.1074/jbc.M010832200. [DOI] [PubMed] [Google Scholar]
- 50.Brunner J, Parhofer KG, Schwandt P, Bronisch T. Cholesterol, essential fatty acids, and suicide. Pharmacopsychiatry. 2002;35:1–5. doi: 10.1055/s-2002-19834. [DOI] [PubMed] [Google Scholar]
- 51.Gibbs, T.T., and Farb, D.H. 2000. Dueling enigmas: neurosteroids and sigma receptors in the limelight. Sci. STKE 2000:PE1. http://stke.sciencemag.org/cgi/reprint/sigtrans;2000/60/pe1.pdf. [DOI] [PubMed]
- 52.Maurice T, Urani A, Phan VL, Romieu P. The inter-action between neuroactive steroids and the sigma1 receptor function: behavioral consequences and therapeutic opportunities. Brain Res Brain Res Rev. 2001;37:116–132. doi: 10.1016/s0165-0173(01)00112-6. [DOI] [PubMed] [Google Scholar]
- 53.Shackleton, C., Roitman, E., Guo, L., Wilson, W.K., and Porter, F.D. 2002. Identification of 7(8) and 8(9) unsaturated adrenal steroid metabolites produced by patients with 7-dehydrosterol-7-reductase deficiency (Smith-Lemli-Optiz syndrome). J. Steroid Biochem. Mol. Biol. In press. [DOI] [PubMed]
- 54.Moebius FF, Striessnig J, Glossmann H. The mysteries of sigma receptors: new family members reveal a role in cholesterol synthesis. Trends Pharmacol Sci. 1997;18:67–70. doi: 10.1016/s0165-6147(96)01037-1. [DOI] [PubMed] [Google Scholar]
- 55.Mauch DH, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 56.Hartmann T. Cholesterol, A beta and Alzheimer’s disease. Trends Neurosci. 2001;24(Suppl.):S45–S48. doi: 10.1016/s0166-2236(00)01990-1. [DOI] [PubMed] [Google Scholar]
- 57.Eichner JE, et al. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. Am J Epidemiol. 2002;155:487–495. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- 58.Papassotiropoulos A, et al. 24S-hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J Psychiatr Res. 2002;36:27–32. doi: 10.1016/s0022-3956(01)00050-4. [DOI] [PubMed] [Google Scholar]
- 59.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 60.Fassbender K, et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:5856–5861. doi: 10.1073/pnas.081620098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Refolo LM, et al. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol Dis. 2001;8:890–899. doi: 10.1006/nbdi.2001.0422. [DOI] [PubMed] [Google Scholar]
- 62.Greeve I, et al. The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress. J Neurosci. 2000;20:7345–7352. doi: 10.1523/JNEUROSCI.20-19-07345.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]