

Short abstract
Protein families and phylogeny represent complex data with statistical outliers and special cases. The JEvTrace implementation of the evolutionary trace method in a JAVA graphical interface allows detailed mining and graphical visualization of evolutionary sequence relationships.
Abstract
Background
Details of functional speciation within gene families can be difficult to identify using standard multiple sequence alignment (MSA) methods. The evolutionary trace (ET) was developed as a visualization tool to combine MSA, phylogenetic and structural data for identification of functional sites in proteins. The method has been successful in extracting evolutionary details of functional surfaces in a number of biological systems and modifications of the method are useful in creating hypotheses about the function of previously unannotated genes. We wish to facilitate the graphical interpretation of disparate data types through the creation of flexible software implementations.
Results
We have implemented the ET method in a JAVA graphical interface, JEvTrace. Users can analyze and visualize ET input and output with respect to protein phylogeny, sequence and structure. Function discovery with JEvTrace is demonstrated on two proteins with recently determined crystal structures: YlxR from Streptococcus pneumoniae with a predicted RNA-binding function, and a Haemophilus influenzae protein of unknown function, YbaK. To facilitate analysis and storage of results we propose a MSA coloring data structure. The sequence coloring format readily captures evolutionary, biological, functional and structural features of MSAs.
Conclusions
Protein families and phylogeny represent complex data with statistical outliers and special cases. The JEvTrace implementation of the ET method allows detailed mining and graphical visualization of evolutionary sequence relationships.
Background
Whole-genome analyses have allowed the study of gene families both within species and in different species. Computational and experimental studies of genomes and gene families are providing new perspectives on our understanding of the evolution of specificity and cellular metabolic organization. These efforts remain limited, however, by our ability to annotate gene function accurately. In yeast, the number of open reading frames (ORFs) with functions assigned by sequence-similarity-based methods is around 43% [1]. With the inclusion of extensive experimental data this value is approaching 70% [2]. Meanwhile, a search of the Protein Data Bank (PDB) for the keyword 'unknown function' retrieved 31 protein structures. Many of these are the result of structural genomics initiatives. As this number is likely to grow, it has become more important to develop computational tools to deduce function from analysis of sequence information in the context of structure.
Assigning function by sequence homology alone is subject to a number of caveats, including the occurrence of structurally homologous enzymes that catalyze different reactions [3] and the propagation of error through successive rounds of sequence annotation [4]. Conversely, assigning function by structure alone can also be daunting, even if one ignores the implicit selection bias in structure databases relative to sequence databases. Analysis of the CATH database revealed that whereas function was conserved in nearly 51% of enzyme families, function had diverged considerably in highly populated families [5]. This has direct implications for structure-based function predictions using threading algorithms [6,7]. Another serious complication in structure-based deduction of function is the intrinsic limit on our ability to compare distantly related sequences and to recognize the role of specific residue subsets in multifunctional proteins. It can be difficult to recognize whether a distantly related homolog belongs to a superfamily with one functional site in common [8] or whether that particular structural scaffold accommodates multiple functional sites, as with the G proteins [9].
It follows that similarity-free function-prediction methods are especially desirable. Marcotte et al. [10] used correlated evolution, correlated mRNA profiles and patterns of domain fusion for genome-wide function prediction. A method based on local gene order of orthologous genes has been proposed [11]. Protein-protein interactions have been used to assign function with surprising success [12] and functional descriptors have been used to search structure space [13]. However, the individual function-prediction capabilities of current methods remain limited, judging by the gene annotation content of public databases.
ET presumes that the branchpoints separating subclades of a phylogenetic tree can specify molecular speciation events, and hence evolutionary selection of amino acids. Thus, nodes can mark points in evolution where a protein gains, modifies or loses a binding or catalytic function [14]. The original ET method relies on a partitioning of the phylogeny. This procedure results in sets of nodes at different levels of percent (sequence) identity cutoff (PIC) [15]. However, as phylogenies often contain extreme branches as a result of distant homologs or rapid speciation, pairs of protein family members are not represented uniformly across the sequence-identity range. This is reflected in a skewed topology of the phylogeny, see, for example, the Pseudomonas aeruginosa and Streptococcus pyogenes hypothetical proteins at the bottom of Figure 1. Hence, the PICs correspond to intervals of percent sequence identity, and the greater the number and/or magnitude of outliers in the family, the larger the percent identity interval (see Figure 1). The presence of these outliers affects multiple alignment and phylogenetic models and in ET analysis can misrepresent the functional variability at the presumed PIC level. This issue has been addressed by normalizing the score in the ET method with sequence variability and sequence uniqueness measures [16]. However, numerical normalization reduces the problem to numerical analysis, in effect disregarding evolutionary aspects. In the case of distant subclades, ET analysis of appropriately chosen subclades of the phylogeny will have the desired normalization effect. This approach can be used to correct for positional variability, sequence representation bias and non-uniform phylogenetic topologies.
Figure 1.
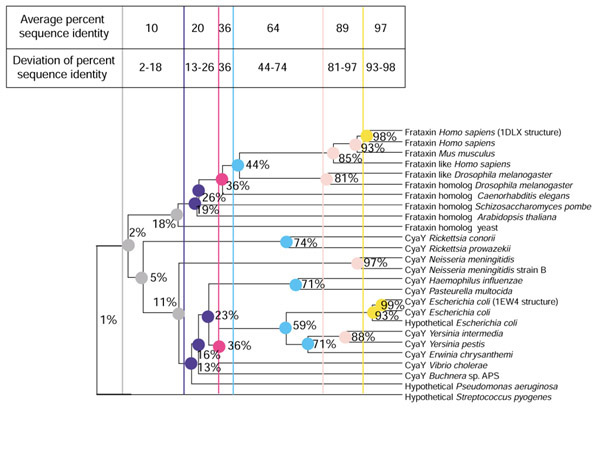
Phylogenetic partitions and sequence outliers in a protein family. A phylogenetic tree of the frataxin family [55] detailing the presence of distant sequence homologs within a phylogeny. The MSA and dendogram of the family were constructed as described in Materials and methods. Partitions of the phylogeny are shown as colored vertical bars. Each partition of the phylogeny corresponds to an interval of percent sequence identity. The percent sequence identity for the selected subclades is shown on the phylogeny. A series of colors is used to indicate distinct partitions from left to right.
Another limitation of the original ET method was the definition of invariant and neutral position types. Uchtarge et al. [14] recognized that with growing sequence databases, the strict definitions of invariance as a total lack of variance, and neutrality as a one-residue variation in an aligned position in even one family, were destined to require amendment. Inherently, the functional resolution in the ET method relies on an optimization based on ET results from multiple partitions, each corresponding to unique definitions of subclade invariance. Although two automated ET methods exist, notably a public ET server by Innis et al. [17] and an implementation by Aloy et al. [18], optimization of the original ET method has evaded automation thus far. The result is that users have to resort to manipulation of the underlying data and cycles of ET analysis and visual inspection of the results mapped to protein structures.
Aside from manually filtering and pruning the data, there has been no simple way of controlling which subclades of the protein family are used in the analysis. An elegant solution to this problem is to allow the user to access all possible subclades represented by the phylogenetic tree. In this way a number of ET variations can be performed, extending the analysis to multiple views of protein family evolutionary data.
JEvTrace is one possible implementation of protein family analysis. Such analyses, which include experimental techniques such as alanine scanning [19] and computational techniques such as MSA coloring schemes [20], attempt to organize the massive amounts of sequence and structure data. The results introduce the problem of choice of strategies to identify biologically meaningful patterns. In general, sequence and structure alignments are frequently used to sort features of gene-family data. Analysis of alignments provides coherence to the understanding of biological data, especially from the perspective of distinct features that may explain the unique functional attributes of an individual entry. As is common in ET analysis, these features may extend over sequentially or spatially clustered sets of nucleotides or amino acids, and patterns are frequently difficult to identify without a form of color coding. Obviously, color coding exploits our cognitive pattern-recognition skills - skills that have been difficult to replicate algorithmically.
Results
An example of a protein with unknown function is PDB 1G2R, representing the ylxR gene from Streptococcus pneumoniae [21]. YlxR belongs to the putative nusA/infB operon in S. pneumoniae. The operon contains seven genes, three of which are conserved in other bacteria: rbfA, nusA (with its well characterized gene product IF2 [22]) and infB. These three proteins are involved in translation and ribosomal function during cold-shock response [23]. The YlxR protein sequence has been assigned to the 'cluster of orthologous groups' (COG) 2740 [24], which contains the conserved amino-acid motif GRGA(Y/W) (in the single-letter amino-acid code). The proteins of COG 2740 are predicted to be nucleotide-binding proteins implicated in transcription termination. Several features of the structure of YlxR, including the conserved and appropriately spaced arginines that could form a characteristic positively charged surface patch (colored red in Figure 2b,2c) and a large bent groove reminiscent of other RNA-binding structures, support the argument that YlxR is an RNA-binding protein.
Figure 2.
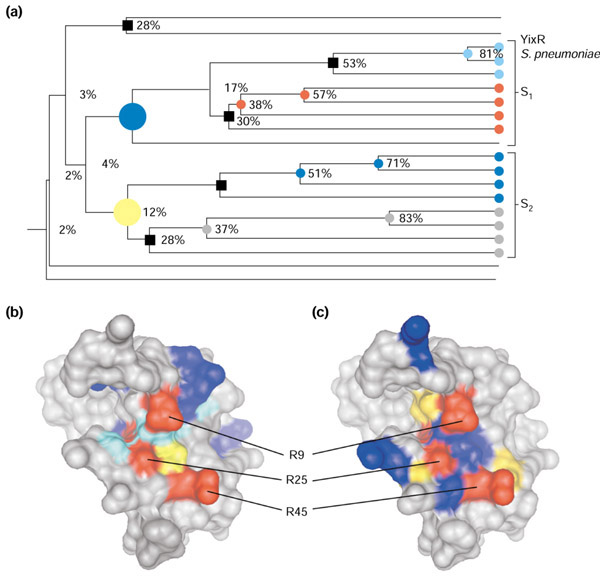
JEvTrace analysis of the YlxR protein family. (a) Phylogeny of the 20 protein sequences identified by PSI-BLAST as members of the YlxR family (see text). The two major subclades are labeled S1 and S2 and the location of the S. pneumoniae YlxR (PDB 1G2R) in the phylogeny is shown (YlxR S). The large blue and yellow circles indicate the two major subclades; minor subclades are indicated by black squares; small colored circles represent minor subclades belonging to major subclades. (b) Results of analysis of the five minor subclades mapped onto the three-dimensional structure of YlxR. The position score corresponds to the color coding in Figure 4. (c) Sequence conservation comparison between the two major subclades. Residues are coloured blue or yellow according to their conservation in either subclade. In all cases positions conserved across all sequences are colored red. Graphics of the molecular surfaces were created with Chimera [49] and MSMS [56] using the SCF format to import JEvTrace results. Graphics of the phylogeny were created with JEvTrace.
Structures of proteins in complexes with small molecules have led to and confirmed predictions of protein function. The structure of YlxR is complexed with three sulfate ions, two of which are bound to the arginines R25 and R45, and the third to a lysine pair K62 and K63. It has been observed that the distances between the sulfate ions correspond to distances between phosphate groups in an RNA duplex [21]. The predicted binding site also fully encompasses two out of three of the sulfate ions and borders the third.
The protein family retrieved by PSI-BLAST [25] consists of 20 unique. The hypothetical ancestor sequence has three conserved arginines. Although this is a relatively small family for ET analysis, a simple MSA suggests that only the arginine of the conserved GRGA(Y/W) motif is absolutely conserved. However, as the key arginines associated with the predicted function are absolutely conserved, this implies that the predicted RNA-binding function is conserved across this family.
JEvTrace subclade trace analysis was performed on all the subclades of this family ranging from 28% to 53% sequence identity (Figure 2a). After filtering out residues that are inaccessible to the solvent, the following residues were identified in the vicinity of the conserved arginines (the conserved arginines are in parentheses): K10, V12, V13, S14, K20 (r9), G40 (R25), G46 (R25), 48Y (R25, R45) and K30 and E31 (R45). At least eight residues form a spatial cluster in the vicinity of the conserved arginines. The residues K10, the backbone of S11, V12, V12, S14, V17, G40, E55, K63 and 64V, from the kinked carboxy-terminal helix, a beta-turn, and parts of the beta-sheet, define a putative binding epitope (Figure 2b). The epitope includes a collection of hydrophobic interactions that have been correlated with the evolution of residues forming a surface epitope in the vicinity of r9 (Figure 2b).
There appear to be two distinct subclades within the YlxR sequence family, both consisting of proteins with unknown or uncertain function (S1 and S2, Figure 2a). A subclade comparison was carried out to analyze the conserved residues. Such a comparison is useful when a protein family has few representatives or limited evolutionary diversity, that is, few subclades. In this family, it appears that independent sets of residues are conserved, all in the vicinity of one or more of the conserved arginines. These amino acids define a slightly larger and differently oriented surface epitope (Figure 2c) than in the JEvTrace subclade trace analysis (Figure 2b). We propose that the conserved residues modulate the specificity of the predicted RNA interaction, and that the two subclades correspond to specificity subtypes within the larger family, possibly with unique functional features. The residues not identified by subclade trace analysis but appearing in the subclade comparison are responsible for a finer level of molecular specificity. In this case of a predicted protein function, JEvTrace analysis presented direct evidence for additional binding functions and highlighted the presence of potential subtypes in the RNA-binding specificity.
Another interesting family of unknown function is the bacterial YbaK proteins. A structure of the homolog from H. influenzae has been solved by Zhang et al. [26]. This gene product has been proposed to serve as a regulatory protein [27,28]. Analysis of the sequence family in the context of the structure revealed one conserved residue, K46, in a small putative binding site [26]. The YbaK fold is related to a circular permutation and truncation of the C-lectin fold. However, a saccharide binding function for YbaK is unlikely, because of a small putative binding site and lack of saccharide binding residues [26]. Zhang et al. discuss the possibility of an oxyanion hole formed by backbone nitrogens of the two residues following conserved G101 (with the exception of an arginine in an unknown protein from Mycobacterium. smegmatis (AAD41809)).
The YbaK family is composed of three large subclades, related by an absolutely conserved lysine, K46. The three subclades are YbaK (S2 in Figure 3a), an insertion domain in the acceptor stem of prokaryotic prolyl-tRNA synthetases (S1 in Figure 3a) and a prokaryotic family of hypothetical proteins (S3 in Figure 3a). Seventy-one sequences were used in the JEvTrace analysis. Of these, 23 formed a distinct subclade containing the H. influenzae YbaK sequence.
Figure 3.
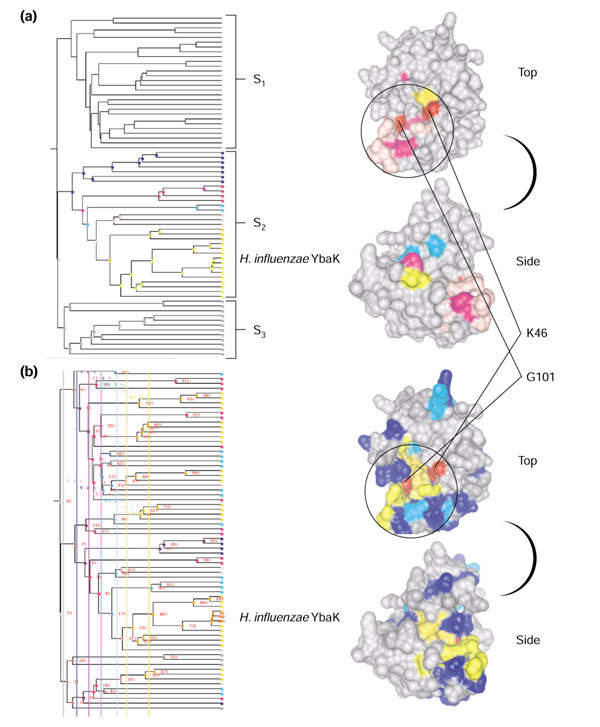
JEvTrace analysis of the YbaK protein family. The three major subclades are labeled S1, S2 and S3, and the location of H. influenzae YbaK (PDB 1DBX) in the phylogeny is shown. The 3D structure of YbaK viewed from the top and the side is given to the right of each phylogeny. (a) Results of parent tracing through seven consecutive parent subclades. Color coding corresponds to the colors of the subclades in the phylogeny. (b) Partition trace results for six partitions of the phylogeny, ranging from 7-50% average sequence identity. Coloring of residues in the 3D structures in (a) and (b) corresponds to the color-coded scores in Figure 4. Black circles in the top views of (a) and (b) represent the approximate location of the putative binding site. Graphics of the molecular surfaces were created with Chimera [49] and MSMS [56] using the SCF format to import JEvTrace results. Graphics of the phylogeny were created with JEvTrace.
JEvTrace parent trace analysis, which relies on tracing the conservation of the progenitor sequences of a single selected node (Figure 3a), identified a number of neutral polar and hydrophobic amino acids conserved in the YbaK subclade on the conserved lysine face of YbaK (Figure 3a, top). This was consistent with the analysis of Zhang et al. [26]. Among these conserved positions, JEvTrace identified a cluster of solvent-accessible residues above and beyond the proposed oxyanion hole, including Y20, H22, D23, E32 and R132. Together, these residues form a polar surface patch and the wall of the putative binding site. D23 and E32 contribute to the negative face identified by Zhang et al.
The phylogenetic partition JEvTrace algorithm was carried out using six partitions from the 7-50% average percent sequence identity range (Figure 3b). The resulting highest-scoring position is S104 (magenta), and then T47, T96, Y98, G102, I103 and S129 (orange). Most of these positions are partially shielded from solvent and/or contribute main-chain hydrogen-bonding interactions. Eliminating solvent-accessible residues left S129, a position that belongs to the neutral polar patch of the putative binding site. Considering residues with less prominent scores (gray, blue, cyan, yellow), the size of the epitope identified by JEvTrace increases considerably and encompasses nearly half of the K46 face (Figure 3b, top). Together these positions form a partially buried cluster that defines the bottom and walls of the putative ligand-binding site spanning the K46 face. From the fifth level on (Figure 4) all identified positions are on the conserved lysine face of YbaK. Significantly, the loop immediately above the oxyanion hole is disordered in the YbaK PDB 1DBX structure. This loop, formed by residues 26-30, is not conserved nor does it conserve chemical properties across the phylogeny. However, a number of subclades express invariance at these positions. Because of its proximity to the conserved G101 and one branch of the J-shaped putative binding site, this disordered region is predicted to contribute to the functional interaction and specificity of YbaK. A number of structural studies by NMR have shown that RNA-binding proteins are flexible and undergo conformational changes upon binding [29,30,31].
Figure 4.
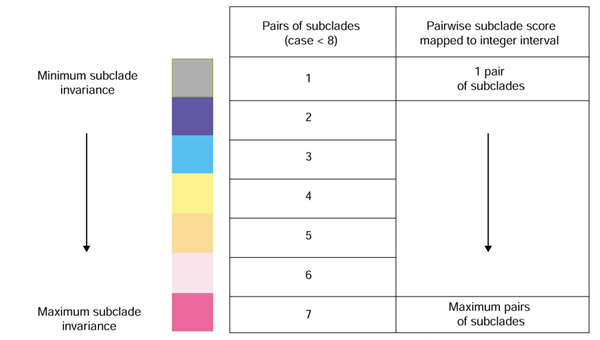
A description of the scoring scheme and coloring scale used in JEvTrace. The score for a given position is calculated as the pairwise sum of subclade invariance across partitions or set of nodes. In the event of numerous invariant subclade pairs, the score is normalized to an integer interval corresponding to colors in the color scale.
Using GRASP [32] Zhang et al. [26] predicted a positively charged patch on the face of the protein opposite K46, and a negatively charged patch on a face adjacent to K46. However, the YbaK structure has the interesting feature of a single conserved lysine separated by a ring of hydrophobic or neutral residues from a circular arrangement of mixed charged residues (Asp, Arg, Glu, Lys). These residues line the perimeter of the K46 face. This is reminiscent of numerous examples of protein-protein interaction, where hydrophobic "rings" of residues are observed to surround polar and charged residues, with the proposed purpose of screening ionic interactions from solvent [33,34]. This potential protein-protein interaction feature of YbaK is additionally supported by evidence that the prolyl-tRNA synthetases (S1 in Figure 3a) interact with other proteins involved in protein synthesis. There may be additional surface patches of mixed positively and negatively charged residues in YbaK. However, the positively charged surface identified by Zhang et al. contains the largest number of high scoring positions in the JEvTrace analysis of multiple phylogenetic partitions (Figure 3b). The lysine perimeter patch (Figure 3a,3b, top) and other potential patches are not conserved nor are they identified completely by the partition algorithm (Figure 3a,3b, side), and thus are not expected to be a predominant functional feature of the YbaK family.
JEvTrace analysis suggests that YbaK is involved in a protein-protein interaction requiring a binding site with hydrophobic and polar patches, and an oxyanion hole opposite a conserved lysine. Pursuing the protein-protein interaction hypothesis, it appears that a protruding J-shaped polypeptide volume involving an aspartic or glutamic acid, or a negatively charged cofactor, is a likely ligand for the YbaK-binding site. The face opposite this binding site presents a patch of positively charged residues, supporting the hypothesis of a nucleotide binding function for at least some subclades of the YbaK family. Thus although, the YbaK family subtypes only share one conserved amino-acid across species, the patterns of subclade sequence conservation suggest a main binding function, characterized by unique specificity within multiple clades that is spatially centered around the conserved lysine, K46.
Discussion
JEvTrace
The ET method has been a successful tool for analyzing protein functional surfaces using the additional information present in protein phylogenetic trees. However, this approach has been limited by difficulty in producing a dynamic graphical user interface to analyze the data. The optimization involved in producing ET results has previously relied on manually manipulating the underlying data, while certain paths of analysis were inherently inaccessible. Thus, identification of the dominant spatial cluster of invariant residues has been unwieldy. To improve this operational challenge we have constructed JEvTrace, a JAVA suite of algorithms and objects together with a graphical user interface. The algorithms allow the user to identify evolutionarily relevant positions based on user selections of subclades (Figure 2a,2b) or partitions of the phylogeny (Figure 3b). This approach introduces new features and parameters in ET analysis. Additional algorithms for tracing subclade conservation through parents or children of a specific subclade (Figure 3a) and subclade conservation comparisons (Figure 2a,2c) are also implemented. The user interface produces interactive graphical results, and access to the corresponding sequence, phylogenetic and protein structure data. To map ET results and alignment selections to the structural dimension, JEvTrace is dynamically linked to a 3D-structure JAVA viewer, WebMol [35].
These algorithms (see Materials and methods) allow comparisons of features within a phylogeny in ways that are not directly limited by the topology of the phylogeny, sequence representation bias or sequence distance metrics. The implementation allows an analysis of any possible combination of subclades within the protein phylogeny. The resulting decompositions of evolutionary sequence data allow multiple definitions of sequence, structure and function homology within a protein family, and hence grant new perspectives to family sequence analysis.
The original ET method relies on protein structures to filter phylogenetic results in order to identify predicted functional sites. A recognized limitation of the original method was filtering out buried polar side chains within structural clefts [14]. JEvTrace gives access to the entire set of results before residue solvent-accessibility filtering. Extensive structural filtering, not limited to solvent accessibility can be carried out in WebMol [35].
JEvTrace facilitates the analysis of other features of protein families. Conserved positions can be found for any subclade in the phylogeny, and the conservation and variability between any set of subclades can be analyzed (subclade comparison, Figure 2b,2c). This functionality can be used to distinguish homologous proteins with different functions, as first suggested by Aloy et al. [18]. For a particular subclade, JEvTrace can perform a parent or child trace, identifying the subclade specific conservation within a chain of parent or child subclades of a node (Figure 3a). This method can be used as an ET surrogate if there is lack of significant homology between subclades of a protein family. This was helpful in the analysis of YbaK. JEvTrace can identify the unique residues in a single sequence relative to the considered sequence data. This was useful for our drug-design efforts on a malarial cysteine protease [36]. JEvTrace also serves as a sequence and structure viewer. Any JEvTrace analysis of the MSA or phylogenetic data can be visualized on available protein structures. This can be useful in protein structure homology modeling by highlighting the evolutionary contexts for structural analysis within a protein family [36]. All these informatics features address sequence determinants of specificity and similarity using distinct biological data.
In general, the ET approach is more difficult to apply at lower percent sequence identity, owing to the problem of building accurate sequence alignments, especially in the absence of structural information [37,38]. For example, annotations based on remote homology pairwise alignments were a significant source of errors in the initial yeast genome annotation [1]. Known alignment problems occur at the amino and carboxyl termini of a protein, and even more commonly in loop regions. In addition to an accurate alignment, ET also requires a minimal amount of sequence information and the related parameter of evolutionary diversity within the protein family. Of the algorithms provided in JEvTrace, the parent/child trace and subclade comparisons can be applied with as few as two sequences. The partition trace and subclade trace require more than a pair of subclades, and benefit non-linearly from larger amounts of data. Overall, the two largest effects of limited sequence data are the signal-to-noise ratio for the correlation of invariant sites to functional residues and the ability to identify functional specificity and specific functions. As a corollary, until the sequence space of a protein family has been sampled sufficiently, insight into the full functions and specificities within a phylogeny remain limited.
SCF: an MSA sequence coloring format
With rapidly enlarging biological data sources, there is a clear need for sensible standards. We propose SCF: a file format that will encode any user-defined coloring scheme for protein and nucleotide sequences as well as their secondary and tertiary structures, based on the inherent structure of MSAs. The format is simple, easy to verify manually, and readable as text by any alignment or structure viewer.
An example protein alignment with a selection of residues, including absolutely conserved positions as well as positions forming two structural epitopes in one of the known protein structures, is shown in Figure 5. It is important to note that only selected positions are encoded - which is both a performance and a storage asset. Our implementation of an alignment viewer allows transparent interaction with tertiary structures, using the JAVA applet WebMol [35]. In this setting, the color format serves to annotate protein structures with multiple sequence information, and allows comparisons across multiple sequences and structures. This format should also aid the visualization of experimental results pertaining to biological sequences and structures. Most important, it will allow integration of visualized sequence alignment results under a single representation scheme.
Figure 5.
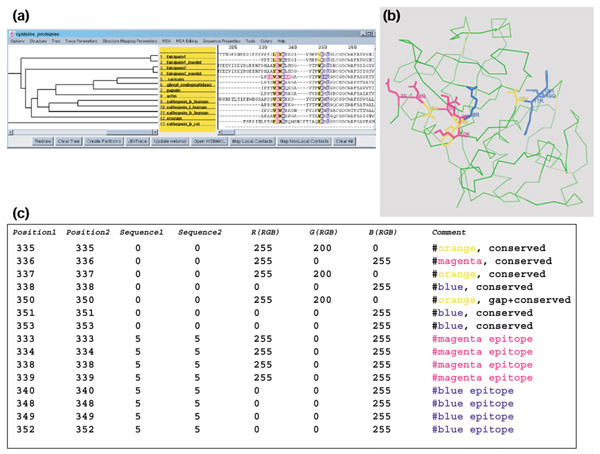
An example of the SCF coloring format. (a) Colored MSA selections; (b) the selections mapped to a representative protein structure; (c) text encoding of the MSA selections in (a) according to the SCF format specification. The MSA graphics were created with JEvTrace and the structure graphics with WebMol [35].
Conclusions
We have designed a JAVA application, JEvTrace, implementing the ET method [14] and its variations. These methods have in common the analysis of protein families through MSAs, phylogenetic trees and protein structures. The ET method has proved a useful tool for understanding the sequential and structural aspects of protein function, including the analysis of variations relevant to molecular specificity. From an evolutionary perspective, the function of proteins within a protein family encompasses both variation, for example, substrate specificity reflected in amino acids lining substrate-binding pockets, and conservation, for example, regions responsible for general enzymatic activity or binding of a common molecular scaffold. While it is trivial to identify absolutely conserved residues, function discovery often requires a context for the predicted or unknown function associated with the absolute conservation pattern.
Using the examples of YlxR and YbaK, JEvTrace identified residues clustering around the putative conserved functional residue(s), thus validating a functional prediction. These findings supported an RNA-binding prediction for YlxR and most likely a protein-protein interaction interface for YbaK. For discovery of additional functional properties, as in the case of a new binding epitope in YlxR or the extensive putative binding site and positively charged epitope in YbaK, JEvTrace provided phylogenetic evidence of clusters of residues on the protein surface.
It is hoped that the JEvTrace implementation will lead to analysis of protein families at varying levels of detail, leading to useful decompositions of the data. One of these decompositions comes from evidence in the evolutionary record of protein sequences. As documented by the biological applications of the ET method, evolutionary data presents evidence allowing the distinction of conserved spatial arrangements of residues versus evolutionary sequence changes with negligible or no effect on function. Together with experimental data, the decompositions of evolutionary data provided by JEvTrace may enable us to make additional distinctions in the molecular specificity, kinetic and dynamic properties of protein function.
Materials and methods
Sequence family retrieval and analysis
We chose two protein structures of unknown function, and retrieved their protein families from the sequence database. The sequence of the structure was used as a query for PSI-BLAST [25] against the GenPept database from the National Center for Biotechnology Information (NCBI). The sequences were then aligned and phylogenetic trees created with CLUSTALW [39] and/or combinations of software from the GCG package [40] including PILEUP [41,42,43] and PAUPSEARCH [44].
Algorithm
The binary phylogenetic tree and MSA data are implemented as JAVA objects. The phylogenetic tree, assumed to be binary, is modeled as branches and nodes along with an ordering such that each branch shares a node with a parent branch and from zero to two child branches. Each node in the phylogeny corresponds to a subset of sequences in the MSA. Every phylogenetic branch is represented with an abstract consensus sequence, used to model the corresponding subclade sequence conservation. The implemented algorithm derives a consensus sequence for every subclade of sequences represented in the phylogenetic tree. This information is used to dynamically generate results with the supplied algorithms based on user-defined subclades or partitions of subclades.
The partition trace variation of the ET method assigns nodes from the tree to a defined partition of the phylogeny. Partitions are perpendicular to the direction of branches in the tree (Figure 1). Sequence conservation in each subclade is compared pairwise to conservation in all other subclades within a given partition. Alternatively, in the subclade trace algorithm, requiring user specification of a set of nodes, the defined nodes are algorithmically treated as a single partition. The subclade trace does not require partitions, and is therefore independent of the topology of the phylogeny. In both algorithms, each position of the MSA is scored by the frequency of conservation of different amino acids in pairs of subclades at that MSA position. In the partition trace, the score is cumulative across partitions. All scores are normalized if there are more than seven pairs of invariant subclades at any alignment position. The numerical scores are mapped to a seven-color scale (Figure 4), limited by graphical interaction with the structure. Scores can include normalization by the sequence variability of the identified invariant subclades.
JEvTrace also provides the ability to perform a single subclade trace. The user-defined subclade is assigned as a parent or child node, and the subclade-specific sequence conservation below or above that node is identified. Subclade-specific conservation is defined by the set of residues that are conserved in a subclade but not in its parent. The results are a chain of related subclades of the phylogeny, with color-coded subclade sequence selections on the MSA and structure. We call this variation of the ET method a parent (or child) trace, and it is especially useful for families with few subclades and cases of highly speciated specificity.
JEvTrace generates results dynamically, displays them on the MSA and enables saving in standard graphics formats or the SCF format. As in the original ET method, absolutely conserved positions are inherently excluded from the analysis. Structural filtering is assigned to the WebMol JAVA program, packaged with JEvTrace. Concurrently with WebMol, JEvTrace reads PDB data and aligns the sequence of the structure with a selected sequence in the MSA. This alignment enables JEvTrace to map results and selections from the MSA to the structural dimension. JEvTrace also presents the option of using the ACCESS program [45], which calculates the static solvent accessibility of a protein structure [46]. This solvent-accessibility data can be used to filter results of the phylogenetic analysis by three states of amino acid solvent accessibility [47].
JEvTrace implementation
The program takes as input an MSA, or an MSA with a corresponding phylogenetic tree. The PILEUP (GCG), CLUSTALW [39] and New Hampshire [48] formats are recognized. Phylogeny is interpreted as a binary tree with a hypothetical root. Protein structure viewing is designated to the JAVA structure viewer WebMol [35]. Alignment selections in JEvTrace can be mapped to the Chimera structure viewer [49] using an earlier version of the SCF format available in JEvTrace. As many proteins lack representative crystal structures, use of structures in JEvTrace analysis is optional. Currently, JEvTrace supports one active WebMol window per session.
Users can select up to seven partitions of the phylogenetic tree, or choose any set of nodes. A number of operations, including the ET method, can be carried out on the selected parts of the phylogeny. The resulting data are independent of structural information and can be viewed and manipulated directly on the MSA of the protein family. To aid interpretation of the phylogenetic data, the tree can be annotated with the percent sequence identity of all the subclades. The identified positions are visualized on the MSA as well as any available structures (Figure 2).
Among the many sequence-structure features of JEvTrace is the ability to highlight the residues in contact with a selected position, using a residue-residue distance calculation and a distance cutoff. A number of sequence-based features are also available, including calculation of alignment position statistics for a variety of physical-chemical properties: molecular volume [50], average pKa [50], hydrogen-bonding potential, number of rotatable bonds and hydrophobicity [51].
JEvTrace consists of three graphical canvases: a binary phylogenetic tree, a list of sequence identifiers (for example, accession codes) and an MSA. The three canvases are aligned by row, such that the terminal nodes (representing individual sequences) of the phylogenetic tree align with their names and amino-acid sequences. The tree and alignment canvases are scrollable in two dimensions, and have a practical capacity of more than 150 sequences of less than 400 amino acids, on a Pentium workstation with 256 M of RAM. All JEvTrace functions are organized into menus and buttons, allowing extensive user interaction with the data. Any results that are represented graphically in JEvTrace or WebMol can be printed or saved.
Sequence coloring format (SCF) implementation
The colored format for the MSA is given as a text file with the file extension '.SCF'. It is accurate with respect to the underlying sequence data, given that the sequence(s) remains unchanged in length and order. As a safeguard for underlying sequence data consistency, the SCF object calculates a MSA checksum variable (see SCF website [52] for details). Relational databases and software environments such as JEvTrace represent dynamic extensions of the format. The coloring data can exist as an individual file or can be appended to the actual data file - Multiple Sequence Format (MSF) (GCG) or CLUSTALW [39] files in the case of MSA, and a PDB file [53] for structural data. Appending the coloring to the underlying data can allow transparent annotation by color.
The residue positions of sequences in an alignment are uniquely indexed from top to bottom, using sequence numbers starting at one as rows, and left to right, using alignment positions starting at zero as columns. The actual file format consists of six columns: sequence number, residue number, three columns for the primary color space (red green blue, RGB) designation of the color, and an optional comment/property column (Figure 1c). The last column can accommodate accepted coloring schemes or can be used to define properties for colors and/or the underlying data. This format accommodates any 24-bit digital color, and allows highlighting of any subset of residues in any subset of sequences of the MSA. The selections are encoded in a hierarchical sorted manner, that is, smallest to largest sequence position, and within this group smallest to largest sequence, and within those groups, smallest to largest color values.
Our JEvTrace implementation of the SCF format in JAVA, allows reading of MSF, CLUSTALW and PDB data files, and interpretation of the SCF coloring data in each of these contexts. In addition, the underlying sequence data are modeled as JAVA objects, whose properties are dynamically updated. In this implementation, it is possible to translate selections between different MSAs that share at least one sequence. Using a single sequence as a 'translator', any selections can be 'translated' from one alignment to another, given that both alignments contain the 'translator' sequence. This feature is useful in bridging analysis of families containing distant homologs, performing independent analysis of multiple subclades of a protein family, or updating MSA data.
Hardware
The JEvTrace and SCF JAVA packages have been tested on SGI MIPS, Pentium Pro (Windows and Linux) and Macintosh systems. Both JAVA packages are compatible with 1.2 and higher versions of JAVA.
Availability
A JAVA executable package and manual for JEvTrace v1.0 is available on the web [54]. A description and JAVA source for the SCF v1.0 format are also available on the web [52].
Acknowledgments
Acknowledgements
We are deeply grateful for the help of Dietlind Gerloff, Dirk Walther, Jonathan Blake, John-Marc Chandonia, Wally Novak, Anthony Lau and Chern-Sing Goh during the development of the application. Anthony Lau and Elaine Meng provided invaluable comments on the manuscript.
References
- Mewes HW, Albermann K, Heumann K, Liebl S, Pfeiffer F. MIPS: a database for protein sequences, homology data and yeast genome information. Nucleic Acids Res. 1997;25:28–30. doi: 10.1093/nar/25.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M, Morgenstern B, Munsterkotter M, Rudd S, Weil B. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 2002;30:31–34. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt JA, Babbitt PC. Can sequence determine function? Genome Biol. 2000;1:reviews0005.1–0005.10. doi: 10.1186/gb-2000-1-5-reviews0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner SE. Errors in genome annotation. Trends Genet. 1999;15:132–133. doi: 10.1016/s0168-9525(99)01706-0. [DOI] [PubMed] [Google Scholar]
- Pearl F, Todd AE, Bray JE, Martin AC, Salamov AA, Suwa M, Swindells MB, Thornton JM, Orengo CA. Using the CATH domain database to assign structures and functions to the genome sequences. Biochem Soc Trans. 2000;28:269–275. doi: 10.1042/bst0280269. [DOI] [PubMed] [Google Scholar]
- Jones DT, Tress M, Bryson K, Hadley C. Successful recognition of protein folds using threading methods biased by sequence similarity and predicted secondary structure. Proteins. 1999;Suppl 3:104–111. doi: 10.1002/(sici)1097-0134(1999)37:3+<104::aid-prot14>3.3.co;2-g. [DOI] [PubMed] [Google Scholar]
- Panchenko A, Marchler-Bauer A, Bryant SH. Threading with explicit models for evolutionary conservation of structure and sequence. Proteins. 1999;Suppl13:133–140. doi: 10.1002/(sici)1097-0134(1999)37:3+<133::aid-prot18>3.3.co;2-4. [DOI] [PubMed] [Google Scholar]
- Russell RB, Sasieni PD, Sternberg MJ. Supersites within super-folds. Binding site similarity in the absence of homology. J Mol Biol. 1998;282:903–918. doi: 10.1006/jmbi.1998.2043. [DOI] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, Cohen FE. Evolutionarily conserved Gαβγ binding surfaces support a model of the G protein-receptor complex. Proc Natl Acad Sci USA. 1996;93:7507–7511. doi: 10.1073/pnas.93.15.7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D. A combined algorithm for genome-wide prediction of protein function. Nature. 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- Kolesov G, Mewes HW, Frishman D. SNAPping up functionally related genes based on context information: a colinearity-free approach. J Mol Biol. 2001;311:639–656. doi: 10.1006/jmbi.2001.4701. [DOI] [PubMed] [Google Scholar]
- Hishigaki H, Nakai K, Ono T, Tanigami A, Takagi T. Assessment of prediction accuracy of protein function from protein-protein interaction data. Yeast. 2001;18:523–531. doi: 10.1002/yea.706. [DOI] [PubMed] [Google Scholar]
- Di Gennaro JA, Siew N, Hoffman BT, Zhang L, Skolnick J, Neilson LI, Fetrow JS. Enhanced functional annotation of protein sequences via the use of structural descriptors. J Struct Biol. 2001;134:232–245. doi: 10.1006/jsbi.2001.4391. [DOI] [PubMed] [Google Scholar]
- Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- Du P, Alkorta I. Sequence divergence analysis for the prediction of seven-helix membrane protein structures: I. Comparison with bacteriorhodopsin. Protein Eng. 1994;7:1221–1229. doi: 10.1093/protein/7.10.1221. [DOI] [PubMed] [Google Scholar]
- Landgraf R, Fischer D, Eisenberg D. Analysis of heregulin symmetry by weighted evolutionary tracing. Protein Eng. 1999;12:943–951. doi: 10.1093/protein/12.11.943. [DOI] [PubMed] [Google Scholar]
- Innis CA, Shi J, Blundell TL. Evolutionary trace analysis of TGF-beta and related growth factors: implications for site-directed mutagenesis. Protein Eng. 2000;13:839–847. doi: 10.1093/protein/13.12.839. [DOI] [PubMed] [Google Scholar]
- Aloy P, Querol E, Aviles FX, Sternberg MJ. Automated structure-based prediction of functional sites in proteins: applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J Mol Biol. 2001;311:395–408. doi: 10.1006/jmbi.2001.4870. [DOI] [PubMed] [Google Scholar]
- Wells JA. Systematic mutational analyses of protein-protein interfaces. Methods Enzymol. 1991;202:390–411. doi: 10.1016/0076-6879(91)02020-a. [DOI] [PubMed] [Google Scholar]
- Taylor WR. Residual colours: a proposal for aminochromography. Protein Eng. 1997;10:743–746. doi: 10.1093/protein/10.7.743. [DOI] [PubMed] [Google Scholar]
- Osipiuk J, Gornicki P, Maj L, Dementieva I, Laskowski R, Joachimiak A. Streptococcus pneumoniae YlxR at 1.35 Å shows a putative new fold. Acta Crystallogr D Biol Crystallogr. 2001;57:1747–1751. doi: 10.1107/s0907444901014019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grill S, Moll I, Hasenohrl D, Gualerzi CO, Blasi U. Modulation of ribosomal recruitment to 5'-terminal start codons by translation initiation factors IF2 and IF3. FEBS Lett. 2001;495:167–171. doi: 10.1016/s0014-5793(01)02378-x. [DOI] [PubMed] [Google Scholar]
- Bae W, Xia B, Inouye M, Severinov K. Escherichia coli CspA-family RNA chaperones are transcription antiterminators. Proc Natl Acad Sci USA. 2000;97:7784–7789. doi: 10.1073/pnas.97.14.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Huang K, Li Z, Banerjei L, Fisher KE, Grishin NV, Eisenstein E, Herzberg O. Crystal structure of YbaK protein from Haemophilus influenzae (HI1434) at 1.8 Å resolution: functional implications. Proteins. 2000;40:86–97. doi: 10.1002/(sici)1097-0134(20000701)40:1<86::aid-prot100>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Burns DM, Beacham IR. Identification and sequence analysis of a silent gene (ushA0) in Salmonella typhimurium. J Mol Biol. 1986;192:163–175. doi: 10.1016/0022-2836(86)90358-x. [DOI] [PubMed] [Google Scholar]
- Bensing BA, Dunny GM. Cloning and molecular analysis of genes affecting expression of binding substance, the recipient-encoded receptor(s) mediating mating aggregate formation in Enterococcus faecalis. J Bacteriol. 1993;175:7421–7429. doi: 10.1128/jb.175.22.7421-7429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani L, Gunderson SI, Mattaj IW, Kay LE, Neuhaus D, Varani G. The NMR structure of the 38 kDa U1A protein - PIE RNA complex reveals the basis of cooperativity in regulation of polyadenylation by human U1A protein. Nat Struct Biol. 2000;7:329–335. doi: 10.1038/74101. [DOI] [PubMed] [Google Scholar]
- Feng W, Tejero R, Zimmerman DE, Inouye M, Montelione GT. Solution NMR structure and backbone dynamics of the major cold-shock protein (CspA) from Escherichia coli: evidence for conformational dynamics in the single-stranded RNA-binding site. Biochemistry. 1998;37:10881–10896. doi: 10.1021/bi980269j. [DOI] [PubMed] [Google Scholar]
- Markus MA, Hinck AP, Huang S, Draper DE, Torchia DA. High resolution solution structure of ribosomal protein L11-C76, a helical protein with a flexible loop that becomes structured upon binding to RNA. Nat Struct Biol. 1997;4:70–77. doi: 10.1038/nsb0197-70. [DOI] [PubMed] [Google Scholar]
- GRASP: Graphical Representation and Analysis of Structural Properties http://btcpxx.che.uni-bayreuth.de/COMPUTER/Software/GRASP/
- Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- Thorn KS, Bogan AA. ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics. 2001;17:284–285. doi: 10.1093/bioinformatics/17.3.284. [DOI] [PubMed] [Google Scholar]
- Walther D. WebMol-a Java-based PDB viewer. Trends Biochem Sci. 1997;22:274–275. doi: 10.1016/s0968-0004(97)89047-0. [DOI] [PubMed] [Google Scholar]
- Joachimiak MP, Chang C, Rosenthal PJ, Cohen FE. The impact of whole genome sequence data on drug discovery - a malaria case study. Mol Med. 2001;7:698–710. [PMC free article] [PubMed] [Google Scholar]
- Wilson CA, Kreychman J, Gerstein M. Assessing annotation transfer for genomics: quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J Mol Biol. 2000;297:233–249. doi: 10.1006/jmbi.2000.3550. [DOI] [PubMed] [Google Scholar]
- Devos D, Valencia A. Practical limits of function prediction. Proteins. 2000;41:98–107. [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng DF, Doolittle RF. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol Evol. 1987;25:351–360. doi: 10.1007/BF02603120. [DOI] [PubMed] [Google Scholar]
- Feng DF, Doolittle RF. Progressive alignment of amino acid sequences and construction of phylogenetic trees from them. Methods Enzymol. 1996;266:368–382. doi: 10.1016/s0076-6879(96)66023-6. [DOI] [PubMed] [Google Scholar]
- Higgins DG, Sharp PM. Fast and sensitive multiple sequence alignments on a microcomputer. Comput Appl Biosci. 1989;5:151–153. doi: 10.1093/bioinformatics/5.2.151. [DOI] [PubMed] [Google Scholar]
- Rogers JS, Swofford DL. Multiple local maxima for likelihoods of phylogenetic trees: a simulation study. Mol Biol Evol. 1999;16:1079–1085. doi: 10.1093/oxfordjournals.molbev.a026197. [DOI] [PubMed] [Google Scholar]
- Protein sequence and structure utilities - ACCESS http://www.cmpharm.ucsf.edu/~srp/utils.html
- Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- Defay TR, Cohen FE. Multiple sequence information for threading algorithms. J Mol Biol. 1996;262:314–323. doi: 10.1006/jmbi.1996.0515. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics. 1989;5:164–166. [Google Scholar]
- Huang CC, Couch GS, Pettersen EF, Ferrin TE. Chimera: an extensible molecular modeling application constructed using standard components. Pac Symp Biocomput. 1996;1:724. [Google Scholar]
- Creighton TE. Proteins: Structures and Molecular Properties. New York: WH Freeman; 1992. [Google Scholar]
- Karplus PA. Hydrophobicity regained. Protein Sci. 1997;6:1302–1307. doi: 10.1002/pro.5560060618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCF sequence coloring format description and source code download http://www.cmpharm.ucsf.edu/~marcinj/SCF/
- Bernstein FC, Koetzle TF, Williams GJ, Meyer EF, Jr, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The protein data bank: a computer-based archival file for macromolecular structures. Arch Biochem Biophys. 1978;185:584–591. doi: 10.1016/0003-9861(78)90204-7. [DOI] [PubMed] [Google Scholar]
- JEvTrace manual and executable JAVA package download http://www.cmpharm.ucsf.edu/~marcinj/JEvTrace/
- Cho SJ, Lee MG, Yang JK, Lee JY, Song HK, Suh SW. Crystal structure of Escherichia coli CyaY protein reveals a previously unidentified fold for the evolutionarily conserved frataxin family. Proc Natl Acad Sci USA. 2000;97:8932–8937. doi: 10.1073/pnas.160270897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]