

Short abstract
High-quality full-insert sequence for 8,921 putative full-length cDNA clones in the Drosophila Gene Collection has been generated and compared to the annotated Release 3 genomic sequence. More than 5,300 cDNAs have been identifieed that contain a complete and accurate protein-coding sequence, corresponding to at least one splice form for 40% of the predicted D. melanogaster genes.
Abstract
Background
A collection of sequenced full-length cDNAs is an important resource both for functional genomics studies and for the determination of the intron-exon structure of genes. Providing this resource to the Drosophila melanogaster research community has been a long-term goal of the Berkeley Drosophila Genome Project. We have previously described the Drosophila Gene Collection (DGC), a set of putative full-length cDNAs that was produced by generating and analyzing over 250,000 expressed sequence tags (ESTs) derived from a variety of tissues and developmental stages.
Results
We have generated high-quality full-insert sequence for 8,921 clones in the DGC. We compared the sequence of these clones to the annotated Release 3 genomic sequence, and identified more than 5,300 cDNAs that contain a complete and accurate protein-coding sequence. This corresponds to at least one splice form for 40% of the predicted D. melanogaster genes. We also identified potential new cases of RNA editing.
Conclusions
We show that comparison of cDNA sequences to a high-quality annotated genomic sequence is an effective approach to identifying and eliminating defective clones from a cDNA collection and ensure its utility for experimentation. Clones were eliminated either because they carry single nucleotide discrepancies, which most probably result from reverse transcriptase errors, or because they are truncated and contain only part of the protein-coding sequence.
Background
One of the goals of the Berkeley Drosophila Genome Project is to define experimentally the transcribed portions of the genome by producing a collection of fully sequenced cDNAs. We have previously reported the construction of cDNA libraries from a variety of tissues and developmental stages; these libraries were used to generate over 250,000 expressed sequence tags (ESTs), corresponding to approximately 70% of the predicted protein-coding genes in the Drosophila melanogaster genome [1,2]. We used computational analysis of these ESTs to establish a collection of putative full-length cDNA clones, the Drosophila Gene Collection (DGC) [1,2]. Here, we describe the process by which we sequenced the full inserts of 8,921 cDNA clones from the DGC, describe the methods by which we assess each clone's likelihood of containing a complete and accurate protein-coding region, and illustrate how these data can be used to uncover additional cases of RNA editing. We have confirmed the identification of 5,375 cDNA clones that can be used with confidence for protein expression or genetic complementation.
Results and discussion
Sequencing strategy
Current approaches to full-insert sequencing of cDNA clones include concatenated cDNA sequencing [3], primer walking [4], and strategies using transposon insertion to create priming sites [5,6,7,8,9]. We adopted a cDNA sequencing strategy that relies on an in vitro transposon insertion system based on the MuA transposase, combined with primer walking (see Materials and methods for details).
The production of full-insert sequences from DGC cDNAs is summarized in Tables 1 and 2. For DGCr1, clones were sized before sequencing. Small clones (< 1.4 kilobases (kb)) were sequenced with custom primers and larger clones were sequenced using either mapped or unmapped transposon insertions. For DGCr2, clones were not sized and a set of unmapped transposon insertions was sequenced to generate an average of 5× sequence coverage. For both DGCr1 and r2, custom oligonucleotide primers designed using Autofinish [10] were used to bring the sequences to high quality. To date, we have completed sequencing 93% of the DGCr1 clone set and 80% of the DGCr2 clone set. The strategy used for sequencing DGCr1 clones appears to be more efficient, because on average they required fewer sequencing reads than DGCr2 clones. However, we were able to reduce cycle time and increase throughput using the shotgun strategy adopted for sequencing the DGCr2 clones. The average insert size of the 8,770 high-quality cDNA sequences that have been submitted to GenBank is 2 kb and they total 17.5 megabases (Mb) of sequence. The largest clone (SD01389) is 8.7 kb and is derived from a gene (CG10011) that encodes a 2,119-amino-acid ankyrin repeat-containing protein.
Table 1.
Status of DGCr1 and DGCr2 clones
| DGCr1 | DGCr2 | Total | |
| Clones in each release | 5,849 | 5,061 | 10,910 |
| Clones stopped while in progress* | 148 | 739 | 887 |
| Incorrect clone | 0 | 40 | 40 |
| Co-ligated inserts | 13 | 493 | 506 |
| No poly(A) | 9 | 97 | 106 |
| Transposable element (TE) | 11 | 71 | 82 |
| Incomplete coding sequence | 115 | 38 | 153 |
| Candidate clones to be sequenced | 5,701 | 4,322 | 10,023 |
| Submitted to GenBank† | 5,291 | 3,479 | 8,770 |
| Clones in progress | 410 | 843 | 1,253 |
*Quality-control analysis was carried out on clones during the sequencing process. Initial quality-control analysis was carried out for DGCr1 clones before full-length sequencing and for DGCr2 clones during the initial shotgun phase. This difference accounts for the different frequencies of error types observed in the DGCr1 and DGCr2. For example, the DGCr1 3' ESTs were generated before adding the clones to the sequencing pipeline allowing us to eliminate co-ligated clones and clones without poly(A) tails. Conversely, the DGCr2 has fewer clones with incomplete coding sequences because the DGCr2 clones were selected by aligning ESTs to the annotated genomic sequence, providing a more reliable way of selecting clones with complete ORFs than the inter se clustering of ESTs used to select the DGCr1. Clones were removed from finishing if they: were the incorrect clone as revealed by their 5'-end sequence; consisted of two cDNA molecules ligated into the same plasmid vector, as indicated by their 5'- and 3'-end reads aligning more than 300 kb apart in the genome; did not contain a poly(A) tract at their 3' end; corresponded to a member of the transposable element data set [20]; or did not extend to the ATG start site of the corresponding predicted protein in the Release 2 CDS data set. †Each clone submitted to GenBank has a contiguous sequence with a phrap estimated error rate of not more than one error per 50,000 bases. Additionally, each individual base has a phred [32,33] quality score of 25 or higher. An exception to these rules was made for 475 clones from the DCGr1 clone set that were submitted to GenBank before we increased our error rate standard from one in 10,000 to one in 50,000. These clones are undergoing additional sequencing to improve their quality to meet the higher standard.
Table 2.
Finished clone statistics
| DGCr1 | DGCr2 | Total | |
| Number submitted to GenBank | 5,291 | 3,479 | 8,770 |
| Percentage of clones finished without custom primers* | 88% | 88% | 88% |
| Average number of reads/kb for finished clones | 12.9 | 19.4 | 15 |
| Average number of primers to finish* | 3.7 | 2.4 | 3.4 |
| Average insert size of finished clones (kb) | 2.23 | 1.67 | 2.01 |
| Sequence (Mb) | 11.8 | 5.7 | 17.5 |
*Excludes clones <1.4 kb in size.
Evaluating the coding potential of each cDNA on the basis of its full-insert sequence
For many potential uses in proteomics and functional genomics [11,12,13], it is important to establish cDNA collections comprised only of cDNAs with complete and uncorrupted open reading frames (ORFs). To determine which of our sequenced clones meet this standard, we compared them to the annotated Release 3 genome sequence [14,15] using a combination of BLAST [16] and Sim4 [17] alignments (see Materials and methods for details).
We grouped the cDNAs into four categories (Table 3). The first category contains a total of 5,916 cDNA clones, or 68% of the sequenced clones. We are confident that 5,375 of these clones contain a complete and accurate ORF, as they precisely match the Release 3 predicted protein for the corresponding gene. An additional 541 clones are from the SD, GM and AT libraries, which were generated from fly strains that are not isogenic with the strain used to produce the genome sequence. The predicted ORFs from clones from these libraries were required to be identical in length to the Release 3 predicted protein with less than 2% amino-acid difference to be placed in this category. We cannot at present distinguish whether these differences result from strain polymorphisms or reverse transcriptase (RT) errors. However, our own internal estimates of RT errors (see below), based on the observed nucleotide substitution rate in cDNAs derived from the same strain as the genomic sequence, and published estimates of strain polymorphisms [18] lead us to believe that the majority of these changes are the result of strain polymorphism.
Table 3.
cDNA analysis
| DGCr1 | DGCr2 | Total | |
| Clones that encode complete ORFs | |||
| ORFs identical to the Release 3 predicted proteins* | 3,429 | 1,946 | 5,375 |
| ORFs with 1-2% differences to Release 3 proteins† | 235 | 306 | 541 |
| Total | 3,664 | 2,252 | 5,916 |
| Clones known to be compromised‡ | |||
| Nucleotide discrepancies | 485 | 829 | 1314 |
| 5' short | 618 | 150 | 768 |
| 3' truncated | 57 | 26 | 83 |
| Co-ligated inserts | 23 | 54 | 77 |
| ORFs with less than 50 amino acids | 49 | 21 | 70 |
| Antisense transcripts | 53 | 58 | 111 |
| Transposable elements | 12 | 9 | 21 |
| Bacterial contaminants | 2 | 4 | 6 |
| Total | 1,299 | 1,151 | 2,450 |
| Clones that may represent alternative transcripts§ | |||
| 5' short with upstream in-frame stop codon | 32 | 4 | 36 |
| 3' truncated with downstream in-frame stop codon | 55 | 17 | 72 |
| Putative missed micro-exon in Release 3 annotation | 23 | 7 | 30 |
| Total | 110 | 28 | 138 |
| Unclassified clones¶ | 257 | 160 | 417 |
Summary of analysis of the 8,770 clones in GenBank plus 151 clones for which we do not have accession numbers yet. *The ORF predicted from the cDNA sequence is identical to the corresponding Release 3 predicted protein; 4,620 of these clones are from the LD, GH, HL, LP, RE or RH cDNA libraries, which were made from the same strain that was sequenced. Thus, we required their ORFs to be identical to those of the predicted Release 3 proteins. An additional 755 clones with ORFs identical to Release 3 proteins are from the AT, GM or SD libraries. †The ORF predicted from the cDNA sequence is the same length as the Release 3 predicted protein with less than 2% amino-acid difference. These clones are derived from the AT, GM or SD cDNA libraries, which were made from strains or cell lines that are not isogenic with the strain that was sequenced. ‡See text for explanation of the individual subclasses of compromised clones. §These clones have structures that are inconsistent with the corresponding Release 3 predicted gene. The 5'-short and 3'-truncated clones may reflect alternative splice products or promoters, or perhaps more likely, incompletely processed primary transcripts with retained introns. Additional experimental work will be required to distinguish these possibilities. Those clones referred to as putative missed micro-exons in Release 3 annotations are cases in which the cDNA clone contains additional nucleotides that are a multiple of 3, relative to the Release 3 predicted mRNA, and maintains the ORF. We expect that most of these discrepancies result from a failure of Sim4 to align micro-exons and that these cases will be resolved by modifying the Release 3 gene model; see [15] for more discussion. ¶The predicted ORF from the cDNA clone does not match a Release 3 predicted protein, but the underlying cause could not be classified into one of the above categories. We expect that very few of these clones accurately reflect actual gene transcripts.
The second category represents 2,450 clones that are known to be compromised in one of a number of ways. The sequences of the largest class of compromised clones (1,314) align to the Release 3 predicted transcripts, but have nucleotide discrepancies that are most likely the result of errors generated by RT during library construction. These include missense and frameshift (+/-1 or +/-2 nucleotide difference) changes in the predicted ORF relative to the Release 3 predicted protein. Clones placed in this class can show up to 2% amino acid differences from the Release 3 peptide for isogenic libraries, and up to 4% difference for non-isogenic libraries. We estimated the error rate of an RNAseH-deficient RT (SuperScriptII, Invitrogen, Carlsbad, CA) by comparing the nucleotide sequence of cDNAs from isogenic libraries to the genomic sequence. For the GH, HL, LD, and LP libraries [1], we observed an error rate of 1 in 4,000; for the RE and RH libraries [2], we observed an error rate of 1 in 1,000. This difference is likely due to the different RT reaction conditions used in these two library construction protocols [1,2]. Although these numbers are higher than the 1 in 15,000 figure reported for SuperScriptII (Taurai Nenguke, personal communication), the in vitro assay used to obtain this error rate is based on assaying a single site for mutations that revert an amber codon.
The next largest class of compromised clones (768) consists of clones apparently truncated at their 5' ends, as judged by comparison to the Release 3 predicted ORFs of the corresponding genes. The 768 5'-short clones represent 757 distinct Release 3 annotated transcripts. For 151 of the 5'-short clones, 143 from DGCr1 and eight from DGCr2, we were able to identify clones with longer ORFs by additional EST sequencing. The remaining 606 clones are assumed to be 5' short because they do not possess a 5' in-frame stop codon and the corresponding annotated ORF in Release 3 extends further 5'. This class of clones represents approximately 9% of all finished clones, consistent with our original estimates that 80-94% of the DGC clones would contain the full ORF [1,2].
The remaining six classes of compromised clones consist of a total of 368 cDNAs (4% of all finished clones, see Table 3). Eighty-three clones encode ORFs that are truncated at their carboxy-termini and are most likely the result of priming from internal poly(A) tracts. Seventy-seven clones contain two unrelated ORFs and are almost certainly the result of two cDNAs being cloned into the same plasmid vector during library construction. Seventy clones contain ORFs of less than 50 amino acids. One hundred and eleven clones overlap a Release 3 predicted gene but are transcribed from the opposite strand from that of the mRNA encoding the Release 3 predicted protein and are considered anti-sense transcripts; a number of such cases were documented in the reannotation of the genome [15] and have been reported in many organisms [19]. Twenty-one clones correspond to transcripts of transposable elements on the basis of their sequence similarity to identified Drosophila transposons [20]. Finally, six clones contain a bacterial transposable element (Tn10, IS1 or IS2) that most likely inserted into the clone during propagation in Escherichia coli (bacterial contaminants).
The third and fourth categories consist of clones that may represent alternative transcripts (138) and clones that are currently computationally unclassified (417), respectively. The summary of the analysis of these clones is described in Table 3.
Improving the Drosophila cDNA resource
We have identified and sequenced cDNA clones that contain a complete and accurate ORF for 40% of all predicted Drosophila genes. We plan on extending this project in two ways. First, we intend to increase the number of genes represented in this set of fully vetted cDNA clones using a combination of experimental approaches. We can use site-directed mutagenesis to correct clones that carry single nucleotide changes or other small, localized defects. For the majority of the compromised clones, we have candidate replacement clones available that were identified as part of our EST sequencing and analysis efforts [2]. Generation of the Release 3 annotation of the genome made extensive use of our full-insert sequence data [15]. In the course of that effort, human curators identified a total of 2,013 clones that have become the DGCr3. The DGCr3 currently includes 309 clones chosen to replace clones with truncated ORFs, 543 clones for genes that are not currently represented in the DGC, and 833 clones that represent alternative splicing forms. To identify cDNAs for the remaining genes, we plan on using a combination of additional EST sequencing, reverse transcriptase PCR (RT-PCR) and cDNA library screening. Second, we plan on transferring ORFs to a universal cloning system (see [21,22] for examples) in order to generate a standard reagent for proteomics and other functional genomic experiments. In collaboration with Orbigen [23], we have already generated 72 baculovirus expression clones from a set of Gateway (Invitrogen, Carlsbad, CA) clones encoding transcription factors.
mRNA editing
RNA editing is a well-documented mechanism of generating nucleotide diversity beyond that directly encoded by the genome. Adenosine deaminase (ADAR) targets double-stranded regions of nuclear-encoded RNAs, catalyzing the deamination of adenosine (A) to inosine (I) [24]. Inosine mimics guanosine (G) in its base-pairing properties, and the translational machinery of the cell interprets I as G. In this way, an A-to-I conversion in the mRNA can alter the genetic information and, consequently, protein function. Null mutations in the single ADAR gene in Drosophila (dADAR) suggest that the function of pre-mRNA editing is to modify adult behavior by altering signaling components in the nervous system [25,26]. Among the mRNAs known to be edited in Drosophila are those encoded by cacophony (a calcium channel gene) [27], paralytic (a sodium channel gene) [28] and GluCla (a chloride channel gene) [29], all of which have multiple editing sites in their coding sequences.
In the course of evaluating the quality of the DGCr1 and DGCr2 cDNAs, described above, we compared their translation products to those of the recently completed Release 3 genomic sequence. Such comparisons should reveal cases of RNA editing. In cases in which the predicted protein sequences disagreed, we examined the corresponding nucleotide sequences in search of site-specific A-to-G variation between cDNA and genomic sequences. We identified over 30 candidates consistent with RNA editing; however, additional cDNA or EST data will be required to distinguish RNA editing from RT errors or strain polymorphisms. In a few cases we had enough cDNA and EST data to indicate that RNA editing is the most likely explanation for the observed variation. One such example is shown in Figure 1. The gene CG18314 encodes a G-protein-coupled receptor of the rhodopsin family, containing a seven-transmembrane protein domain with similarity to β2-adrenergic receptors of mouse and human [30,31]. Ten potential sites of RNA editing were revealed by comparison of the genomic sequence with those of two cDNAs and three ESTs. We validated these 10 sites by gene-specific RT-PCR using RNA isolated from heads of isogenic animals and identified 15 new sites (see legend to Figure 1). We are now in the process of a more rigorous and thorough analysis of potential RNA-editing targets.
Figure 1.
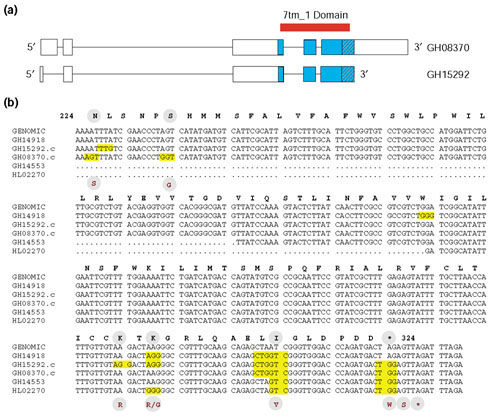
A putative example of RNA editing as revealed by comparison of cDNA and genomic DNA sequences. (a) Gene models for CG18314 based on sequence of two DGCr1 full-length cDNA clones (GH15292.c, GH08370.c) that differ at their 5' and 3' termini. Although the cDNAs have alternative 5' and 3' UTRs and are alternatively spliced, they share the same protein-coding potential (shown in blue). CG18314 encodes a G-protein-coupled receptor of the rhodopsin family, containing a seven-transmembrane protein domain (7tm_1; the red bar shows the extent of the domain) with similarity to β2-adrenergic receptors of mouse (X15643, E value = 9e-23) and human (M15169, E = 8e-22). Shown hatched is a 310-bp portion of cDNA sequences with A-to-G nucleotide variation. (b) Sequence alignments of this 310-bp portion of genomic sequence, two cDNA and three EST sequences (GH14918, GH14553, HL02270). Shown in yellow are codons with A-to-G nucleotide variation. Above the genomic nucleotide sequence is its translated amino-acid sequence starting at amino acid 224 of the protein. Comparing the cDNA nucleotide sequence to the genomic sequence identifies 10 A-to-G nucleotide variations. Two are silent, seven result in amino-acid changes, and one alters the stop codon, allowing two additional amino acids to be encoded. The amino acids that are affected are shown below the nucleotide sequence (red letters in a gray circle). Two of the amino-acid changes (N224S and S229G) map to the conserved seven-transmembrane protein domain. The Anopheles gambiae genomic draft contains sequence encoding this protein (gi|21299606|gb|EAA11751.1| (AAAB01008960) agCP5433) which is highly conserved at the amino-acid sequence level (E = e-168) and also encodes N and S at these sites. To sample additional transcripts of this gene, we performed gene-specific RT-PCR to amplify the region shown in (b). From a total of 64 independent transcripts we confirmed the 10 cases of editing diagrammed above, and identified 15 new sites of A-to-G nucleotide variations. A list of these putative editing sites showing the resulting amino-acid change and the number of times this change was observed, given in parentheses, is as follows: N224D (2), N224S (12), L225L (9), N227S (1), S229G (9), H230R (1), M231V (1), L236L (16), A239A (1), P246P (2) E254G (1), I272I (1), I275M (1), I281V (1), S286G (1), K306R (16), K308R (5), K308G (8), Q312Q (1), A313A (1), L315L (31), I316V (52), *323W (44) and S324G (4).
Materials and methods
Sequencing strategy
The Drosophila Gene Collection (DGC) consists of two releases, DGCr1 and DGCr2. A process flow diagram of our sequencing strategies is available online [32] and is summarized below. The clones in DGCr1 were arrayed by insert size [1] and sequenced accordingly; clones in DGCr2 were not arrayed by size. DGCr1 clones less than 1.4 kb were assembled using phrap [33] and analyzed with custom scripts to determine whether they were complete. Autofinish (part of the consed computer software package) was used to automatically design custom primers [10] for clones that needed quality improvement. Clones that did not finish in the first two rounds of Autofinish were sent to a manual finishing queue for more sophisticated finishing. cDNA clones larger than 1.4 kb were divided into three groups: 1.4 to 3 kb, 3 to 4.5 kb, and greater than 4.5 kb. All clones were sequenced using the in vitro Template Generation System (TGStm Finnzyme). Clones 3 to 4.5 kb in size, were sequenced using a minimal path of transposon-bearing clones. Clones, 1.4 to 3 kb and those greater than 4.5 kb, were sequenced with 24 and 48 unmapped transposon-bearing clones, respectively. After the initial cycle of transposon sequencing, the clones were analyzed using in-house scripts and Autofinish to determine their state of completeness and quality. DGCr2 clones were sequenced using 24 unmapped transposon-bearing clones. After an initial cycle of transposon sequencing, the clones were analyzed for completeness and quality as described above for DGCr1 clones, using in-house scripts and Autofinish. DGCr2 clone sequences were screened for transposable element sequences, cases of co-ligation, and presence of a poly(A) tail before any finishing work was ordered.
In vitro transposition and mapping insertion sites
Transposon insertion reactions were carried out in 96-well format using the Template Generation System (TGStm) according to the manufacturer's recommendations (Finnzyme). Transposon reactions consisted of 1 μl (50-150 ng) plasmid DNA isolated from Qiagen or Revprep DNA isolation robots, 1.6 μl 5× reaction buffer, 8 ng Entranceposon (KanR), 0.4 μl MuA transposase, and deionized water to bring the final volume to 8 μl. Reactions were carried out in PCR plates and incubated in an ABI thermocycler according to the manufacturer's instructions. After heat inactivation of the MuA transposase, 2 μl of the reaction were used to transform 17 μl of DH5α chemically competent cells (Invitrogen) in 96-well format. Following incubation at 37°C for 1 h in 183 μl SOC medium, cells were plated onto appropriate medium selecting for vector and Entranceposon antibiotic resistance. Plates were incubated at 37°C overnight. Colonies were picked into 1.2-ml polypropylene titer tubes (E&K Scientific) containing 0.5 ml LB medium supplemented with 7.5% glycerol and the appropriate antibiotics and incubated at 37°C overnight. These stocks were then used to inoculate 1.2 ml 2XYT medium in 96-well square deep-well plates (E&K Scientific) for culture and DNA plasmid preps. Transposon insertion sites were mapped relative to the vector ends by PCR essentially as described [34]. Forty-eight transposon-bearing clones were picked for PCR mapping using the Mu-End primer (present at both ends of the tranposon) in combination with vector-specific primers, resulting in 96 PCR products. Agarose gels were imaged using custom software developed in-house (Earl Cornell, LBNL) and analyzed using an algorithm, Supertramp [35,36], to identify a minimal path of transposon-bearing clones to be re-arrayed and sequenced.
DNA sequencing
Purified plasmid DNA from transposon-bearing clones was sequenced using 2 μl ABI BigDye II Dye terminator mix (Applied Biosystems) in a 10-μl reaction. Sequencing reactions were processed through 96-well Sephadex G-50 SF plates (Multiscreen filter plates; Millipore) and loaded onto ABI Prism 3700 DNA Analyzer. Sequencing primers specific for each end of the Entranceposon were used in the reactions (5'-ATCAGCGGCCGCGATCC-3' and 5'-TTATTCGGTCGAAAAGGATCC-3'). Sequencing of 5' and 3' cDNA ends was carried out as previously described [2]. The sequencing reported here was carried out over a 2-year period during which we made several major modifications to the strategy; for example, switching from sequencing mapped transposon insertions to random transposons. These changes improved throughput and cycle time, but made the process less efficient in terms of the required number of sequencing reads. Because of these changes, it is not possible to give a meaningful single efficiency estimate; however, our overall efficiency is comparable to other efforts using a similar strategy [8,9].
Data processing and assembly
cDNA clone data management relied on custom scripts and an Informix database. Sequences were processed using phred [37,38] and assembled using phrap [33]. 5' and 3' EST end-reads were combined with the transposon-based reads to generate cDNA clone assemblies. We adopted the sequence quality-control standards defined for the Mammalian Gene Collection project [39]. Custom scripts evaluated assemblies for: 5' and 3' EST reads in a single contig in the proper orientation; at least 10 bases of 3' poly(A) tail; phrap estimated error rate of less than one in 50,000 bases; and individual base quality of at least q25. Double-stranded coverage was not a criterion for a clone to be considered finished; however, we have determined that 96.2% of all submitted bases are double-stranded and 48% of clones had complete double-stranded coverage. Autofinish [10] was used to design primers to improve quality or extend sequence from multiple sequence contigs. cDNA clones with an estimated error rate greater than one in 50,000 bp were automatically identified and processed with additional rounds of Autofinish designed finishing work. If Autofinish could not design primers, custom primers were designed manually using consed. Custom scripts were used to manually order primers to generate a further round of sequencing.
The sequence data described in this paper have been submitted to the GenBank data library under accession numbers:
AF132551-AF132560, AF160903-AF160904,
AF132562-AF132563, AF160906, AF160909,
AF132565-AF132567, AF160911-AF160913,
AF145594-AF145621, AF160916-AF160917,
AF145623-AF145684, AF160921, AF160923,
AF160879, AF160882, AF160933-AF160934,
AF160889-AF160891, AF160938-AF160944,
AF172635-AF172637, AY071209-AY071211,
AF181622-AF181650, AY071213-AY071216,
AF181652-AF181657, AY071218-AY071250,
AF184224-AF184230, AY071252-AY071266,
AY047496-AY047580, AY071268-AY071288,
AY050225-AY050241, AY071290-AY071313,
AY051411-AY052150, AY071315-AY071320,
AY058243-AY058797, AY071322-AY071331,
AY059433-AY059459, AY071333-AY071342,
AY060595-AY061633, AY071347-AY071381,
AY061821-AY061834, AY071383-AY071385,
AY069759-AY069867, AY071389-AY071406,
AY070491-AY070597, AY071408-AY071436,
AY070599-AY070602, AY071438-AY071445,
AY070604-AY070608, AY071447-AY0 71450,
AY070610-AY070623, AY071452-AY071454,
AY070625-AY070628, AY071456-AY071461,
AY070632-AY070634, AY071463-AY071476,
AY070646-AY070651, AY071545-AY071557,
AY070653-AY070656, AY071559-AY071564,
AY070658-AY070662, AY071566-AY071577,
AY070664-AY070667, AY071579-AY071581,
AY070671-AY070692, AY071583-AY071606,
AY070694-AY070716, AY071608-AY071632,
AY070777-AY070805, AY071634-AY071661,
AY070807-AY070830, AY071663-AY071664,
AY070832-AY070909, AY071666-AY071672,
AY070915-AY070920, AY071681-AY071683,
AY070922-AY070951, AY071685-AY071692,
AY070953-AY070954, AY071694-AY071703,
AY070957-AY070964, AY071705-AY071711,
AY070975-AY070985, AY071726-AY071727,
AY070987-AY071000, AY071729-AY071731,
AY071004-AY071006, AY071743-AY071745,
AY071008-AY071056, AY071747-AY071764,
AY071058-AY071064, AY071767-AY071768,
AY071066-AY071072, AY075158-AY075228,
AY071074-AY071084, AY075230-AY075262,
AY071086-AY071090, AY075264-AY075441,
AY071094-AY07H36, AY075453-AY075473,
AY071138-AY071140, AY075475-AY075524,
AY071142-AY071154, AY075526-AY075588,
AY071156-AY071157, AY084089-AY084152,
AY071159-AY071197, AY084154-AY084214,
AY071199-AY071203, AY089215-AY089229,
AY071205-AY071207, AY089231-AY089329,
AY089331-AY089461, AY118273-AY118672,
AY089463-AY089564, AYn8674-AYn8713,
AY089566-AY089601, AY118715-AY119132,
AY089603-AY089615, AY119134-AY119287,
AY089617-AY089700, AY119441-AY119665,
AY094627-AY094871, AY121612-AY121684,
AY094873-AY094970, AY121686-AY121700,
AY094996-AY095100, AY121702-AY121717,
AY095172-AY095206, AY122061-AY122270,
AY095508-AY095533, AY128413-AY128506,
Analysis of finished cDNA sequences
cDNA sequence was submitted to GenBank with a preliminary annotation of the longest ORF and a gene assignment based on a high BLASTN similarity score to the Release 2 genome annotations. Subsequent processing was used to determine a more detailed analysis of the clone quality. Using BLASTN, sequence from each cDNA clone was compared to genomic sequence, predicted genes, predicted coding sequences (CDSs), known Drosophila transposable elements, and Escherichia coli transposable elements. Using BLASTP, the translation of the longest ORF was compared to the predicted Release 3 translations [15]. Custom scripts were used to parse the BLAST output and record similarity results. We also compared the nucleotide sequence of each clone to the Release 3 genome sequence [14] using Sim4 and to the Release 3 predicted CDS with the highest BLAST score.
mRNA editing
We confirmed the sequence quality of the genomic region encompassing CG018314 (12,731 bp) by independently assembling an 18,284 bp contig consisting solely of whole-genome shotgun (WGS) traces. The assembled sequence contig has an average of 8.6× sequence coverage. The phrap estimated error rate for each genomic base corresponding to a mRNA edited base is q90. Similarly, we determined the phrap estimated error rate for each mRNA edited base to be q90. We manually inspected chromatograms for high-quality discrepancies in the genomic sequence and found none, indicating that the edited bases are not due to population heterozygosity. To validate the editing sites, total RNA was isolated from heads from a mixed population of male and female adult flies from the isogenic strain y1; cn1 bw1 sp1 using the Concert™ Cytoplasmic RNA isolation reagent according to the manufacturer's guidelines (Invitrogen). Nine independent gene-specific RT-PCR reactions were performed using the Superscript™ one-step RT-PCR kit according to the manufacturer (Invitrogen) and PCR products were cloned into the PCR2.1 vector. Twenty-four independent subclones from each of four independent RT-PCR products were sequenced and twelve independent subclones from an additional five independent RT-PCR products were sequenced; we considered amplicons to represent independent transcripts if they arose from different RT-PCR reactions or if they differed in sequence. The gene-specific primers used in the RT-PCR experiments were 5'-GTGCAGACGAAAACGAGATGCCAATG-3' and 5'-TGTAGTTCTTCTCAAAGGGATTACG-3'.
Acknowledgments
Acknowledgements
We thank Catherine Nelson for critically reading and improving the manuscript, Sandeep Patel for help during the manual finishing phase of cDNA sequencing, Michelle Chew for excellent technical assistance in the early stages of this project, and the entire staff of the BDGP sequencing center. We also thank the FlyBase curators for their help in identifying the DGCr3, and Erwin Frise and Eric Smith for system administration. This work was supported by NIH Grant P50-HG00750 (GMR), Department of Energy Grant nos. DE-FG03-98ER62625 and DE-FG03-99ER62739 (GMR), and performed under Department of Energy Contract DE-AC0376SF00098, University of California.
This article is part of a series of refereed research articles from Berkeley Drosophila Genome Project, FlyBase and colleagues, describing Release 3 of the Drosophila genome, which are freely available at http://genomebiology.com/drosophila/.
References
- Rubin GM, Hong L, Brokstein P, Evans-Holm M, Frise E, Stapleton M, Harvey DA. A Drosophila complementary DNA resource. Science. 2000;287:2222–2224. doi: 10.1126/science.287.5461.2222. [DOI] [PubMed] [Google Scholar]
- Stapleton M, Liao G, Brokstein P, Hong L, Carninci P, Shiraki T, Hayashizaki Y, Champe M, Pacleb J, Wan K, et al. The Drosophila Gene Collection: identification of putative full-length cDNAs for 70% of D. melanogaster genes. Genome Res. 2002;12:1294–1300. doi: 10.1101/gr.269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Andersson B, Worley KC, Muzny DM, Ding Y, Liu W, Ricafrente JY, Wentland MA, Lennon G, Gibbs RA. Large-scale concatenation cDNA sequencing. Genome Res. 1997;7:353–358. doi: 10.1101/gr.7.4.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann S, Weil B, Wellenreuther R, Gassenhuber J, Glassl S, Ansorge W, Bocher M, Blocker H, Bauersachs S, Blum H, et al. Toward a catalog of human genes and proteins: sequencing and analysis of 500 novel complete protein coding human cDNAs. Genome Res. 2001;11:422–435. doi: 10.1101/gr.154701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathmann M, Hamilton BA, Mayeda CA, Simon MI, Meyerowitz EM, Palazzolo MJ. Transposon-facilitated DNA sequencing. Proc Natl Acad Sci USA. 1991;88:1247–1250. doi: 10.1073/pnas.88.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine SE, Boeke JD. Efficient integration of artificial transposons into plasmid targets in vitro: a useful tool for DNA mapping, sequencing and genetic analysis. Nucleic Acids Res. 1994;22:3765–3772. doi: 10.1093/nar/22.18.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapa S, Suomalainen S, Eerikainen S, Airaksinen M, Paulin L, Savilahti H. An efficient DNA sequencing strategy based on the bacteriophage mu in vitro DNA transposition reaction. Genome Res. 1999;9:308–315. [PMC free article] [PubMed] [Google Scholar]
- Shevchenko Y, Bouffard GG, Butterfield YS, Blakesley RW, Hartley JL, Young AC, Marra MA, Jones SJ, Touchman JW, Green ED. Systematic sequencing of cDNA clones using the transposon Tn5. Nucleic Acids Res. 2002;30:2469–2477. doi: 10.1093/nar/30.11.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield YS, Marra MA, Asano JK, Chan SY, Guin R, Krzywinski MI, Lee SS, MacDonald KW, Mathewson CA, Olson TE, et al. An efficient strategy for large-scale high-throughput transposon-mediated sequencing of cDNA clones. Nucleic Acids Res. 2002;30:2460–2468. doi: 10.1093/nar/30.11.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D, Desmarais C, Green P. Automated finishing with autofinish. Genome Res. 2001;11:614–625. doi: 10.1101/gr.171401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout AJ, Boulton SJ, Vidal M. Yeast two-hybrid systems and protein interaction mapping projects for yeast and worm. Yeast. 2000;17:88–94. doi: 10.1002/1097-0061(20000630)17:2<88::AID-YEA20>3.0.CO;2-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M. A biological atlas of functional maps. Cell. 2001;104:333–339. doi: 10.1016/s0092-8674(01)00221-5. [DOI] [PubMed] [Google Scholar]
- Zhu H, Snyder M. "Omic" approaches for unraveling signaling networks. Curr Opin Cell Biol. 2002;14:173–179. doi: 10.1016/s0955-0674(02)00315-0. [DOI] [PubMed] [Google Scholar]
- Celniker SE, Wheeler DA, Kronmiller B, Carlson JW, Halpern A, Patel S, Adams M, Champe M, Dugan SP, Frise E, et al. Finishing a whole shotgun sequence assembly: Release 3 of the Drosophila euchromatic sequence. Genome Biol. 2002;3(12):research0079.1–0079.14. doi: 10.1186/gb-2002-3-12-research0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell K, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, et al. Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol. 2002;3:research0083.1–0083.22. doi: 10.1186/gb-2002-3-12-research0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JR. Progress and prospects in evolutionary biology: the Drosophila model. New York: Oxford University Press. 1997. pp. 344–420.
- Shendure J, Church GM. Computational discovery of sense-antisense transcription in the human and mouse genomes. Genome Biol. 2002;3:research0044.1–0044.14. doi: 10.1186/gb-2002-3-9-research0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminker JS, Bergman C, Kronmiller B, Carlson J, Svirskas R, Patel S, Frise E, Wheeler DL, Lewis SE, Rubin GM, et al. The transposable elements of the Drosophila melanogaster euchromatin - a genomics perspective. Genome Biol. 2002;3:research0084.1–0084.20. doi: 10.1186/gb-2002-3-12-research0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout AJ, Temple GF, Brasch MA, Hartley JL, Lorson MA, van den Heuvel S, Vidal M. GATEWAY recombinational cloning: application to the cloning of large numbers of open reading frames or ORFeomes. Methods Enzymol. 2000;328:575–592. doi: 10.1016/s0076-6879(00)28419-x. [DOI] [PubMed] [Google Scholar]
- BD Creator™ Gene Expression Systems http://www.clontech.com/products/families/creator/index.shtml
- Orbigen http://www.orbigen.com
- Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O'Connell MA, Reenan RA. A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell. 2000;102:437–449. doi: 10.1016/s0092-8674(00)00049-0. [DOI] [PubMed] [Google Scholar]
- Ma E, Tucker M, Chen Q, Haddad G. Developmental expression and enzymatic activity of pre-mRNA deaminase in Drosophila melanogaster. Brain Res Mol Brain Res. 2002;102:100–104. doi: 10.1016/s0169-328x(02)00186-9. [DOI] [PubMed] [Google Scholar]
- Smith LA, Peixoto AA, Hall JC. RNA editing in the Drosophila DMCA1A calcium-channel α1 subunit transcript. J Neurogenet. 1998;12:227–240. doi: 10.3109/01677069809108560. [DOI] [PubMed] [Google Scholar]
- Reenan RA, Hanrahan CJ, Barry G. The mlenapts RNA helicase mutation in Drosophila results in a splicing catastrophe of the para Na+ channel transcript in a region of RNA editing. Neuron. 2000;25:139–149. doi: 10.1016/s0896-6273(00)80878-8. [DOI] [PubMed] [Google Scholar]
- Semenov EP, Pak WL, Related Articles, Links Diversification of Drosophila chloride channel gene by multiple posttranscriptional mRNA modifications. J Neurochem. 1999;72:66–72. doi: 10.1046/j.1471-4159.1999.0720066.x. [DOI] [PubMed] [Google Scholar]
- Allen JM, Baetge EE, Abrass IB, Palmiter RD. Isoproterenol response following transfection of the mouse β2-adrenergic receptor gene into Y1 cells. EMBO J. 1988;7:133–138. doi: 10.1002/j.1460-2075.1988.tb02792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobilka BK, Dixon RA, Frielle T, Dohlman HG, Bolanowski MA, Sigal IS, Yang-Feng TL, Francke U, Caron MG, Lefkowitz RJ. cDNA for the human-adrenergic receptor: a protein with multiple β2 membrane-spanning domains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet-derived growth factor. Proc Natl Acad Sci USA. 1987;84:46–50. doi: 10.1073/pnas.84.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- cDNA production process flow http://www.fruitfly.org/DGC/FLSWorkflow.html
- The Phred/Phrap/Consed system home page http://www.phrap.org
- Kimmel B, Palazzolo MJ, Martin CH, Boeke JD, Devine SE. Transposon-mediated DNA sequencing. In: Birren B, Green ED, Klapholz S, Myers RM, Roskams J, editor. In Genome Analysis: a laboratory manual Analyzing DNA. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 455–532. [Google Scholar]
- Veklerov E, Martin CH, Theil EH. TRAMP: a software package for generating transposon maps. Comput Appl Biosci. 1995;11:173–179. doi: 10.1093/bioinformatics/11.2.173. [DOI] [PubMed] [Google Scholar]
- Cawley SE, Speed TP. DNA sequencing with transposons. J Comput Biol. 2000;7:717–729. doi: 10.1089/106652701446161. [DOI] [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Mammalian Gene Collection http://mgc.nci.nih.gov