

Short abstract
Apollo was developed to enable curators to inspect genome annotations closely and edit them. FlyBase biologists successfully used Apollo to annotate the Drosophila melanogaster genome.
Abstract
The well-established inaccuracy of purely computational methods for annotating genome sequences necessitates an interactive tool to allow biological experts to refine these approximations by viewing and independently evaluating the data supporting each annotation. Apollo was developed to meet this need, enabling curators to inspect genome annotations closely and edit them. FlyBase biologists successfully used Apollo to annotate the Drosophila melanogaster genome and it is increasingly being used as a starting point for the development of customized annotation editing tools for other genome projects.
Rationale
Unadorned genomic sequence data is simply a string of As, Ts, Gs, and Cs, with perhaps an associated confidence value for each base. In this raw state, sequence data provides very little biological insight. To utilize any sequence it must be interpreted in the context of other biological knowledge. This is the process of annotation, the task of adding explanatory notations to the sequence text. We define an annotation as the biological evaluation and explanation of a specific region on a nucleic acid sequence that includes, but is not limited to, gene transcripts. Any feature that can be anchored to the sequence - for example, an exon, a promoter, a transposable element, a regulatory region, or a CpG island - is an annotation. The genomic sequence will stabilize and reach a finite endpoint, but the annotations will continue to evolve indefinitely, as biological knowledge increases. To understand the genetic legacy of an organism we must interpret its genomic sequence, translating the information it contains in molecular form into human-readable annotations.
Part of this process is purely computational, and in its simplest terms can be described as a process of recognition: can anything be located that is somehow already familiar? The first obvious tactic is to collect sequences that may represent interesting biological features and to search the genomic sequence in order to discover the presence or absence of similar sequences. The principle is the same whether the sequences used in this comparison are expressed sequence tags (ESTs), full-length cDNAs, repeated elements or highly conserved sequences, and whether the sequences come from the same species, a closely related species or a distantly related species; it is only the alignment algorithm that will vary appropriately, for example, BLAST [1], Sim4 [2], or BLAT [3]. The second general tactic is to utilize what is already known about specific classes of sequence features to construct an abstract model representing this combined knowledge and to search the genomic sequence to identify locations that match this model, for example, hidden Markov models (HMM) [4,5,6] and stochastic context-free grammars (SCFG) [7,8].
While these computational examinations reveal the general character of the primary sequence the results are still of limited use for further functional biological research [9,10,11,12]. For example, BLAST provides alignment results composed of high-scoring pairs (HSPs) indicating local regions of similarity, but in total these typically do not provide the best global alignment over the entire length of the peptide to the genomic sequence. Similarly gene-prediction programs can generate quite different results depending on the length of the genomic sequence analyzed and the gene density of the surrounding sequence. Even systems that strive to make adjustments and collate the results of all the analyses are unable to consistently provide accurate biological models (see [13] for a more detailed discussion). Our current computational analysis capabilities are useful for primary annotation, but the biological imprecision of the results, especially within eukaryote genomes, makes expert evaluation absolutely essential to ensure the correctness of the annotations. In addition, this step is required to deepen our understanding by connecting these annotations to all the experimentally known biology of the organism.
Software developers are familiar with debugging tools and techniques. Apollo is an analogous tool for debugging genomic feature descriptions derived from automated analyses and computational pipelines. It is also a tool to facilitate connecting and comparing these annotations with existing biological data. As an introspective tool, Apollo makes it possible for researchers to closely examine and compare explicit details and contrast them with their own biological knowledge and apply any necessary corrections. This evaluation is absolutely essential for progress in genomics for three reasons.
First, we must improve our current algorithms and recognition techniques for protein-coding genes. Several groups have augmented standard approaches based solely on protein-coding potential by exploiting exact alignments to cDNA and EST data and similarities to sequences from the same or different genomes (see, for example [14,15]). However, we are not yet at the stage where we can make intelligent use of all the existing data automatically. Resolving the conflicts that exist between EST evidence, BLAST similarities and computational predictions can be extremely complicated and is still best done by manual inspection. The simplifications that existing automated annotation pipelines must make lead to a number of systematic errors, including under- or over-predicting genes as well as merging the exons from two genes in one gene model or splitting the exons from one gene into two gene models. For example, if the software discounts unspliced EST evidence (a common simplification made to reduce the effect of errors in EST databases), small genes can easily be omitted. In addition, issues such as determining alternative transcripts, understanding promoter usage, identifying polyadenylation sites, distinguishing genes within the intron of another gene, and recognizing dicistronic genes, are all outstanding problems in the annotation of protein-coding genes. Yet, as more genomes become available, we cannot hope to provide a high level of manual examination and correction to describe the genes present on these genomes precisely. It is essential that we utilize the knowledge we are gaining from these initial 'well-annotated' genomes to improve the existing computational methods, both by generating more comprehensive training sets as well as by refining the algorithms themselves.
Second, we must expand the available computational repertoire to include methods for annotating other biologically important features. Current computational analyses are primarily only capable of describing simple protein-coding gene models and these are therefore the sole targets of most automated strategies. There are other useful predictive programs for tRNA genes [16] and promoters [17,18,19,20], but a large number of biological features still cannot be recognized by computational methods alone. For example, there are no fully automatic computational methods to detect biologically important annotations such as pseudogenes and non-coding RNA genes. Until a day arrives when we know all the feature types that we are searching for, and precisely how to find them in the genomic sequence, we will need to rely upon expert inspection to augment automated analyses.
Third, we must deliver the annotations to the community as quickly and as accurately as possible because the annotations themselves are an important resource. Researchers depend on the availability of complete peptide sets, gene structures and other data. Moreover, to be truly useful these data must be correct. Many people have noted and remarked upon the current fallibility of first-pass protein-coding gene predictions and other annotations (see, for example [9,10,11,12,21,22,23]). The technology is not yet available to provide highly reliable datasets. This impedes research that relies on these data. If researchers in the community were provided with a means of viewing the supporting data for each annotation as well as for making corrections to annotations, then the accurate data they require would become available much sooner.
Origins of Apollo
In the year 2000, within the space of six months, both the Drosophila genome [24] and the draft human genome [25] were published. Both the Berkeley Drosophila Genome Project (BDGP) working with Celera Genomics and other researchers, and the Ensembl [26] project working on the human genome provided the community with annotations in a very short time. However, to those who were working closely with the data it was obvious that in the longer term this initial analysis would have to be refined, improved and extended by manual curators. The genomics community required tools that would provide more than simply a view of these data, but that would enable professional curators, and ultimately every researcher, to easily modify and correct the annotations on an ongoing basis through direct interaction. Both FlyBase-BDGP [27] and Ensembl shared this understanding, and in the summer of 2000 entered into a collaborative agreement to achieve this aim.
Apollo has roots reaching back more than a decade, and builds on previous generations of software and domain knowledge. In the early 1990s the C. elegans project was underway at the Sanger Institute and the Drosophila genome project was beginning at Berkeley. Both projects share the same informatics root because both began by using ACeDB (documentation, code and data available by anonymous FTP from [28,29,30]). Berkeley developed a variant called FlyDB [31] and was actively involved in porting AceDB to the Macintosh computer platform. The Sanger Institute coordinated the overall development of ACeDB and has relied upon ACeDB for the ongoing annotation of C. elegans and human genome sequence.
In the mid-1990s, first Perl [32] and then Java [33] were becoming widespread as programming languages for developing bioinformatics tools. Our groups explored the use of both languages. Berkeley created bioTkPerl [34,35] and became one of the early participants in BioPerI [36]. BioTkPerl was used in a number of applications, but most notably in this context it was the underpinning for the annotation tool Genotator [37]. Genotator has been widely distributed and in turn formed the basis for other Perl-based genomic tools such as Genquire [38].
Initially we had hopes that Java would enable us to distribute both the data and the application software dynamically over the internet, and thus solve the problem of providing a far-flung community with both the freshest data and the latest application releases simultaneously. In practice, both the Berkeley and Ensembl groups use Perl for internet-based data access [39] and Java for locally resident applications. Our first Java applications were Artemis at the Sanger Institute [40] and CloneCurator at Berkeley. Artemis is widely used for annotating microbial and other smaller genomes. CloneCurator was used to annotate the 2.9 million base (Mb) Adh region of Drosophila [41] and provided the baseline that was used to evaluate the results of the Genome Annotation Assessment Project [9] trial. Although both provided useful tools for the tasks they were originally developed for, we needed a more sophisticated, and easier to use, annotation editor for a large, complex eukaryotic genome. There have been two commercial products that originated with CloneCurator and built upon its intellectual legacy, AnnotationStation (Neomorphic Software for TIGR) and Genome-Browser (Celera), but neither was freely available. Apollo is the most recent undertaking and builds upon all of this accumulated experience.
Several general concepts were learned over this period as we gained experience in working with genomic sequence data. One was the importance of distinguishing the presentation of the information from the underlying biological data model. Searls [42] stated this concept most clearly as the need to "drive semantics out" of the graphical presentation. The second key concept was an appreciation of well-understood and standard issues in data management and portability. We needed a graphical annotation tool that could be used easily at multiple sites with different data-management environments. Taken together, these issues led to the development of Apollo.
Overview of Apollo from the curatorial perspective
To quote Edward Tufte: "There are only two industries which refer to their customers as users, drugs and computers" [43]. In software engineering, a great divide has arisen separating the people writing the code from the people using the code. We sought to narrow this divide and wish to emphasize the degree to which people with a strong biological research incentive were integral to our team. Apollo's development was highly interactive and was entirely motivated by the scientific needs of the curators. The curators had a fundamental role in determining the design, requirements, and testing of Apollo. This section provides an overview of Apollo and its capabilities from a user's perspective.
The primary Apollo user interface is superficially familiar to anyone accustomed to genomic browsers; in addition, however, Apollo offers researchers the ability to probe, manipulate and alter the interpretation of the underlying data. Within the various views, annotations can be created, deleted, merged, split, classified and commented upon. For example, Apollo enables exon-level refinements for creating, deleting, splitting and merging exons, as well as adjusting splice edges for individual exons. Unlike a simple browser, Apollo is a tool with full editorial capabilities.
Viewing the data
The curator can load data by specifying a region on a selected chromosome. Depending on the source of the data this region may be determined by absolute sequence position, a file name, or, for Drosophila, gene name, cytological region (map position), scaffold accession or sequence (which will be mapped to a genomic location by BLAST similarity). After data from the desired region have been loaded, a navigation bar allows the user to easily access other regions of the genome - for example, the region immediately upstream or downstream of the currently displayed sequence. Apollo can transparently access data across the network from remote machines, as well as reading files that reside locally.
The main Apollo screen shows genomic features layered on a segment of DNA, where each genomic feature appears as a colored shape (typically rectangles, as shown in Figure 1). Features are grouped, meaning that the separate local alignments of subsequences derived from the same source sequence (HSPs) are treated as a single composite feature, and likewise the distinct exons from a prediction are grouped together into a single feature (composite features are typically drawn as lines connecting the rectangles representing member features). By default, the display shows features on both the forward and reverse strands, with the forward strand on top and the reverse strand on the bottom, separated by a coordinate bar. For each strand, the inner (light blue) panel contains annotations, showing exons and introns for each transcript of a protein-coding gene. The outer (gray) panel displays computational evidence to support these annotations, such as gene predictions generated by programs such as GenScan [44] and Genie [45], and other biological data, such as ESTs aligned using the program Sim4 [2] and protein homologies revealed by BLASTX [1].
Figure 1.
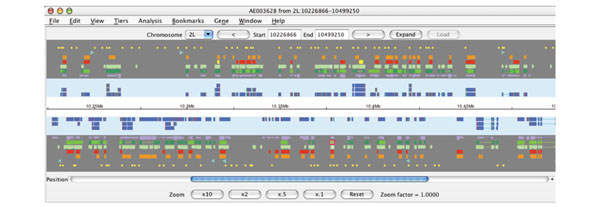
A standard view in Apollo, showing a 250 kb segment of Drosophila chromosome arm 2L. The background colors for the result (gray), annotation (light blue) and sequence (white) panels are configurable. The features and annotations on the forward strand are shown above the white sequence panel, and those on the reverse strand are shown below it. There are seven tiers (or rows) of data presented here in the results panel (the number of tiers is configurable). On the forward strand from top to bottom these results are promoter predictions (yellow), P-element insertions (turquoise triangles), peptide homologies to other species (orange), peptide homologies to Drosophila (red), Drosophila EST alignments (light green), Drosophila mRNA alignments (bright and dark green) and gene prediction results (lavender). The genomic region navigation bar is above the data displays and the navigational and zoom controls for the current genomic region in the display are below. The user may increase the genomic region visible in the display by using the 'Expand' button. They may also move to adjacent regions by clicking on the '<' (5' upstream) and '>' (3' downstream) buttons or move to entirely new regions by changing the chromosome arm and the start and end positions. Within the display the most basic movement operations (zooming and scrolling) are available from the controls at the bottom of the display.
The features representing the supporting evidence in the outer panel are organized into tiers and types. A tier is a horizontal row of supporting evidence placed at a specified position on the vertical axis that can be labeled and controlled as a single collection. A tier might be organized, for example, to contain all gene prediction results from various programs, or all BLASTX results from comparisons to several different species databases. In addition to the tier, each feature is associated with a particular data 'type'. A feature type is typically an association of that feature to the results produced by a particular analysis, for example, all features derived from a TBLASTX run against all mouse ESTs in dbEST might be given the type 'Mouse'. Each data type can be individually assigned a shape and a color (Figure 2). Users can control the maximum number of rows shown in a tier and set a score threshold so that only results above that threshold are visible. Operations on individual tiers include setting a vertical coordinate position or setting whether the tier is visible or hidden. The user can view the data within a tier in a collapsed style, in which all features within a tier are collapsed into one line (Figure 3), or in an expanded style, in which each individual feature is positioned to avoid overlapping any other feature. These two organizing principles of layout and category simplify viewing the massive amounts of data.
Figure 2.

Some of the feature appearance options that are available. Each feature's shape, visibility, color and panel position is configurable. The result panel can display the complete portfolio of computational results. The top three rows of this figure use small vertical green lines to indicate the position of start codons in the genomic sequence in all three possible translation frames. The second set of three rows does the same for stop codons (red). This enables curators to easily discern ORFs. Beneath this, in orange, are BLASTX hits to other species. Every row represents a separate alignment in this expanded view (see Figure 3 for a discussion of the expanded view). In each alignment the high-scoring pairs (HSP) are separated by two parallel lines to indicate alignment gaps. Seeing what features share edges is important during annotation for adjusting exon-intron boundaries. When an item is selected, corresponding edges of other items that end at the same nucleotide are highlighted with a white line (arrow A). The small turquoise triangle indicates the site of a P-element insertion. The rows of light green rectangles are Sim4 alignments of Drosophila ESTs to the genome. When a 5' and a 3' EST are derived from the same cDNA clone we connect them with a dashed line (arrow B). This contrasts with the solid lines used to represent the gaps, corresponding to introns, introduced into an EST to permit its alignment to the genome. The final piece of computational evidence shown in this figure is the gene prediction, shown in lavender. In this example, we have intentionally represented introns in two different ways - as straight or peaked lines - as an illustration of Apollo's configurable graphics. In the annotation panel, the curator has created two alternative transcripts for this gene, each of which is supported by multiple pieces of EST evidence. Individual exons that the curator has selected are outlined in yellow. The translation start and stop sites are shown as green and red vertical lines, respectively. Each individual curator has a signature color; any annotation that this particular curator creates is shown in bright blue. In the sequence panel the scale is drawn in red to indicate that this gene is on the reverse strand.
Figure 3.
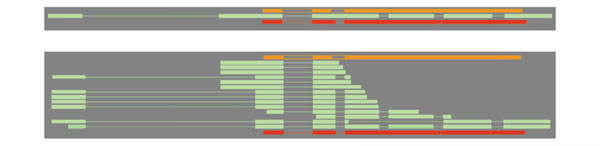
A tier organizes a collection of one or more feature types into a single horizontal row in the view. One of the viewing options is whether the features within that tier are 'collapsed' or 'expanded'. In the collapsed view (top) features lie on top of one another and thus appear along a single line. In this view the EST alignments (shown in light green) are collapsed. In the expanded view (below) the tier disallows overlapping features and thus as many lines as needed are used. The individual EST alignments (one line per EST) now appear separately. The tiers that flank the EST tier, alignments of peptide from other species (orange) or from Drosophila (red), are shown collapsed in both the top and bottom panels.
A feature can be selected by clicking on it, at which point detailed information such as name, sequence range and score appear in the text panel at the bottom of the screen (Figure 4). 'Rubber-banding' regions allows one to select multiple entries and view abbreviated information about each in the text display table, which can be sorted by any field (for example, name or score).
Figure 4.
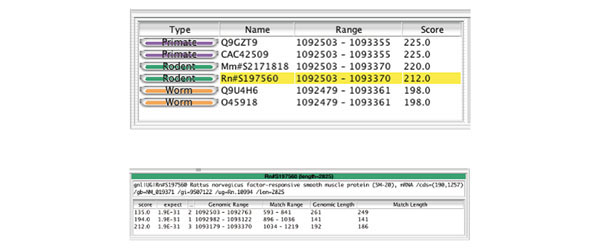
This figure shows the feature description tables. Each row in the top panel describes a single feature. The columns provide the feature type (here designated as 'organism' name), the aligned accession, the genomic position of the alignment and the BLASTX score. Clicking on any column heading will sort the rows according to the values held in that column (the initial sorting order is specified in the configuration file). Detailed information for the selected feature in this table (highlighted in yellow) appears in a second table, shown in the lower panel. The table header displays the name of the aligned sequence, its full length and a full description. The columns are configurable, and for each feature type only those columns that have been specified for that type will be shown. In this example of BLASTX alignments to other species, the available information includes the BLASTX score, BLASTX expectation and the location and length of the alignment on both query and subject sequences.
Further information about an annotation that is locally maintained can be displayed by bringing up the text window, which contains descriptive information about an annotation (Figure 5). Many features originate from other databases (for example, SWISS-PROT [46], EMBL [47] and GenBank [48]), and we can obtain extra information by looking up those features in their parent databases. Apollo allows the user to access this information using a context-dependent menu. By right-clicking a feature, the relevant report is retrieved and shown in an internet browser.
Figure 5.

A textual display provides detailed information for individual annotations. Annotations can be selected either from the menu on the left or by selecting an annotation in any other window. The types of annotations that can be generated by a curator are part of the initial configuration and appear in the menu shown above. A list of comments in a controlled vocabulary that can be used by the curator to add remarks about an annotation is also provided as part of the configuration (one example is shown above in the lower middle portion of the panel). These standardized comments facilitate future querying.
Apollo allows the view to be scaled using zoom buttons that magnify by 10×, 2×, 0.5×, and 0.1×. The magnification can also be targeted to fit a selected feature. Apollo provides 'semantic zooming': some features that would clutter the display at low zoom levels appear only when the user zooms in. At a sufficiently high zoom level, start and stop codons and the underlying genome sequence appear. Likewise, the sequence level information is displayed for each evidence type and annotation, either as nucleotides or amino-acid residues (Figure 6).
Figure 6.

Zooming provides additional information. In the top panel, which shows an intermediate level of zoom, the start and stop codons in all three frames appear as green and red tick marks, then the BLASTX similarities to other species (orange bars) and finally BLASTX similarities to Drosophila peptides (red bars). When the level of zoom is increased as shown as the lower panel, the sequence (either nucleic acid or peptide) of any aligned sequences is loaded and displayed to enable curators to directly compare predicted peptides. An individual start or stop codon, such as the start codon shown selected in this example, can be dragged onto the annotation to explicitly specify translation start and end sites for protein coding transcripts in the curator's annotation (blue).
A view is also available that displays the translation of each of the three frames of the genomic sequence. In this, start and stop codon positions are colored green and red respectively. Also when a transcript is selected, the specified coding frame of each of its exons is highlighted.
In addition to the horizontal scroll bar, curators can move to a specific position by specifying a coordinate, gene name, or short sequence string (Figure 7). Users can also bookmark genes of interest to find them easily again. The sequence of any feature or set of features can be saved in FASTA format, either as the raw DNA sequence or as a virtual cDNA, coding sequence or translated amino-acid sequence. Apollo also provides a restriction enzyme finder, which allows the user to find known restriction enzyme sites within a sequence, and a GC base frequency analyzer, which displays a plot of GC content from an adjustable sliding window over a sequence.
Figure 7.

Apollo provides a variety of search capabilities, including moving to an absolute base pair position, locating an annotation or aligned sequence by name or accession, or finding an exact sequence match (above). The graphical interface centers the view over the selected result. The interface also makes it possible to bookmark locations (below). This allows curators to quickly return to areas they are actively working on.
The most recent addition to Apollo is the synteny viewer and editor (Figures 8,9), which displays features on two different genomes at the same time. The need for this additional tool was precipitated by the recent release of the mouse genome. The synteny display is being used to improve annotations by closely examining the mouse-to-human comparisons. We are currently working to add editing capabilities to this view and to present other genome comparisons, such as between Drosophila and Anopheles.
Figure 8.
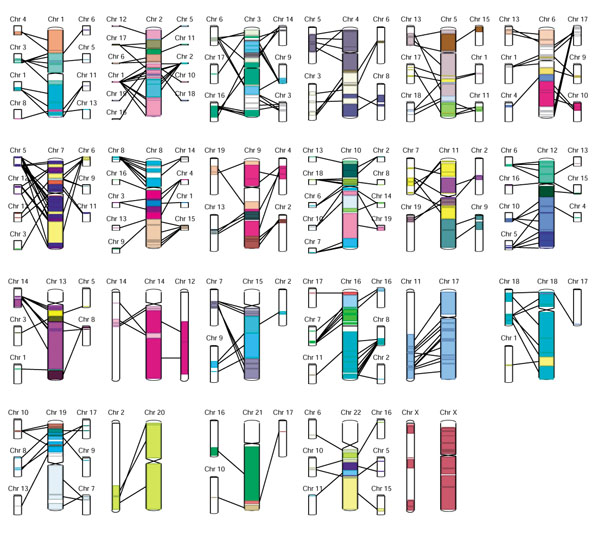
An overview of the synteny between mouse and human is shown in Apollo's synteny display. The central (larger) chromosomes are the human chromosomes, which are flanked by their syntenic mouse chromosomes. Apollo clusters DNA-DNA matches into syntenic regions, eliminating any short paralogous DNA-DNA hits. Selecting a segment displays a menu showing the chromosomal coordinates of the match in the genome sequence of both organisms and offering the user two options. The first opens a gene-level synteny view, illustrated in Figure 9. The second option reads the raw DNA-DNA matches that were used to produce the synteny and displays a sequence-level view of the similarities between the two genomes.
Figure 9.
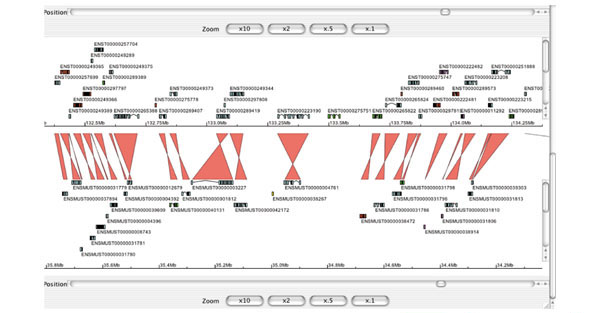
A detailed synteny view in Apollo showing part of human chromosome 20 at the top and part of mouse chromosome 2 at the bottom is illustrated. In the middle are links between orthologous genes as identified by the Ensembl synteny-generating software. The links are crossed because the genes in the top panel and in the bottom panel are in different orientations on their respective chromosomes. Although this figure only shows gene links, other types of link may also be displayed. Information about the genes from both genomes are read from Ensembl databases and information about orthologous gene pairs can either be read from an Ensembl compara database or from a flat file. The central panel can be used to scroll back and forth along the syntenic region or to center the display on a particular region by clicking on one of the colored matches. The top and bottom Apollo panels behave just as a normal, single Apollo panel and can be zoomed, scrolled, collapsed and also link out to a web page (as configured by the user).
Editing the data
Apollo's editing functionality allows curators to manually create and modify gene annotations. Annotation in Apollo is supported by the ability to detect and display edge matches, start and stop codons and potential splice sites. New gene models can be created easily by dragging a feature from the evidence panel into the annotation zone. When a new gene model is created, Apollo automatically creates an annotation using the longest possible open reading frame (ORF) for the transcript. Curator comments can then be attached to the gene annotation or to an individual transcript annotation. Modifications are time-stamped and associated with the curator's name, providing a complete history of the annotation.
As alternatively spliced transcripts are added to a gene model, they are sequentially named and numbered to associate them with that annotation. Apollo automatically creates alternatively spliced transcripts for a gene whenever the ORFs of transcripts overlap. In Drosophila overlapping untranslated regions (UTRs) are observed frequently between adjacent transcripts, but this does not necessarily indicate that the transcripts are for the same gene, so Apollo offers the option to make these new transcripts into distinct overlapping genes. Curators may add exons to existing transcripts by dragging the selected evidence down until the features are positioned directly over an existing transcript. Curators may set a splice site by selecting an exon and one evidence feature and choosing either the 'Set as 5' end' or 'Set as 3' end' option from the annotation pop-up menu.
Sequence-level adjustments are possible with Apollo's exon editor. This allows the curator to modify intron-exon boundaries base by base, split and merge genes, and export genomic or translated sequence. The exon editor (Figure 10) allows one to view the genomic sequence of a single strand and its three-frame conceptual translation, with start and stop codons highlighted.
Figure 10.

Fine control of editing requires close examination of the gene structure. This is accomplished in the exon editor panel, which provides a sequence-level view of gene models. When the exon editor is launched, the region displayed is highlighted in the main Apollo display (blue rectangle in lower panel), and this main display rectangle can be moved along the sequence to control the region shown in the exon editor. Using the contextual menu in the exon editor panel, changes can be made to introns and exons, as well as to precise splice junctions and translation starts. Annotations appear in the viewer with alternating colors for individual exons, and clicking on a transcript selects it and causes a graphical representation (or glyph) of the transcript to appear at the bottom of the window. This transcript glyph shows the translation start and stop sites, and can be displayed with or without introns. In the case illustrated, the intron-exon structure of the gene is shown; the numbers in the exons indicate which frame of the translated genomic sequence is utilized in that exon. Clicking on the different portions of the glyph allows one to quickly navigate to specific regions of the transcript. A sequence search query is also available, as is retrieval of genomic and translated sequence.
Overview of Apollo from the developer's perspective
In the future we anticipate that Apollo will be available to any researcher who is interested in annotating particular areas of biological interest. For this to be possible, researchers must be able to readily configure and customize Apollo as needed, and to access data from remote database sites as well as incorporate local information. This section discusses the different means of customizing and extending Apollo so that it is optimized for a particular environment and can meet the requirements of individual researchers. The first part of the section simply describes how to configure Apollo and personalize it. The second part briefly describes how one might add an adaptor to interact directly with an internal database server, a personal file format, or an external source over the network. The third part focuses on how to add a class to draw features in a new shape or style. The final part is an overview describing the addition of entirely new views of the data in independent frames. To present these topics an understanding of Apollo's Java class hierarchy is needed.
Apollo has three main packages: external data adaptors; data models; and the graphical user interfaces. In addition, there are some auxiliary packages from external sources and minor additional packages within Apollo: seq, analysis, util and io. The seq package contains utilities for handling FASTA files. The analysis package will allow various analyses of the sequence and the annotations to be carried out by direct command of the user. The util package provides general utilities, such as sequence translation and simple statistical functions. Similarly, the io package handles basic input and output functions.
The data models fall roughly into two categories: sequence models and feature models (Figure 11). In a sense, feature models may be viewed as a superclass of sequences, because every feature in fact defines a sequence, but within Apollo they are treated as distinct and separable classes.
Figure 11.

Categories of data models. The Apollo data models fall into one of two broad categories (or are a descriptive auxiliary class, for example, Comment): (a) a location on a sequence; or (b) a sequence. The corresponding Java superclasses are Range and AbstractSequence, respectively. The inheritance hierarchy from these two central classes is shown here, but some minor classes and relationships are omitted to simplify this description. Each class or interface is drawn as a rectangle, and interfaces have the suffix 'I' included in their class name (an interface specifies the methods that the class is required to implement). Lines ending with an open-headed arrow-point indicate the superclass and subclass relationships and a dotted line connects an interface to a class that implements that interface. Thus, both GenomicRange and SeqFeature are subclasses of the base class Range (which implements the Range I interface) and in turn FeatureSet, GenericAnnotation, FeaturePair, and AssemblyFeature are specializations of SeqFeature. Similarly an SRSSequence is a subclass of AbstractLazySequence, which in turn is a subclass AbstractSequence. Each of these subclasses inherits all the methods of their parent class and may extend the model's behavior either by adding new methods or by overriding the inherited methods. In addition to inheritance, the connecting lines also depict other types of relationships, with the terminus indicating the potential cardinality. Thus, a GenericAnnotationSet must have at least one, but may possibly have more, pieces of Evidence associated with it (drawn as a single crossbar to indicate 'at least one' and a triangular tripod to represent 'many'), one Identifier, which maintains synonyms and database cross references (a single crossbar indicates 'one and only one'), and may have zero or more optional Comments (drawn as a triangular tripod to represent 'many' and a single circle to indicate 'none'). Thus, a FeatureSet is both itself a SeqFeature that is composed of one or more component SeqFeatures. This enables Transcripts to be composed of a set of Exons, or an alignment to be composed of a set of high-scoring pairs. Likewise a CurationSet (across two different species) may contain component CurationSets (for the individual species) to enable comparative analysis.
Sequences have obvious attributes, including the residues themselves, a name, a unique identifier, length, database cross-references and a description, but internally, the abstract sequences are much more complex. It is impracticable for an application to load an entire annotated chromosome into memory, because it is far too large. Therefore, the actual sequence residues are only retrieved and loaded when the magnification level of the view is high enough to limit the required length of sequence to a tractable amount. The logic to determine when it is appropriate to retrieve more sequence (or replace the current sequence) is hidden within the sequence classes, and thus Apollo's performance and memory footprint is kept within reasonable bounds.
The simplest feature class is reduced to a range on a sequence consisting of just four attributes: a low end for the range; a high end for the range; the sequence that this range falls on; and a strand. The features further split into three subclasses: genomic ranges; results; and annotations. Genomic ranges include a chromosome and organism, and are used to support comparisons of syntenic genomic regions within Apollo. Results support alignments for viewing pairs of ranges along two similar sequences and contain information describing how the analysis was carried out. Annotations reference the results as evidence, but also contain information supplied by the individual carrying out the editing, such as their name, their comments, the date of each edit, and processing flags for feedback to the computational pipeline.
Installation and configuration
Apollo runs under the Windows, Mac OS X, Linux and Solaris operating systems and requires Java 1.2 or 1.3. In our memory tests with a 300 kilobase (kb) sequence with several thousand features, the memory heap size hovered around 100 Mbytes (MB). Therefore Apollo will run on a machine with 128 MB of available memory, but it will do better with 256 MB.
Apollo is highly configurable in terms of appearance, connectivity and extensions. Users can specify the color, graphical representation and placement of any feature (both annotations and computational results). The configuration file describes both the tiers and the types of data that are to be displayed. The file describes each tier by a label (for example 'Prediction'), a visible flag (either true or false), and a layout flag (if true then all data in this tier is arranged to avoid overlap, that is, the tier is expanded). The file also describes for each feature type: the tier it is to appear in; a label for that feature type; a color; the list of attributes to appear in the tabular report; initial score thresholds; and the drawable class to be used to display this feature. URLs for connecting features to outside data sources are also configurable. Note that the separation of tiers and types allows multiple feature types to be displayed in the same tier. For example the results of all gene-prediction programs can be shown in a single 'Prediction' tier. The aim is provide as much flexibility as possible in the presentation of the data. The information included in this file is incorporated when Apollo data are loaded.
The user can change some of the 'style' attributes of a feature type - for example, its color and tier position - interactively. A 'types' panel pops up as a separate window and shows all the tier names and colors. Right-clicking on a type lets the user adjust the color used for displaying features of that type; middle clicking allows the score threshold to be adjusted (features with scores below the threshold are not shown in the display). The user can adjust the order of tiers by selecting and dragging tiers with the mouse and the control key. The new tier options can then be saved to a tiers file, allowing each user to store personal style preferences.
External data adaptors: connecting Apollo to a database
Apollo is populated with data via a set of adaptors. Typically (for data coming from an external data source) these consist of two Java classes: a dialog box and the adaptor itself. The dialog box allows the user to tell the system what sequence they wish to examine. The adaptor provides a method for translating between the structure of the external data and the internal Apollo data models.
The existing adaptors allow data to be loaded from the Ensembl CGI server, GAME XML files, direct connections to either the Ensembl or Gadfly databases, DAS servers [49], and the Ensembl variant of GFF. Once edited the data may be stored as GAME XML files, GFF files, or GenBank table format [50]. We recognize a need for additional adaptors to read other variants of GFF (particularly that generated by Artemis), and GenBank and EMBL flat-file entries.
The class Apollo.gui.DataLoader manages the fetching and recording of annotation session data, which are held in the CurationSet data model. This class is instantiated whenever a new request is received for an additional feature or sequence data from an external source. The DataLoader class is quite straightforward; it provides two publicly available methods for loading and saving the data utilized during an annotation session. If a specific data adaptor is not provided among the arguments in the initial request, then the DataLoader will bring up a dialog box (org.bdgp.swing.widget.DataAdapterChooser) to allow the user to select an adaptor for an external data source (or sink). When the DataLoader instantiates a DataAdapterChooser it provides it with an org.bdgp.io.DataAdapterRegistry. This DataAdapterRegistry is populated with the list of all available adaptors from the configuration file and is initialized during Apollo's startup. These adaptors are one of the two pieces of software that must be provided to add a new external adaptor.
A data adaptor extends the org.bdgp.io-AbstractDataAdapter class and implements the following methods of the ApolloDataAdapterI interface:
GetUI: The DataAdapterChooser requires an interface to interact with the user and collect any necessary information. The DataAdapterChooser obtains this interface via this DataAdapter method, including as an argument the type of input/output operation (for example, IOOperation.READ or IOOperation.WRITE). This user interface is the other class (in addition to the DataAdapter itself) that the software developer must provide to add an external adaptor.
GetCurationSet: his returns a CurationSet model (from which all annotations and features can be accessed). A CurationSet is the hub for all sequence and feature models contemplated in an annotation session.
CommitChanges:The argument passed to this is a CurationSet. The purpose is to record the curator's annotation in a persistent form, whether this is a file or a database.
GetStateInformation: Following a load this method captures the current query parameters so that they may be saved and then restored in subsequent sessions.
setStateInformation: This method restores the parameters needed to access a CurationSet that was under examination in an earlier session. It is an alternate to explicitly setting load parameters using a dialog.
There are other required methods as well, but those described above provide the essential functionality for a DataAdapter.
The explicit DataAdapterUI that must be written provides an implementation of the org.bdgp.swingAbstractIntDataAdapUI interface. It provides the DataAdapter with any user information that is entered into the dialog window. The key method is doOperation, which performs the data transformation by delegating it to its partner data adaptor. The input and outputs to this method are entirely dependent on the operation the data adaptor is supposed to perform.
In summary, the mechanics of adding a new external data source are straightforward. This allows a developer to focus solely on writing the software to handle the semantic mapping between their own data representation and the Apollo data models.
Adding new Drawables
Several graphical elements are already available for representing features (Figure 2). Developers can incorporate new Drawable classes by simply extending the existing base classes and implementing relevant methods. The following describes some of the methods that can be overridden.
DrawSelected, drawUnselected, or simply draw. These methods actually paint the graphics for the primary representation of the sequence.
AddHighlights can be used to draw graphical indications that a feature shares start or end positions with other features on the sequence.
AddDecorations is used to draw any additional characteristics, for example the sites of start and stop codons within the feature.
A single feature type in fact requires two Drawable classes, one for a set of those features and another for an individual feature. This is useful for drawing gene structures because the draw method for the set can be used to draw a single line to illustrate the introns, whereas the draw method for the individual exons can be used to draw the rectangles to illustrate the exons. Once a developer has written the Drawable software it can be added by including the name of the class in the configuration file.
Adding new views
This is undoubtedly the most ambitious way of extending Apollo. A developer must understand how events are passed between windows because it is these events that indicate some change has occurred in the state of the shared underlying data models. While there are many other steps that are involved in adding a new view, this section will focus on crucial elements of event handling inside Apollo.
Windows that are interested in receiving a particular type of event must implement the appropriate listener interface for that event. When the listening window is instantiated it is provided with the central Controller object and must add itself to the global list of listeners held in the Controller object by calling the Controller's addListener method with itself as the only argument. When the Controller receives notification of a particular event it notifies every listener on this list.
Events themselves are provided to the central event Controller. The Controller implements an individual handler method for every type of event within Apollo. These Controller handler methods notify all registered Listeners that an Event of that type has occurred. Some examples of the current list of events for broadcasting changes involving feature and sequence selection, movement to a different sequence position, changes to feature content or appearance, window operations, and the availability of new data are briefly described below.
Feature selection: This is the most basic of events and indicates that one or more of the sequence features has been selected, usually by a mouse click, but also from the text search dialog, menus and scrolling lists.
Base focus: This event is used to center a display of the genomic sequence on a particular base. This may be used to position a display on a feature, or because a user has typed in a particular position, or because a given sequence string occurs at that position.
Region change: This event indicates that a completely new region of the genome is replacing the current sequence.
Annotation change: The AnnotationEditor is the sole generator of these events. To track changes to these important features and ensure consistency this task is highly centralized. Thus, for example, when editing from the synteny view, the modification requests will be relayed to the AnnotationEditor. While the user interacts with the synteny display, the non-graphical AnnotationEditor class directly manages the edits. The AnnotationEditor carries out the additions, deletions and changes to start and end positions for exons, transcripts, genes and any other annotation features. The Controller then passes these events on to the listeners so that the annotations can be redrawn accordingly.
Result change: This event is used only under unusual circumstances. Features are occasionally placed on the incorrect strand because of problems the alignment software has in identifying the correct strand (EST sequences that do not contain any splice junctions are the common source of this problem). In these cases we allow the curator to move the computed feature from one strand to the other so that it may be used during annotation.
Appearance change: This event is used to issue a request for a change in the appearance of a feature, for example, if the chosen color is altered.
Discussion
Apollo was first put into practical use as an editor in November 2001 for the re-annotation of the complete Drosophila genome. Ten full-time curators located at two sites (FlyBase-BDGP and FlyBase-Harvard) used Apollo over the course of seven months to revise and correct the annotations for the complete genome [51]. During the course of this exercise many improvements and capabilities were made to the software, culminating in a well-exercised code-base for Apollo. The Ensembl group provides Apollo to the community for browsing human and mouse annotations.
Other groups, both commercial and academic, are also using Apollo. The bioinformatics group at Biogen has been using Apollo to examine alignments, executed with BLAT, between their internal data and the public human genomic sequence (X. Zhijun, personal communication). Their group wrote a custom data adaptor to load their data that uses either chromosome and range, or a sequence, or a sequence name to query their internal data source and then display the specified region in Apollo. The data adaptor retrieves the data from an internal web service. The Fugu genomics group at the UK HGMP-RC in Cambridge has been using Apollo to display scaffolds, that is, draft sequences of the Fugu genome (T. Vavouri, personal communication). Annotation has been generated in GFF format and loaded into Apollo. The Fugu group is currently working on modifying Apollo to enable the interactive manipulation and validation of the ordered scaffolds.
The best annotation makes use of every available piece of information. This has already been seen in the Drosophila project's use of results from external pipelines and of the internal sequence reads (individual sequences from the cDNA that are unassembled) available from the full-length cDNA sequencing project [52]. As new data become available, Apollo must be able to display them effectively. Today, this means we need to add displays to Apollo that exploit comparative data. By aligning the genomic regions of related species and looking for conserved sequence regions we will gain insight into both fine gene structure (by identifying microexons) and into regulatory and other conserved regions.
Motivated both by our experience over the past several months and by new data that are becoming available we are actively working on the following new features. A splice junction view and editor will provide a tabular view of donor and acceptor splice pairs together with predicted splice junctions from standard weight matrices [53]. It will allow curators to fine-tune splice sites to generate the most biologically likely peptide. Data adaptors for GenBank and EMBL flat files should be particularly useful for a broader audience of biologists. In addition, we will allow existing entries from these public sequence archives to be overlaid onto the genomic sequence that is being annotated.
Finally, we have developed external analysis adaptors which will be useful in two ways. First they will enable the curator to dynamically reanalyze a specific sub-section of the sequence. This is important because, despite best efforts, the results of a gene prediction or an alignment over a large genomic region are often not as accurate as those produced when the sequence analysis is more targeted. Second, on-demand analysis will allow the curator to dynamically evaluate the peptides generated by protein-coding genes. This peptide evaluation is essential to the process of annotation if one wishes to produce a high-quality peptide set. The annotated peptides must be examined to verify their biological fidelity. This includes both consideration of the motifs that are present on the peptide and a comparison with known peptides. Downstream processing currently carries out this validation of the annotations in bulk, but this delays feedback and problems with the peptides are only cycled back to the curators for correction after some time has passed. Making corrections will be much more effective once the results of the evaluation are immediately available to the curator while they are working with that gene.
There are many other features that could be added to Apollo and we look forward to working with the open source community in bioinformatics to develop Apollo further. The Generic Model Organism Database project (GMOD) [54], whose aim is to provide tools for managing organism databases, has adopted Apollo as its annotation workbench. Software developers who are interested in contributing to this project and customizing Apollo for their own genome annotation projects can find more details at [55] and can obtain the source code from the Concurrent Versions System (CVS) [56] repository maintained on the GMOD Sourceforge site at [57]. Biologists who are interested in using Apollo for genomic annotation will find the detailed user's guide at [58] and the most recent release of Apollo for local installation at [59].
Acknowledgments
Acknowledgements
This work was supported by NIH grant HG00750 to G.M.R., by NIH Grant HG00739 to FlyBase (to W.M. Gelbart), by the Howard Hughes Medical Institute, and by Programme Grant G822559 from the Wellcome Trust. We want to heartily thank the newer curators - Brian Bettencourt, Kathryn Campbell, Pavel Hradecky, Yanmei Huang and Andrew Schroeder - who did not have a chance to participate in the design, but without whom we could not have come this far. They took Apollo and provided essential feedback as alpha testers while simultaneously doing the real work of creating the annotations. James Cuff and Erwin Frise provided great systems support at the Sanger Institute and Berkeley, respectively, and we could not have done this without them. We also thank David Emmert and Michael Ashburner for their valuable comments on a draft of this paper. We also thank everyone who made contributions and suggestions to the Apollo mailing list; your patience is outstanding and we will do our best to keep improving.
This article is part of a series of refereed research articles from Berkeley Drosophila Genome Project, FlyBase and colleagues, describing Release 3 of the Drosophila genome, which are freely available at http://genomebiology.com/drosophila/.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Florea L, Hartzell G, Zhang Z, Rubin G, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent JW. BLAT: the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA. Stochastic models for heterogeneous DNA sequences. Bull Math Biol. 1989;51:79–94. doi: 10.1007/BF02458837. [DOI] [PubMed] [Google Scholar]
- Baldi P, Chauvin Y, Hunkapiller T, McClure MA. Hidden Markov models of biological primary sequence information. Proc Natl Acad Sci USA. 1994;91:1059–1063. doi: 10.1073/pnas.91.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R, Eddy S, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge University Press; 1998. [Google Scholar]
- Dong S, Searls DB. Gene structure prediction by linguistic methods. Genomics. 1994;23:540–551. doi: 10.1006/geno.1994.1541. [DOI] [PubMed] [Google Scholar]
- Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Hartzell G, Harris NL, Ohler U, Abril JF, Lewis SE. Genome annotation assessment in Drosophila melanogaster. Genome Res. 2000;10:483–501. doi: 10.1101/gr.10.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M, Burge CB, Park E, Colinas J, Pelletier J. Assessment of the total number of human transcription units. Genomics. 2001;77:71–78. doi: 10.1006/geno.2001.6620. [DOI] [PubMed] [Google Scholar]
- Harrison PM, Kumar A, Lang N, Snyder M, Gerstein M. A question of size: the eukaryotic proteome and the problems in defining it. Nucleic Acids Res. 2002;30:1083–1090. doi: 10.1093/nar/30.5.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Salzberg SL. Computational gene finding in plants. Plant Mol Biol. 2002;48:39–48. [PubMed] [Google Scholar]
- Mungall CJ, Misra S, Berman BP, Carlson J, Frise E, Harris N, Marshall B, Shu S, Kaminker JS, Prochnik SE, et al. An integrated computational pipeline and database to support whole genome sequence annotation. Genome Biol. 2002;3:research0081.1–0081.11. doi: 10.1186/gb-2002-3-12-research0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, Volfovsky N, Town CD, Troukhan M, Alexandrov N, Feldmann KA, Flavell RB, White O, Salzberg SL. Full-length messenger RNA sequences greatly improve genome annotation. Genome Biol. 2002;3:research0029.1–0029.12. doi: 10.1186/gb-2002-3-6-research0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I, Flicek P, Duan D, Brent MR. Integrating genomic homology into gene structure prediction. Bioinformatics. 2001;Suppl 1:S140–S148. doi: 10.1093/bioinformatics/17.suppl_1.s140. [DOI] [PubMed] [Google Scholar]
- Lowe T, Eddy SR. tRNAscan-SE: a program for improved detection of transfer rna genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. Escherichia coli promoter sequences: analysis and prediction. Methods Enzymol. 1996;273:30–42. doi: 10.1016/s0076-6879(96)73004-5. [DOI] [PubMed] [Google Scholar]
- Pedersen AG, Baldi P, Chauvin Y, Brunak S. The biology of eukaryotic promoter prediction - a review. Comput Chem. 1999;23:191–207. doi: 10.1016/s0097-8485(99)00015-7. [DOI] [PubMed] [Google Scholar]
- Ohler U, Niemann H, Liao GC, Rubin GM. Joint modeling of DNA sequence and physical properties to improve eukaryotic promoter recognition. Bioinformatics. 2001;Suppl 17:199–206. doi: 10.1093/bioinformatics/17.suppl_1.s199. [DOI] [PubMed] [Google Scholar]
- Ohler U, Liao G, Niemann H, Rubin GM. Computational analysis of core promoters in the Drosophila genome. Genome Biol. 2002;3:research0087.1–0087.12. doi: 10.1186/gb-2002-3-12-research0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Bergman A, Gentles AJ. Genomics. Annotation of the Drosophila genome. Nature. 2001;411:259–260. doi: 10.1038/35077152. [DOI] [PubMed] [Google Scholar]
- Gopal S, Schroeder M, Pieper U, Sczyrba A, Aytekin-Kurban G, Bekiranov S, Fajardo JE, Eswar N, Sanchez R, Sali A, Gaasterland T. Homology-based annotation yields 1,042 new candidate genes in the Drosophila melanogaster genome. Nat Genet. 2001;27:337–340. doi: 10.1038/85922. [DOI] [PubMed] [Google Scholar]
- Mounsey A, Bauer P, Hope IA. Evidence suggesting that a fifth of annotated Caenorhabditis elegans genes may be pseudogenes. Genome Res. 2002;12:770–775. doi: 10.1101/gr208802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Bentley DR. Decoding the human genome sequence. Hum Mol Genet. 2000;9:2353–2358. doi: 10.1093/hmg/9.16.2353. [DOI] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, et al. Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FlyBase Consortium. The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 2002;30:106–108. doi: 10.1093/nar/30.1.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- A C. elegans database http://www.acedb.org/Documentation
- A C. elegans database http://www.acedb.org/Software/Downloads/supported.shtml
- AceDB http://www.acedb.org/
- Lewis SE, Harmon C. The Encyclopaedia of Drosophila Berkeley, CA: Berkeley Drosophila Genome Project. 1995.
- Perl Mongers - The Perl advocacy people http://www.perl.org
- The Source for Java™ Technology http://java.sun.com
- Helt G. bioTkperl: Data visualization and gene discovery in Drosophila melanogaster PhD thesis University of California at Berkeley. 1997.
- Helt GA, Lewis S, Loraine AE, Rubin GM. BioViews: Java-based tools for genomic data visualization. Genome Res. 1998;8:291–305. doi: 10.1101/gr.8.3.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervitz SA, Fuellen G, Dagdigian C, Brenner SE, Birney E, Korf I. BioPerl: standard Perl modules for bioinformatics. Objects in Bioinformatics Conference. 1998. http://www.bitsjournal.com/bioperl.html
- Harris NL. Genotator: a workbench for sequence annotation. Genome Res. 1997;7:754–762. doi: 10.1101/gr.7.7.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genquire http://bioinformatics.org/Genquire
- Stein LD, Mungall CJ, Shu S-Q, Caudy M, Mangone M, Day A, Nickerson E, Stajich J, Harris TW, Arva A, Lewis S. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook-Horsnell JT, Rice P, Rajandream M-A, Barrell B. Artemis: sequence visualisation and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Misra S, Roote J, Lewis SE, Blaze JR, Davis T, Doyle C, Galle R, George R, Harris N, et al. An exploration of the sequence of a 2.9-Mb region of the genome of Drosophila melanogaster : the Adh region. Genetics. 1999;153:179–219. doi: 10.1093/genetics/153.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searls DB. bioTk: componentry for genome informatics graphical user interfaces. Gene. 1995;163:1–16. doi: 10.1016/0378-1119(95)00424-5. [DOI] [PubMed] [Google Scholar]
- Edward R Tufte: Computer Literacy Bookshops Interview, 1994-1997 http://www.ercb.com/feature/feature.0008.2.html
- Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Reese MG, Kulp D, Tammana H, Haussler D. Genie - gene finding in Drosophila melanogaster. Genome Res. 2000;10:529–538. doi: 10.1101/gr.10.4.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoesser G, Sterk P, Tuli MA, Stoehr PJ, Cameron GN. The EMBL nucleotide sequence database. Nucleic Acids Res. 1997;25:7–14. doi: 10.1093/nar/25.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Boguski MS, Lipman DJ, Ostell J, Ouellette BF. GenBank. Nucleic Acids Res. 1998;26:1–7. doi: 10.1093/nar/26.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- About Biodas http://biodas.org
- Submission of complete genomes or other large sequence records http://www.ncbi.nlm.nih.gov/Sequin/table.html
- Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, et al. Annotation of the Drosophila euchromatic genome: a systematic review. Genome Biol. 2002;3:research0083.1–0083.22. doi: 10.1186/gb-2002-3-12-research0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton M, Carlson J, Brokstein P, Yu C, Champe M, George R, Guarin H, Kronmiller B, Pacleb J, Park S, et al. A Drosophila full-length cDNA resource. Genome Biol. 2002;3:research0080.1–0080.8. doi: 10.1186/gb-2002-3-12-research0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science. 2002;297:1007–1013. doi: 10.1126/science.1073774. [DOI] [PubMed] [Google Scholar]
- Generic Model Organism Database construction set http://gmod.sourceforge.net
- Apollo genome browser http://www.ensembl.org/apollo
- Concurrent versions system http://www.cvshome.org
- gmod/apollo http://cvs.sourceforge.net/cgi-bin/viewcvs.cgi/gmod/apollo
- Apollo user guide http://www.fruitfly.org/annot/apollo/userguide.html
- Apollo developer guide http://www.fruitfly.org/annot/apollo