

Abstract
Age-related macular degeneration (AMD) is a major cause of blindness in the elderly. We report a genome-wide screen of 96 cases and 50 controls for polymorphisms associated with AMD. Among 116,204 single-nucleotide polymorphisms genotyped, an intronic and common variant in the complement factor H gene (CFH) is strongly associated with AMD (nominal P value <10−7). In individuals homozygous for the risk allele, the likelihood of AMD is increased by a factor of 7.4 (95% confidence interval 2.9 to 19). Resequencing revealed a polymorphism in linkage disequilibrium with the risk allele representing a tyrosine-histidine change at amino acid 402. This polymorphism is in a region of CFH that binds heparin and C-reactive protein. The CFH gene is located on chromosome 1 in a region repeatedly linked to AMD in family-based studies.
Age-related macular degeneration (AMD) is the leading cause of blindness in the developed world. Its incidence is increasing as the elderly population expands (1). AMD is characterized by progressive destruction of the retina’s central region (macula), causing central field visual loss (2). A key feature of AMD is the formation of extracellular deposits called drusen concentrated in and around the macula behind the retina between the retinal pigment epithelium (RPE) and the choroid. To date, no therapy for this disease has proven to be broadly effective. Several risk factors have been linked to AMD, including age, smoking, and family history (3). Candidate-gene studies have not found any genetic differences that can account for a large proportion of the overall prevalence (2). Family-based whole-genome linkage scans have identified chromosomal regions that show evidence of linkage to AMD (4–8), but the linkage areas have not been resolved to any causative mutations.
Like many other chronic diseases, AMD is caused by a combination of genetic and environmental risk factors. Linkage studies are not as powerful as association studies for the identification of genes contributing to the risk for common, complex diseases (9). However, linkage studies have the advantage of searching the whole genome in an unbiased manner without presupposing the involvement of particular genes. Searching the whole genome in an association study requires typing 100,000 or more single-nucleotide polymorphisms (SNPs) (10). Because of these technical demands, only one whole-genome association study, on susceptibility to myocardial infarction, has been published to date (11).
Study design
We report a whole-genome case-control association study for genes involved in AMD. To maximize the chance of success, we chose clearly defined phenotypes for cases and controls. Case individuals exhibited at least some large drusen in a quantitative photographic assessment combined with evidence of sight-threatening AMD (geographic atrophy or neovascular AMD). Control individuals had either no or only a few small drusen. We analyzed our data using a statistically conservative approach to correct for the large number of SNPs tested, thereby guaranteeing that the probability of a false positive is no greater than our reported P values.
We used a subset of individuals who participated in the Age-Related Eye Disease Study (AREDS) (12). From the AREDS sample, we identified 96 case subjects and 50 control subjects as described (13). Because there can be many precursors to the development of either geographic atrophy or choroidal neovascularization, we purposely selected the group of study participants who had both large drusen and sight-threatening AMD as cases. All individuals identified themselves as ‘‘white, not of Hispanic origin.’’ To the extent possible, we kept the proportions of males/females and smokers/nonsmokers the same in cases and controls. Controls were purposely chosen to be older than the cases to increase the probability that they would remain without AMD (table S1).
All 146 individuals were genotyped as described (13). A summary of genotyping quality can be found in table S2. Of the 116,204 SNPs genotyped, 105,980 both were informative and passed our quality-control checks. We then proceeded to analyze the 103,611 of these SNPs that lie on the 22 autosomal chromosomes.
Single-marker associations
For each SNP, we tested for allelic association with disease status. To account for multiple testing, we used the Bonferroni correction and considered significant only those SNPs for which P < 0.05/103,611 = 4.8 × 10−7. This correction is known to be conservative and thus ‘‘over-corrected’’ the raw P values (14). Of the autosomal SNPs, only two, rs380390 and rs10272438, are significantly associated with disease status (Bonferroni-corrected P = 0.0043 and P = 0.0080, respectively) (Fig. 1A).
Fig. 1.
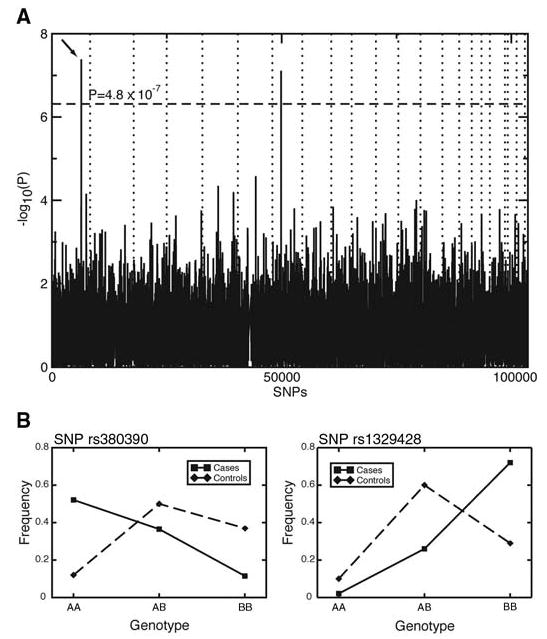
(A) P values of genome-wide association scan for genes that affect the risk of developing AMD. –log10(p) is plotted for each SNP in chromosomal order. The spacing between SNPs on the plot is uniform and does not reflect distances between SNPs on the chromosomes. The dotted horizontal line shows the cutoff for P = 0.05 after Bon-ferroni correction. The vertical dotted lines show chromosomal boundaries. The arrow indicates the peak for SNP rs380390, the most significant association, which was studied further. (B) Variation in genotype frequencies between cases and controls.
One criticism of case-control association studies such as ours is that population stratification can result in false-positive results. If the cases and controls are drawn from populations of different ancestry, with different allele frequencies, we might detect these population differences instead of loci associated with the disease. All individuals in this study self-identify their ethnicity as non-Hispanic white, and all of the case and control individuals are drawn from the same AREDS population. There was some differential recruiting of cases from office practices and recruiting of controls from radio and newspaper advertising (3). Finding two SNPs out of >100,000 implied the absence of genetic stratification, but we nonetheless used genomic control methods to control for this possibility (15). We consistently found that the significance of the tests was not inflated and that, therefore, these two SNPs are significantly associated with disease.
SNP rs380390 was successfully genotyped in all individuals. In 21 individuals, no genotype was determined for SNP rs10272438, and it appears to be excessively out of Hardy-Weinberg equilibrium (HWE χ2 = 36), indicating possible genotyping errors. Missing genotypes were determined by resequencing (16). After inclusion of these additional genotypes, the association was no longer significant after Bonferroni correction. Furthermore, the SNP with the third-lowest P value, rs1329428 (Bonferroni-corrected P = 0.14), is located 1.8 kb telomeric to rs380390. The genotype frequencies at these two neighboring loci vary between the case and control populations (Fig. 1B). Homozygotes for the C allele of rs380390 and the C allele at rs1329428 have an increased risk of developing AMD (Table 1). The risk conferred by these genotypes accounts for approximately 45% (rs380390) to 61% (rs1329428) of the total population risk (Table 1). We therefore focused on these two SNPs.
Table 1.
Odds ratios and population attributable risks (PARs) for AMD. The dominant odds ratio and PAR compare the likelihood of AMD in individuals with at least one copy of the risk allele versus individuals with no copy of the risk allele. The recessive odds ratio and PAR compare the likelihood of AMD in individuals with two copies of the risk allele versus individuals with no more than one copy of the risk allele. The population frequencies for the risk genotypes are taken from the CEU HapMap population (CEPH collection of Utah residents of northern and western European ancestry).
| Attribute | rs380390 (C/G) | rs1329428 (C/T) |
| Risk allele | C | C |
| Allelic association χ2 nominal P value | 4.1 × 10−8 | 1.4 × 10−6 |
| Odds ratio (dominant) (95% CI) | 4.6 (2.0–11) | 4.7 (1.0–22) |
| PAR (95% CI) | 70% (42–84%) | 80% (0–96%) |
| Frequency in HapMap CEU | 0.70 | 0.82 |
| Odds ratio (recessive) (95% CI) | 7.4 (2.9–19) | 6.2 (2.9–13) |
| PAR (95% CI) | 46% (31–57%) | 61% (43–73%) |
| Frequency in HapMap CEU | 0.23 | 0.41 |
Risk haplotype
These two SNPs lie in an intron of the gene for complement factor H (CFH), located on chromosome 1q31 (GenBank accession NM_000186). Because both SNPs are noncoding and neither appears to alter a conserved sequence, we explored whether the two SNPs are in linkage disequilibrium with a functional polymorphism. Analysis of linkage disequilibrium throughout this chromosomal region (Fig. 2A) revealed that the two SNPs lie in a 500-kb region of high linkage disequilibrium. Because this region is longer than typically observed blocks of high linkage disequilibrium (17) and there are long stretches in this region where there are no SNPs in our data set (Fig. 2B), we referred to other data sources with denser SNP coverage to narrow the region.
Fig. 2.
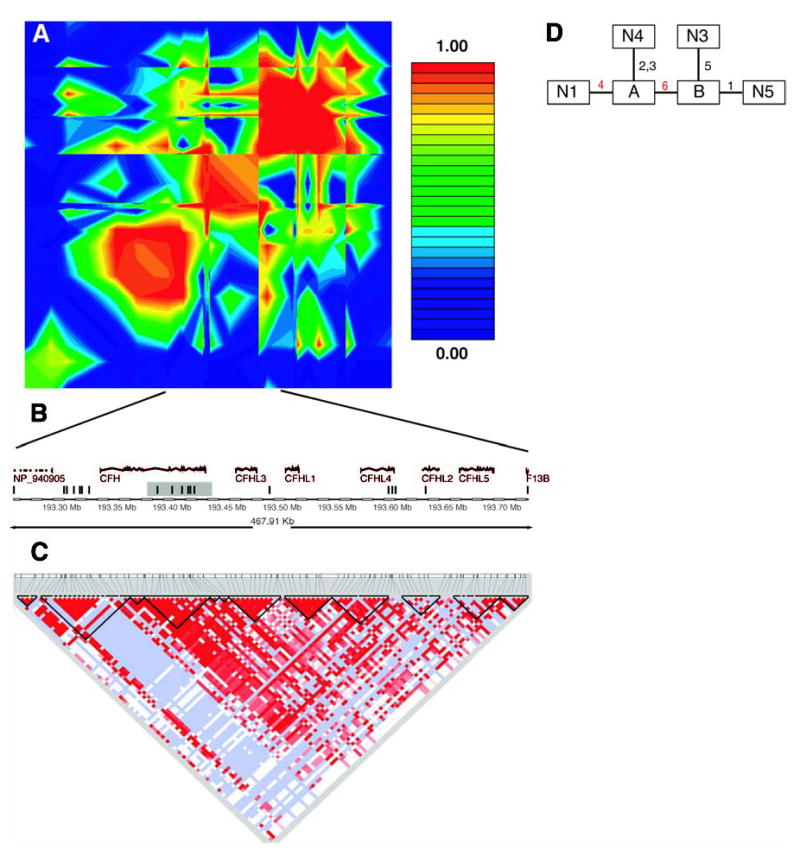
(A) Linkage disequilibrium across the CFH region, plotted as pairwise D′values. The red/orange box in the center of the plot is the region in strong linkage disequilibrium with the two associated SNPs in our data. (B) Schematic of the region in strong linkage disequilibrium with the two associated SNPs in our data. The vertical bars represent the approximate location of the SNPs available in our data set. The shaded region is the haplotype block found in the Hap-Map data. (C) Haplotype blocks in the HapMap CEU data cross the region. Darker shades of red indicate higher values of D′. Light blue indicates high D′with a low logarithm of the odds ratio for linkage (lod score). The dark lines show the boundaries of haplotype blocks. (D) Maximum-parsimony cladogram derived from haplotypes across the 6-SNP region. The number near each line indicates which of the six SNPs changes along that branch. The two red numbers are the two SNPs initially identified as being associated with AMD. SNP 4 is rs380390 and SNP 6 is rs1329428.
We used data from the International HapMap project to look at patterns of linkage disequilibrium in a population of residents of Utah with ancestry from northern and western Europe [the Centre d’Étude du Polymorphisme Humain (CEPH) sample] (18). In the 500-kb region of interest, there were only 19 SNPs in our data set as compared with 152 SNPs in the HapMap data set. Using a standard definition of linkage-disequilibrium blocks (17), we found that the two associated SNPs lie in a block that is 41 kb long and entirely contained within the CFH gene (Fig. 2C).
Six SNPs from our data set were in this 41-kb region. These SNPs form four predominant haplotypes, each with a frequency greater than 1% (table S3). Combined, these four haplotypes represent 99% of the chromosomes in this study. Reconstructing inferred haplotypes and building a phylogenetic tree allowed assessment of the evolutionary relationship between haplotypes (Fig. 2D). Using inferred haplotypes for each individual, we computed the odds ratio of the risk for disease in a nested cladistic framework under both dominant and recessive models (19). The highest risk was conferred by haplotype N1, which is the only haplotype containing the risk allele at SNP rs380390. Being heterozygous for this haplotype increases the likelihood for AMD by a factor of 4.6 [95% confidence interval (CI) 2.0 to 11] in our sample population. Being homozygous for this haplotype increases the likelihood for AMD by a factor of 7.4 (95% CI 3.0 to 19) in our sample population. Therefore, we expected to find the functionally relevant polymorphism in the context of haplotype N1. Most likely, this polymorphism would occur somewhere in the CFH gene, because the 41-kb haplotype block is entirely within CFH.
From markers to candidate functional polymorphism
To identify the polymorphism underlying susceptibility to AMD, we chose 96 individuals for exonic resequencing, including the exon/intron junctions. We sequenced all CFH exons, including those outside of the 41-kb block, as well as the region of SNP rs380390 as a control. SNP rs380390 was successfully resequenced in 93 individuals; the genotype derived from resequencing matched the original genotype in all cases. We identified a total of 50 polymorphisms; 17 of these have a minor-allele frequency of at least 5% (table S4). Of these 17, three represent nonsynonymous polymorphisms. We found a polymorphism in exon 9 of CFH (rs1061170) that is located 2 kb upstream of the 41-kb haplotype block, represents a tyrosine-histidine change, and is the polymorphism most strongly associated with AMD among the non-synonymous SNPs we found. Adding this SNP to the haplotype analysis reveals that 97% of the chromosomes with the highest risk haplotype (N1) also have the risk allele (His).
Human complement factor H
Several lines of evidence support the hypothesis that sequence polymorphisms in CFH can lead to AMD. First, the gene for CFH is located on chromosome 1q31, a region that had been implicated in AMD by six independent linkage scans (4–8, 20). Although one study concluded that mutations in a different gene in this region (HEMICENTIN-1) were responsible for AMD (20), mutations in HEMICENTIN-1 have not been found to be generally associated with AMD in three separate, independent studies (7, 21, 22).
CFH is a key regulator of the complement system of innate immunity (23). The complement system protects against infection and attacks diseased and dysplastic cells and normally spares healthy cells. When C3 convertase is activated, it leads to the production of C3a and C3b and then to the terminal C5b-9 complex. CFH on cells and in circulation regulates complement activity by inhibiting the activation of C3 to C3a and C3b and by inactivating existing C3b.
Various components of the complement cascade, including the C5b-9 complex, have been identified in the drusen of patients with AMD (24, 25). We also examined the eyes of four patients with AMD to look for the presence of C5b-9 (fig. S1). Deposition of activated complement C5b-9 was noted in Bruch’s membrane, in the intercapillary pillars, and within drusen. The observation of complement components in drusen in both humans (24, 25) and mice (26) has led to the hypothesis that AMD results from an aberrant inflammatory process that includes inappropriate complement activation (27).
Both age and smoking, two important risk factors for AMD, influence plasma levels of complement factor H (28). CFH sequences have been observed in an expressed sequence tag library derived from human RPE and choroid (29). We confirmed by immunofluorescence experiments that CFH is present in this region of the eye (Fig. 3). Strong staining was observed in choroid vessels (retinal blood vessels) and in an area bordering the RPE. Drusen of similar composition to that found in AMD are found in the eyes of patients with membranoproliferative glomerulonephritis type II (MPGNII), a kidney disease (30); CFH deficiency can cause MPGNII (23). Our immunostaining experiments (Fig. 3 and fig. S1) suggest that in AMD, the risk variant of CFH may give rise to complement deposition in choroidal capillaries (more severe) and choroidal vessels (less severe), with subsequent leakage of plasma proteins into Bruch’s membrane. Nutritional supplementation with zinc slows down the progression of AMD; biochemical studies have shown that CFH function is sensitive to zinc concentration (12, 31).
Fig. 3.
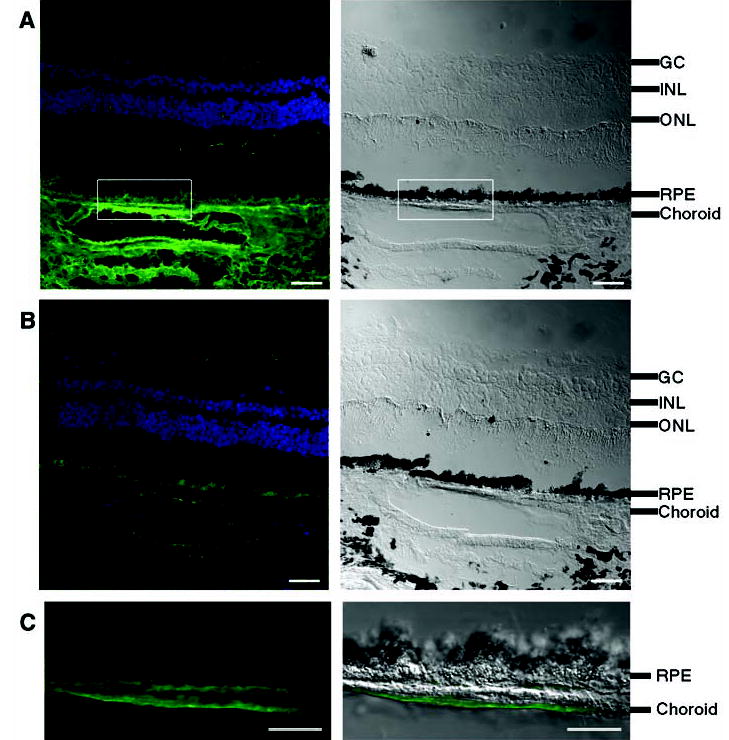
Immunofluorescence localization of CFH protein in human retina. Neighboring human retina sections are stained with (A) antibody to CFH or (B) antibody to CFH preabsorbed with CFH as negative control. (C) High-magnification view of the boxed area in (A). For (A), (B), and (C), left panels are the fluorescence images, with CFH labeling in green and DAPI (4′,6′-diamidino-2-phenylindole)–stained nuclei in blue; right panels are differential interference contrast (DIC) images showing the tissue morphology. In (C), the CFH signal is superimposed onto the DIC image. Labeling of CFH is intense in choroid, including blood vessels and areas bordering RPE [(A) and (C)]; this CFH signal is competed away by purified CFH protein (B), which demonstrates the labeling specificity. The fluorescence signal from RPE arises from lipofuscin autofluorescence, which cannot be competed away with CFH protein [(A) and (B)]. The black spots in DIC images correspond to melanin granules in RPE and choroids. The cell layers are indicated: GC, ganglion cells; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium. Scale bars: 40 μm in (A) and (B), 20 μm in (C).
We identified a tyrosine-histidine polymorphism in which the histidine variant almost always occurs in the context of the AMD risk haplotype. This polymorphism is located in a region of CFH that binds to both heparin and C-reactive protein (CRP) (23). It has been previously suggested that this binding could be altered by the replacement of a neutral tyrosine with a positively charged histidine (23). Elevated serum levels of CRP have been shown to be associated with AMD (32). Further work to establish the causal role of the tyrosine-histidine polymorphism in AMD is warranted.
Supplementary Material
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/1109557/DC1
Materials and Methods
Fig. S1
Tables S1 to S5
References
References
- 1.Friedman DS, et al. Arch Ophthalmol. 2004;122:564. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 2.Tuo J, Bojanowski CM, Chan CC. Prog Retinal Eye Res. 2004;23:229. doi: 10.1016/j.preteyeres.2004.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.AREDS Research Group. Ophthalmology. 2000;107:2224. [Google Scholar]
- 4.Majewski J, et al. Am J Hum Genet. 2003;73:540. doi: 10.1086/378134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seddon JM, Santangelo SL, Book K, Chong S, Cote J. Am J Hum Genet. 2003;73:780. doi: 10.1086/378505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weeks DE, et al. Am J Hum Genet. 2004;75:174. doi: 10.1086/422476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abecasis GR, et al. Am J Hum Genet. 2004;74:482. doi: 10.1086/381652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iyengar SK, et al. Am J Hum Genet. 2004;74:20. doi: 10.1086/380912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Risch N, Merikangas K. Science. 1996;273:1516. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 10.Botstein D, Risch N. Nat Genet. 2003;33(suppl):228. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 11.Ozaki K, et al. Nat Genet. 2002;32:650. doi: 10.1038/ng1047. [DOI] [PubMed] [Google Scholar]
- 12.AREDS Research Group. Arch Ophthalmol. 2001;119:1417. [Google Scholar]
- 13.Materials and methods are available as supporting material on Science Online.
- 14.McIntyre LM, Martin ER, Simonsen KL, Kaplan NL. Genet Epidemiol. 2000;19:18. doi: 10.1002/1098-2272(200007)19:1<18::AID-GEPI2>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 15.Devlin B, Bacanu SA, Roeder K. Nat Genet. 2004;36:1129. doi: 10.1038/ng1104-1129. [DOI] [PubMed] [Google Scholar]
- 16.Klein et al., data not shown.
- 17.Gabriel SB, et al. Science. 2002;296:2225. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 18.The International HapMap Consortium. Nature. 2003;426:789. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 19.Templeton AR, Boerwinkle E, Sing CF. Genetics. 1987;117:343. doi: 10.1093/genetics/117.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schultz DW, et al. Hum Mol Genet. 2003;12:3315. doi: 10.1093/hmg/ddg348. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi M, et al. Ophthalmic Genet. 2004;25:111. doi: 10.1080/13816810490514342. [DOI] [PubMed] [Google Scholar]
- 22.McKay GJ, et al. Mol Vis. 2004;10:682. [PubMed] [Google Scholar]
- 23.Rodríguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sanchez-Corral P. Mol Immunol. 2004;41:355. doi: 10.1016/j.molimm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 24.Johnson LV, Leitner WP, Staples MK, Anderson DH. Exp Eye Res. 2001;73:887. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- 25.Mullins RF, Russell SR, Anderson DH, Hageman GS. FASEB J. 2000;14:835. [PubMed] [Google Scholar]
- 26.Ambati J, et al. Nat Med. 2003;9:1390. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 27.Hageman GS, et al. Prog Retinal Eye Res. 2001;20:705. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- 28.Esparza-Gordillo J, et al. Immunogenetics. 2004;56:77. doi: 10.1007/s00251-004-0660-7. [DOI] [PubMed] [Google Scholar]
- 29.Wistow G, et al. Mol Vis. 2002;8:205. [PubMed] [Google Scholar]
- 30.Mullins RF, Aptsiauri N, Hageman GS. Eye. 2001;15:390. doi: 10.1038/eye.2001.142. [DOI] [PubMed] [Google Scholar]
- 31.Blom AM, Kask L, Ramesh B, Hillarp A. Arch Biochem Biophys. 2003;418:108. doi: 10.1016/j.abb.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 32.Seddon JM, Gensler G, Milton RC, Klein ML, Rifai N. JAMA. 2004;291:704. doi: 10.1001/jama.291.6.704. [DOI] [PubMed] [Google Scholar]
- 33.The Raymond and Beverly Sackler Fund for Arts and Sciences’ generous support made this project possible. We thank Raymond Sackler, J. Sackler, and E. Vosburg for their input and encouragement. We also thank AREDS participants and investigators; G. Gensler, T. Clemons, and A. Lindblad for work on the AREDS Genetic Repository; S. Westman and A. Evan for assistance with the microarrays; R. Fariss for the human retinal sections and advice on confocal microscopy; E. Johnson for assistance with immunostaining; and J. Majewski for constructive comments on the manuscript. Partially funded by NIH-K25HG000060 and NIH-R01EY015771 (J.H.), Macula Vision Research Foundation and the David Woods Kemper Memorial Foundation (C.B.), NIH-R01MH44292 (J.O.), and NIH-K01RR16090 and Yale Pepper Center for Study of Diseases in Aging (C.Z.). This work also benefited from the International HapMap Consortium making their data available prior to publication.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.