

Short abstract
The extent to which protein subcellular localization and functional categories established from clustering of orthologous genes agree with gene-expression data has been investigated in Saccharomyces cerevisiae. An increase in the resolution of biologically meaningful classes is observed upon the combination of experiments under different conditions. Differences and similarities in the expression between cytoplasmic-mitochondrial and interspecies translation machineries complement evolutionary information from sequence similarity.
Abstract
Background
Genome projects have provided a vast amount of sequence information. Sequence comparison between species helps to establish functional catalogues within organisms and to study how they are maintained and modified across phylogenetic groups during evolution. Microarray studies allow us to determine groups of genes with similar temporal regulation and perhaps also common regulatory upstream regions for binding of transcription factors. The integration of sequence and expression data is expected to refine our current annotations and provide some insight into the evolution of gene regulation across organisms.
Results
We have investigated how well the protein subcellular localization and functional categories established from clustering of orthologous genes agree with gene-expression data in Saccharomyces cerevisiae. An increase in the resolution of biologically meaningful classes is observed upon the combination of experiments under different conditions. The functional categories deduced by sequence comparison approaches are, in general, preserved at the level of expression and can sometimes interact into larger co-regulated networks, such as the protein translation process. Differences and similarities in the expression between cytoplasmic-mitochondrial and interspecies translation machineries complement evolutionary information from sequence similarity.
Conclusions
Combination of several microarray experiments is a powerful tool for the identification of upstream regulatory motifs of yeast genes involved in protein synthesis. Comparison of these yeast co-regulated genes against the archaeal and bacterial operons indicates that the components of the protein translation process are conserved across organisms at the expression level with minor specific adaptations.
Background
During the past few years sequencing projects have provided the whole genomes of several bacterial [1,2], archaeal [3,4] and eukaryotic [5,6] organisms, including human [7,8]. This genomic information is valuable as, in principle, it encodes all the instructions necessary and sufficient for the life cycle of each organism. Accurate annotation of genes to describe the gene products by their molecular function, subcellular localization and the biological process(es) they are involved in is crucial for the exploitation of genomic data [9]. Sequence annotation by similarity to known genes for which experimental data is available provides a rough initial criterion by which to classify the genes of an organism into functional catalogs. These classifications have been shown to be useful, for example, for computational prediction of common upstream regions that might bind the same transcription factors [10].
Clusters of Orthologous Groups (COGs) is an example of functional classification based only on standard sequence-similarity methods. COGs is an elegant approach that has used all-against-all sequence comparison of proteins in complete genomes to elucidate groups, namely COGs, that contain a set of individual Orthologous proteins or orthologous sets of paralogs from different phylogenetic lineages [11,12,13]. Normally, orthologs are functionally equivalent proteins that arise from vertical evolution, whereas paralogs are the result of duplication events and their function may have diverted from the original ancestor. Each COG is represented by a protein with a characterized function or domain. Individual COGs are assigned to general functional categories, which represent major cellular processes, and in some cases, if known, to more specific pathways or systems. The COG functional categories are identified by one-letter codes (Table 1).
Table 1.
COG codes for general function and pathway/systems
| Code | Genes | Description |
| General function | ||
| 416 | Information storage and processing | |
| J | 242 | Translation, ribosomal structure and biogenesis |
| K | 80 | Transcription |
| L | 94 | DNA replication, recombination and repair |
| 429 | Metabolism | |
| C | 62 | Energy production and conversion |
| G | 71 | Carbohydrate transport and metabolism |
| E | 153 | Amino acid transport and metabolism |
| F | 50 | Nucleotide transport and metabolism |
| H | 54 | Coenzyme metabolism |
| I | 39 | Lipid metabolism |
| 226 | Cellular processes | |
| D | 4 | Cell division and chromosome partitioning |
| O | 98 | Posttranslational modification, protein turnover, chaperones |
| M | 9 | Cell envelope biogenesis, outer membrane |
| N | 6 | Cell motility and secretion |
| P | 71 | Inorganic ion transport and metabolism |
| T | 38 | Signal transduction mechanisms |
| 127 | Poorly characterized | |
| R | 119 | General function prediction only |
| S | 8 | Function unknown |
| Pathway/system | ||
| - | 613 | No pathway/system assigned |
| C1 | 2 | Pyruvate decarboxylation |
| C2 | 18 | TCA cycle |
| C3 | 3 | Glyoxylate bypass |
| E1 | 6 | Arginine biosynthesis |
| E2 | 3 | Phenylalanine/tyrosine biosynthesis |
| E3 | 3 | Tryptophan biosynthesis |
| E4 | 2 | Threonine biosynthesis |
| E5 | 1 | Isoleucine biosynthesis |
| E7 | 19 | Leucine biosynthesis |
| E8 | 7 | Methionine biosynthesis |
| E9 | 4 | Proline biosynthesis |
| E10 | 4 | Histidine biosynthesis |
| F1 | 15 | Purine biosynthesis |
| F2 | 3 | Purine salvage |
| F3 | 12 | Pyrimidine biosynthesis |
| F4 | 2 | Pyrimidine salvage |
| F5 | 7 | Thymidylate biosynthesis |
| G1 | 4 | Glycolysis |
| G2 | 14 | Gluconeogenesis |
| G3 | 6 | Pentose phosphate pathway |
| G4 | 4 | Entner-doudoroff pathway |
| H1 | 8 | Heme biosynthesis |
| H3 | 2 | FAD biosynthesis |
| H4 | 7 | Biotin biosynthesis |
| H5 | 1 | NAD biosynthesis |
| H6 | 7 | Ubiquinone biosynthesis |
| H7 | 3 | Menaquinone biosynthesis |
| H8 | 2 | Thiamine biosynthesis |
| H9 | 1 | Pyridoxal phosphate biosynthesis |
| I1 | 10 | Fatty acid biosynthesis |
| J1 | 26 | Translation factors and enzymes involved in translation |
| J2 | 30 | Aminoacyl-tRNA synthetases, amino acid activation |
| J3 | 47 | Ribosomal proteins - small subunit |
| J4 | 77 | Ribosomal proteins - large subunit |
| K1 | 14 | DNA-dependent RNA polymerase subunits |
| K2 | 7 | Basal transcription factors |
| K3 | 2 | Transcriptional regulators |
| L1 | 20 | Basal replication machinery |
The COG letter-codes for general function and pathway/system categories are given the first column (Code). The Genes column holds the number of genes in the microarray data studied that were found for each functional class. The last column (Description) gives the description for each category.
This functional classification of genes conserved across different organisms has provided new information about how these catalogs of functions are maintained and modified across phylogenetic groups during evolution. However, in overpopulated COGs, the Orthologous relationships between their members are difficult to delineate precisely. Such COGs might contain proteins that evolved new functions with respect to the original ancestor, and even though these proteins still have significant sequence similarity, at the entire sequence or the domain level, they may be part of different cellular processes. This may be a particular problem in the budding yeast COGs as it is the only eukaryotic organism to be included in the database; therefore, those of its proteins involved in biological processes characteristic of eukaryotes may not have the counterparts in bacterial and archaeal genomes required to enable a finer grouping.
To fully understand the dynamic molecular network in any organism, however, the static information provided by sequencing projects will have to be complemented by high-throughput biochemical data from deletion experiments, DNA hybridization arrays, quantitative proteomics, localization experiments and two-hybrid interaction assays (for a review see [14]).
DNA hybridization experiments are a popular tool for monitoring the differential expression of a large number of genes, even complete genomes, under several conditions (reviewed in [15]). Analysis of the data can uncover sets of genes with similar expression profiles. This is achieved either by comparison against a set of genes whose expression behavior is already known for the conditions studied, or by unsupervised classification algorithms that cluster all the genes without imposing any a priori constraints or knowledge [16]. Hints about the function of uncharacterized genes can be deduced from other members of the cluster. DNA microarrays of intergenic regions have also been used for the study of putative binding sites for transcription regulators [17,18].
In the study presented here, we have investigated how well the expression of protein-coding genes in Saccharomyces cerevisiae agrees with the proteins' subcellular localization and functional classification as defined by the S. cerevisiae COG database. Using the available biological information on the yeast genes as the starting point, we have built groups of genes and compared the expression behavior of the proteins within and between these groups. The expression experiments included in our study comprise microarray data [16] of time series analyzing the effects of cell-cycle progression [19], sporulation [20], stress (temperature and reducing shocks) [21] and diauxic shift [22]. The data have been analyzed as a whole and as individual experiments, and an increase in resolution of the classification was observed when several datasets were combined. Although the classes defined by the COGs are, in general, preserved during gene expression, it is possible to divide broad groups into subclasses that reflect the oligomerization state of the proteins, subcellular location and/or more specific functionality, which sometimes clarify the boundaries of the cellular processes they are involved in. The results may be a complementary tool for the COGs, especially for those containing many paralogs that cannot be distinguished by sequence comparison alone but whose expression profiles are clearly different. Finally, the set of genes involved in cytoplasmic protein translation was analyzed in detail and compared to bacterial and archaeal 'ribosomal operons' to investigate the conservation of this key process at the level of gene sequence and expression across phylogenetic groups.
Results
Overview of the experimental data
S. cerevisiae genes present in the COG database were extracted from Eisen's dataset [16]. After removal of genes with low differential expression or too many missing time points, pairwise comparisons of the expression profiles of every possible gene pair were calculated for individual experiments and for the combined dataset by the standard Pearson correlation (see Materials and methods). The value of this comparison, the correlation coefficient (CCF), ranges from -1 to 1, indicating how different or similar, respectively, the compared expression profiles are.
Combining several experiments is expected to improve the separation of genes into more biologically meaningful groups. Some genes not involved in the same pathway or process might, in some experiments, appear to be regulated at the same time just because the time points are not finely scaled enough to separate them, or because they are occasionally required in otherwise independent pathways to maintain a cellular state under certain conditions. If this were true, some gene pairs would be expected to give both positive and negative correlations depending on the experiment considered. In fact, 27% of all the possible gene pairs in the dataset fell into this category. Furthermore, the CCFs derived from a comparison of the whole expression profile of these conflicting pairs can still be significant, that is, above 0.5 or below -0.5 (Figure 1). However, this only affects less than 3% of all the pairs and it is probably due to an unbalanced CCF by experiments with extremely high intensity peaks, as it might be the case with the sporulation data.
Figure 1.

Correlation inconsistency in the expression of some gene pairs between different experiments. For each gene pair, CCF values obtained from combining expression profiles of all experiments (CCFTOTAL, x-axis) are plotted versus those of individual experiments (CCFEXPERIMENT, y-axis). Only gene pairs with a CCFTOTAL value above 0.5 or below -0.5 are shown. x- and y-axis values go from -1 to 1 in steps of 0.5 as depicted in (a). (b-d) Genes involved in the cell cycle: (b) alpha-factor, (c) elutriation and (d) cdc-15 strain. (e) Sporulation. (f, g) Response to stress: (f) heat and (g) cold shocks. (h) Diauxic shift. CCFEXPERIMENT values can spread over a considerable range regardless of their CCFTOTAL (for example, heat shock), even to the point of being significant and with opposite sign to CCFTOTAL (for example, cold stress and diauxic shift). This inconsistency is less pronounced in the sporulation data, perhaps because it contains very high intensity values that may bias the CCFTOTAL to be more similar to CCFspo. Note that only genes whose expression was increased or decreased by 2.3-fold were considered. Those that did not pass the filtering were given a value equal to zero.
Protein subcellular localization versus gene expression
The advantage of using a large number of time points in different conditions to resolve finer relationships was also tested to determine whether the genes for proteins located in the same subcellular compartment tend to be transcribed at the same time.
Figure 2 shows the results for the following compartments: plasma membrane, endoplasmic reticulum, mitochondrion, nucleus and cytoplasm. When a set of proteins of unknown localization (a probable mixture of proteins from different compartments), was analyzed as a control, the CCFs obtained approximated to those expected by chance (Figure 2a). The expression of genes whose products will end up in mitochondria, endoplasmic reticulum or plasma membrane all show similar trends (Figure 2a,2b,2c,2d). The expression of gene pairs from the same compartment shows few negative correlations and many strong positive correlations, especially at very high cutoffs. There is a similar trend for nuclear proteins (Figure 2f); however, its slope is lower, because some nuclear proteins are tightly co-expressed with a considerable number of cytoplasmic proteins involved in protein translation. The cytoplasm behaves as expected with respect to positive correlations, but its proteins correlated negatively more than expected (Figure 2e). This may reflect the fact that, although subcellular compartments are usually populated by proteins involved in specialized interconnected processes [23], the cytoplasm holds a considerable number of different processes that can be independent or mutually exclusive of each other.
Figure 2.
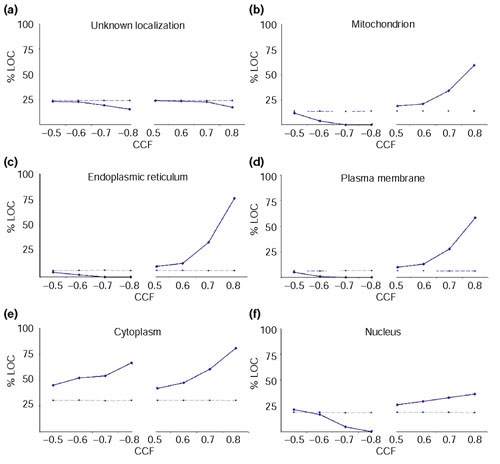
Proteins localized in the same compartment tend to be expressed at the same time. The plots show the trend for the percentage of genes from the same compartment (%LOC, y-axis) with respect to the total number of genes to which they correlate at a given threshold (CCF, x-axis). The solid lines correspond to the values obtained from the experimental data, whereas the dashed lines are the values expected by chance. The expected trend matches with that of a set of proteins with unknown localization (a), in contrast to sets of proteins with identified compartment: (b) mitochondrion, (c) endoplasmic reticulum, (d) plasma membrane, (e) cytoplasm and (f) nucleus.
The trends of Figure 2 were not so obvious when analyzing individual experiments (data not shown). In particular, two datasets for cell division - the dataset synchronized by elutriation and the sporulation experiment - as well as the cold-shock series, showed little discrimination between the expression of proteins from different compartments. This could be due to the reduced number of time points in the experiments, limiting the resolution of the classes. It is also possible that the quality of some experiments was somewhat poor, in particular in the case of the cell-division experiment synchronized by elutriation because of the difficulty of obtaining synchronized cells just by size separation.
COG functional classes of the genes in the expression data
Table 1 summarizes the distribution of the COG functional categories of the S. cerevisiae genes in the microarray data. The highest number of orthologs is found in metabolic processes (CGEFHI), followed by the machinery for storage and processing of information (JKL), especially those genes involved in protein translation (J). Under-represented groups are genes involved in 'Cell division and chromosome partitioning' (D), 'Cell envelope biogenesis' (M), 'Cell motility and secretion' (N), and 'Signal transduction mechanisms' (T). This may reflect a bias in the number of genes for each class in the dataset but also the current bias of the COGs due to the over-representation of bacterial and archaeal organisms compared to eukaryotes. Whereas core processes, such as metabolism and information-handling mechanisms, are conserved in all the phylogenetic lineages, other processes will be representative of their evolutionary group. For example, the proportion of proteins associated with intra- and intercellular communication will be higher in eukaryotes, especially in higher organisms [24].
Table 1 also shows the distribution of the genes with respect to known pathways and functional systems defined in the COGs. Most of them are not assigned to any system/pathway. The populated group corresponding to the protein translation system is split into four finer subclasses.
Overall expression behavior of predefined functional classes
Assuming that three characteristics (COG general function (F), COG pathway/system (P), and subcellular location (L)) can be assigned to every gene, the following combinations were considered to compare the expression behavior of the genes with respect to their annotation: F--, -P-, FP-, F-L, -PL and FPL. For example, class F-- will contain all the gene pairs in which both members have the same general function regardless of their pathway/system and subcellular location. Therefore, the FPL class will hold pairs in which both genes have the same general function, pathway/system (if any) and location. The localization included only two possibilities, mitochondrial or non-mitochondrial genes. Only mitochondrial genes were considered, because this was a large group with a clear positive correlation (Figure 2b). A diagram with detailed examples of some classifications can be found in the additional data files.
Any a priori classification scheme should consider the quality of the resulting classes and the number (and characteristics) of the elements not included. The elements of high-quality classes should show consistent relations between them as a whole; in this case all the genes should have similar expression profiles. Usually, consistent classes will tend to have few elements and thus consideration of which genes are not included may be important because strict classifications may leave out significantly correlating gene pairs. In our study, the overall trend of the gene pairs for each classification was studied in two different ways. These two ways are depicted in Figure 3a.
Figure 3.
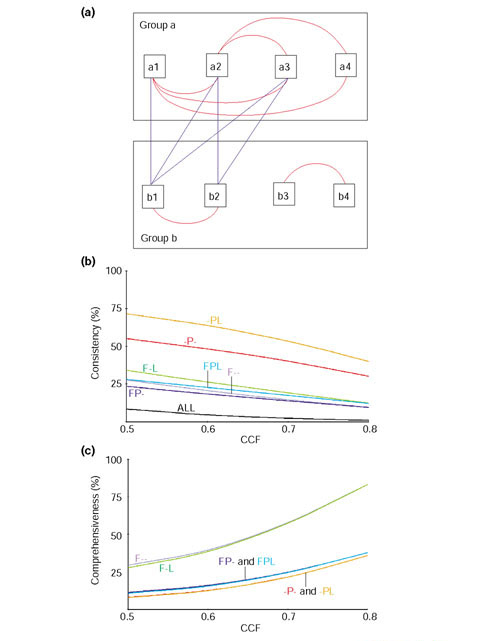
Agreement of functional annotation and expression data of genes. (a) Two groups of genes obtained after an a priori classification by, for example, functional annotation. The red lines connecting the elements within each group represent significant similarity between their expression profiles. The number of red lines with respect to all possible ones gives and indication of the consistency of the group. Thus, group a is very consistent, as all the possible connections but one, a3-a4, are made. On the other hand, the consistency of group b is poorer, as its elements form two subgroups (b1-b2 and b3-b4). The blue lines connecting the elements between each group are relations lost upon the a priori classification used. The higher the number of lost connections the less comprehensive the classification will be. In this case, the subgroup b1-b2 significantly correlates with group a. (b) Consistency of the functional groups established by gene annotation. A decreasing trend implies loss of interactions between members. F--, -P-, FP-, F-L, -PL and FPL indicate functional classes as defined in the text. For example, class F-- contains all gene pairs in which both members have the same general function regardless of their pathway/system (P) or location (L), class FP- contains all gene pairs with the same general function and the same pathway/system but not necessarily the same location, and so on. (c) Comprehensiveness of significant gene-expression pairs in the functional groups established by gene annotation. The increasing trend suggests that genes correlating at high CCF values tend to belong to the same functional class. This is especially obvious when a broad functional classification is used in which nearly all the possible pairs in the experiment are represented at high thresholds. The percentage of the gene pairs in each group with respect to the total number of pairs was: ALL (100%), F-- (8.8%), -P- (1.2%), FP- (4%), F-L (6.7%), -PL (1%) and FPL (3.1%). The group --- was not included in the -P- and -PL classes.
The first reflects the 'consistency' of a class. This measure describes the proportion of all the possible gene pairs in each class that correlate significantly at a given CCF value (see Materials and methods). The consistency of a class in which the expression of all the genes is induced or repressed at the same time will be higher than other classes where only subgroups of genes are significantly co-regulated. Figure 3b shows the results for a range of CCF values, from 0.5 to 0.8. The classes can be grouped into three sets. The one at the bottom (class ALL) gives the poorest consistency and corresponds to all gene pairs without an initial functional pre-grouping. The middle set contains four classes, the ones incorporating the general function, that is, F--, FP-, F-L and FPL. This set presents higher consistency, but further sub-grouping would raise it. The third set, containing -P- and -PL, is at the top, indicating that the original groups defined by this notation are consistent and maintained even at very high thresholds. The group with the highest consistency is that combining pathway and mitochondrial location information. This suggests that processes are best characterized when their compartments are also taken into account (which is true, at least in this case, mainly because it allows the separation of the cytoplasmic and mitochondrial ribosomes). Neither of these classes contains more than 3% of their gene pairs when using an equivalent negative correlation range, from -0.5 to -0.8 (not shown).
The second measure, 'comprehensiveness', tackles the question of how many of the significantly correlated gene pairs are included by these classification schemes. A broad classification will miss less correlating pairs (that is, it is more comprehensive) than other classification in which the groups are very specific (probably very consistent) but in which relationships between some groups may have been left out. The 'comprehensiveness' is assessed by calculating the proportion of correlated gene pairs for each classification with respect to the total number of correlated gene pairs in the whole dataset at a given threshold (Materials and methods). For positive thresholds two sets can be observed (Figure 3c). The set with higher number of pairs contains F-- and F-L, and the second one all the P groups (-P-, FP-, -PL and FPL). The results indicate that for broad classification schemes, F-- and F-L, the higher the threshold, the more similar the expression behavior of the genes with respect to their annotation. On the other hand, when considering groups with 'better defined boundaries' (that is, -P-, -PL, FP-and FPL), the relationships between subclasses are expected to be more important. An example is the protein translation category J. Although many genes in this category can be assigned to a more specific system, for example, initiation and termination factors or ribosomal subunits, they all will work together during protein synthesis. Therefore, by considering very specific groups we will miss the relationships between them.
The next sections tackle the comparison of the expression profiles of gene pairs for each functional group, how their members can sometimes split into more functionally consistent subgroups and which of the resulting subgroups may act together in the same cellular processes.
Subgrouping of genes with the same FPL
The data were divided into sets of genes with the same F, P and L as defined above. Then, the expression profiles of the genes in each group were compared against each other. Genes were split into subgroups according to the similarity of their expression profiles as described in Materials and methods.
Figure 4a. shows an example of three subgroups obtained from genes preassigned to the class 'L - -'; their COG general function is 'DNA replication, recombination and repair' (L), they have not been assigned to any pathway nor are they localized in the mitochondrion. The averaged profiles of the subgroups suggest that they are periodically regulated during the cell cycle. First, a subgroup of genes involved mainly in replication control is expressed, followed by a subgroup containing genes with DNA-repair functions, and finally the histone subgroup. This agrees well with some of the major processes taking place during cellular division: arrest of the cell cycle until everything is ready for division, DNA repair to ensure the fidelity in the transmission of the information to the daughter cells during DNA replication, and then chromosome condensation before mitosis. The periodicity is not observed in the sporulation data, probably because the experiment was not designed to take fine enough points to resolve these groups. In fact, the subgroups split at low thresholds (0.5) when analyzing individual experiments of the cell cycle separately. It is only when combining all the experiments that a higher cutoff (0.7) is needed because of the overlap of peaks in the sporulation series.
Figure 4.
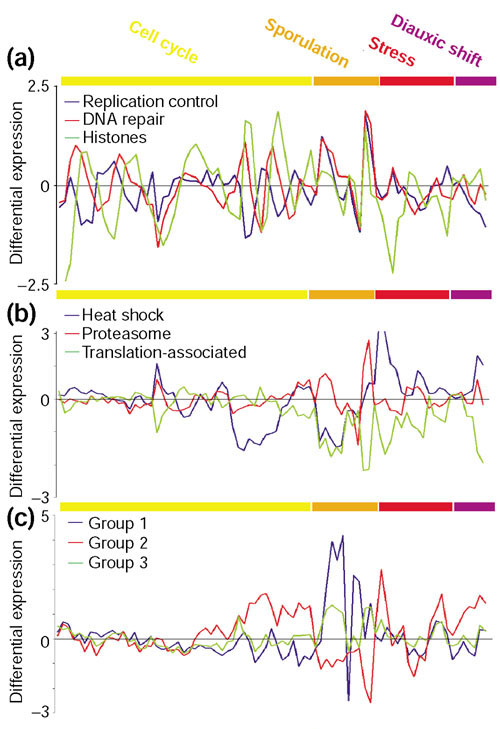
Examples of gene subgrouping within broad functional classes. Expression profiles for several sets of genes after combining experiments under different conditions. The experiments are color-coded (upper bar) as follows: yellow (cell cycle), orange (sporulation), red (stress) and purple (diauxic shift). (a) Subgroups obtained from the 'L - -' class ('DNA replication, recombination and repair'). (b) Subgroups obtained from the 'O - -' class ('post-translational modification, protein turnover and chaperones'). (c) Subgroups obtained from the 'GEPR - -' class, which contains permeases of the major facilitator superfamily. GEPR comprises several functional groups reflecting that their actual function is not clear (R) although they may be involved in the transport of sugars (G), aminoacids (E) and inorganic ions (P). The vertical axis represents the differential expression of genes as the log ratio of the mRNA abundance in experimental versus control samples. At zero values, the mRNA levels are identical. The list of genes included in every subgroup can be found in the additional data files.
Another example is the subgrouping within the set 'O - -' (that is, 'post-translational modification, protein turnover and chaperones'). Figure 4b shows three subgroups with distinguishable expression. The heat-shock protein subgroup is repressed in the cdc-15 and sporulation experiments, but induced under stress and during diauxic shift. On the other hand, the proteasome is only differentially expressed in the sporulation experiments. Finally, the third subgroup contains three proteins: PDR13, SSB2 and FPR4. PDR13 and SSB2 are both Hsp70 homologs. PDR13 interacts with Zu01p to form a ribosome-associated complex [25]. SSB2 is also associated with translating ribosomes and it may bind directly the nascent polypeptide chains [26]. FPR4 is a predicted peptidyl-prolyl cis-trans isomerase. The expression of this subgroup will be shown later to be associated to the cytoplasmic translation machinery and therefore it may have a role in folding of newly synthesized proteins.
Subgrouping of paralogs in populated COGs
At this point it is worth considering some examples of how similar the differential expression of paralogs is. The overpopulation of genes in a COG can be due to the presence of: duplicated genes which may or may not be involved in the same cellular process; functionally equivalent orthologs that are the result of an ancient horizontal gene transfer (for example, mitochondrial ribosomal genes); and promiscuous domains that are found in proteins that are not necessarily functionally related. An example of the latter is COG0515, which holds a number of proteins containing Ser/Thr protein kinase domains. The expression of these genes was diverse and the CCF values for all the different pairs varied between -0.8 and 0.8.
COG0477 is an example of a COG populated with paralogs. This group is a collection of various homologous permeases of the major facilitator superfamily [27]. Figure 4c shows some of the groups obtained by analysis of the expression of the genes in this COG. The proteins naturally form subgroups according to their differential expression. Group 1 contains SEO1, HXT10 and HXT14. SEO1 is a putative permease similar to the allantoate permease family, also called anion: cation symporter. HXT10 and HXT14 belong to the sugar transporter family, although none of them seems to transport glucose [28]. Group 2 contains four of the six major hexose transporters in yeast: HXT3, HXT4, HXT6 and HXT7 [29]. Finally, group 3 contains the genes HXT9, HXT11 and HXT12. None of them is a glucose transporter. In fact, HXT12 is not functional for hexose transport when overexpressed in a mutant lacking all HXT genes [30], whereas hxt9 and HXT11 may be involved in pleiotropic uptake of chemotherapeutic drugs, since a hxt9-hxt11 double-null mutant shows increased resistance to a number of drugs [31].
This could be taken as an example of the potential for finer subgrouping within COGs by comparing functional-expression information of proteins with similar primary structure.
Cross-talk between functional subgroups
The next step was to compare the calculated subgroups to find out how they could be related to each other. For that, the expression profiles of the genes in each selected subgroup were averaged. The averaged profiles of these subgroups, representing all the functional classes, were then correlated and clustered according to their similarity (see Materials and methods).
At 0.71 threshold, a large number of the subgroups did not have a correlating partner, and only three sets were found to contain more than one subgroup. These sets represented two highly conserved processes in living organisms: protein translation (both mitochondrial and cytoplasmic), and DNA replication.
Figure 5 shows the DNA replication set. The four averaged profiles of the functional subgroups, including the 'L - -' subgroup of DNA-repair genes, are all very similar (Figure 5a) and the resulting total average (Figure 5b) clearly retains the periodic features observed already in Figure 4a. In addition to the subgroup involved in DNA repair (genes: MSH2, MSH6, RAD51, OGG1), this class also contains subgroups involved in thymidylate biosynthesis 'F F5 -' (genes: DUT1, RNR1, RNR3, CDC21), basal replication machinery 'L L1 -' (genes: RFA1, POL2, POL30), and cell division and chromosome partitioning 'D - -' (genes: SMC3, RHC18).
Figure 5.
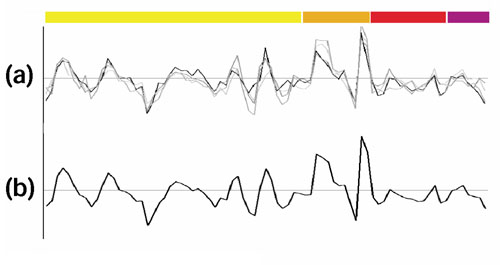
'DNA replication-related' genes. The experiments are color-coded as in Figure 4. (a) The four subgroups with very similar periodic profiles are shown. As mentioned in the text, they correspond to DNA repair and replication, thymidylate biosynthesis, and chromosome partitioning. (b) Profile obtained by averaging those shown in (a).
The mitochondrial translation set only included subgroups with general function J (protein translation), that is, translation factors (J1), aminoacyl-tRNA synthetases (J2), and the ribosomal proteins (J3 and J4). In contrast, the cytoplasmic translation set included several other functional subgroups in addition to the J ones.
Cytoplasmic protein translation set
A quick survey of the COG functional categories of the proteins in the set provides a good overview of the processes related to protein synthesis (Table 2). These are: E (general amino-acid metabolism, including leucine synthesis, E7), F (general nucleotide metabolism, including purine and pyrimidine biosynthesis, F1 and F3 respectively, and purine salvage, F2), G (general carbohydrate metabolism, including glycolysis, G1, gluconeogenesis, G2, and pentose phosphate pathway, G3), H (general coenzyme metabolism, including menaquinone biosynthesis, H7), J (general translation, including translation factors, J1, aminoacyl-tRNA synthetases, J2, and ribosomal proteins for the small subunit, J3, and large subunit, J4), K (general transcription, including DNA-dependent RNA polymerases, K1) and L (general DNA handling, although it mainly contains helicases and RNA-processing proteins).
Table 2.
Common upstream motifs in the genes involved in cytoplasmic protein synthesis
| Presence of regulatory motif | COG general functional category | COG pathway/system | Subcellular localization | GOC number | ORF name | Gene description | ||
| Genes identified experimentally to be regulated by Rap1 | ||||||||
| rap1 | - | - | EH | E7 | - | COG0028 | YLR044C | PDC1 pyruvate decarboxylase, isozyme 1 |
| rap1 | - | - | G | G1 | - | COG0469 | YAL038W | CDC19 pyruvate kinase |
| RAP1 | - | - | G | G2 | - | COG0126 | YCR012W | PGK1 phosphoglycerate kinase |
| RAP1 | - | - | G | G2 | - | COG0057 | YGR192C | TDH3 glyceraldehyde-3-phosphate dehydrogenase 3 |
| RAP1 | - | - | G | G2 | - | COG0588 | YKL152C | GPM1 phosphoglycerate mutase |
| rap1 | - | - | G | G2 | - | COG0148 | YHR174W | ENO2 enolase II (2-phosphoglycerate dehydratase) |
| rap1 | - | - | G | G2 | - | COG0057 | YJR009C | TDH2 glyceraldehyde-3-phosphate dehydrogenase 2 |
| RAP1 | RRPE | - | J | - | - | COG2238 | YOL121C | RPS19A 40S small subunit ribosomal protein S19.E |
| RAP1 | - | - | J | - | - | COG2238 | YNL302C | RPS19B 40S small subunit ribosomal protein S19.E |
| rap1 | - | PAC | J | - | - | COG2451 | YPL143W | RPL33A ribosomal protein L35A.E.C16 |
| rap1 | - | - | J | - | - | COG2451 | YOR234C | RPL33B ribosomal protein L35A.E.C15 |
| RAP1 | - | - | J | J2 | - | COG1190 | YDR037W | KRS1 lysyl-tRNA synthetase, cytosolic |
| rap1 | RRPE | - | J | J2 | - | COG0008 | YOR168W | GLN4 glutaminyl-tRNA synthetase |
| RAP1 | RRPE | PAC | J | J3 | - | COG0199 | YDL061C | RPS29B ribosomal protein S29.E.B |
| RAP1 | RRPE | PAC | J | J3 | - | COG0092 | YNL178W | RPS3 ribosomal protein S3.E |
| RAP1 | RRPE | PAC | J | J3 | - | COG0522 | YPL081W | RPS9A ribosomal protein S9.E.A |
| RAP1 | RRPE | - | J | J3 | - | COG0103 | YDL083C | RPS16B ribosomal protein S16.E |
| RAP1 | RRPE | - | J | J3 | - | COG2004 | YIL069C | RPS24B 40S small subunit ribosomal protein S24.E |
| RAP1 | RRPE | - | J | J3 | - | COG0052 | YLR048W | RPS0B 40S ribosomal protein P40 HOMOLOG B |
| RAP1 | RRPE | - | J | J3 | - | COG0185 | YOL040C | RPS15 40S small subunit ribosomal protein |
| RAP1 | - | PAC | J | J3 | - | COG0052 | YGR214W | RPS0A 40S ribosomal protein P40 HOMOLOG A |
| RAP1 | - | - | J | J3 | - | COG0186 | YBR048W | RPS11B ribosomal protein S11.E.B |
| RAP1 | - | - | J | J3 | - | COG2125 | YBR181C | RPS6B ribosomal protein S6.E |
| RAP1 | - | - | J | J3 | - | COG0522 | YBR189W | RPS9B ribosomal protein S9.E.B |
| RAP1 | - | - | J | J3 | - | COG0100 | YCR031C | RPS14A 40S ribosomal protein S14.E |
| RAP1 | - | - | J | J3 | - | COG0184 | YDR064W | RPS13 ribosomal protein |
| RAP1 | - | - | J | J3 | - | COG1383 | YDR447C | RPS17B ribosomal protein S17.E.B |
| RAP1 | - | - | J | J3 | - | COG0099 | YDR450W | RPS18A ribosomal protein S18.E.C4 |
| RAP1 | - | - | J | J3 | - | COG2004 | YER074W | RPS24A 40S small subunit ribosomal protein S24.E |
| RAP1 | - | - | J | J3 | - | COG0098 | YGL123W | RPS2 40S small subunit ribosomal protein |
| RAP1 | - | - | J | J3 | - | COG0048 | YGR118W | RPS23A 40S small subunit ribosomal protein S23.E |
| RAP1 | - | - | J | J3 | - | COG0051 | YHL015W | RPS20 ribosomal protein |
| RAP1 | - | - | J | J3 | - | COG1471 | YHR203C | RPS4B ribosomal protein S4.E.C8 |
| RAP1 | - | - | J | J3 | - | COG0096 | YJL190C | RPS22A ribosomal protein S15A.E.C10 |
| RAP1 | - | - | J | J3 | - | COG0049 | YJR123W | RPS5 ribosomal protein S5.E |
| RAP1 | - | - | J | J3 | - | COG1471 | YJR145C | RPS4A ribosomal protein S4.E.C10 |
| RAP1 | - | - | J | J3 | - | COG2051 | YKL156W | RPS27A ribosomal protein S27.E |
| RAP1 | - | - | J | J3 | - | COG1890 | YLR441C | RPS1A ribosomal protein S3A.E |
| RAP1 | - | - | J | J3 | - | COG1890 | YML063W | RPS1B ribosomal protein S3A.E |
| RAP1 | - | - | J | J3 | - | COG2125 | YPL090C | RPS6A ribosomal protein S6.E |
| rap1 | RRPE | - | J | J3 | - | COG0100 | YJL191W | RPS14B 40S small subunit ribosomal protein S14.E.B |
| rap1 | RRPE | - | J | J3 | - | COG2053 | YOR167C | RPS28A 40S small subunit ribosomal protein S28.E.C15 |
| rap1 | - | - | J | J3 | - | COG2007 | YBL072C | RPS8A ribosomal protein S8.E |
| rap1 | - | - | J | J3 | - | COG0186 | YDR025W | RPS11A ribosomal protein S11.E |
| rap1 | - | - | J | J3 | - | COG2007 | YER102W | RPS8B ribosomal protein S8.E |
| rap1 | - | - | J | J3 | - | COG0048 | YPR132W | RPS23B 40S small subunit ribosomal protein S23.E |
| RAP1 | RRPE | - | J | J4 | - | COG2058 | YDL130W | RPP1B 60S large subunit acidic ribosomal protein L44PRIME |
| RAP1 | RRPE | - | J | J4 | - | COG0093 | YER117W | RPL23B ribosomal protein L23.E |
| RAP1 | RRPE | - | J | J4 | - | COG0081 | YGL135W | RPL1B 60S large subunit ribosomal protein |
| RAP1 | RRPE | - | J | J4 | - | COG0097 | YGL147C | RPL9A ribosomal protein L9.E |
| RAP1 | RRPE | - | J | J4 | - | COG0198 | YGR034W | RPL26B 60S large subunit ribosomal protein |
| RAP1 | RRPE | - | J | J4 | - | COG1631 | YHR141C | RPL42B ribosomal protein L36A.E |
| RAP1 | RRPE | - | J | J4 | - | COG1552 | YIL148W | RPL40A ubiquitin |
| RAP1 | RRPE | - | J | J4 | - | COG0091 | YJL177W | RPL17B 60S large subunit ribosomal protein L17.E |
| RAP1 | RRPE | - | J | J4 | - | COG1358 | YLL045C | RPL8B 60S large subunit ribosomal protein L7A.E.B |
| RAP1 | RRPE | - | J | J4 | - | COG1632 | YLR029C | RPL15A 60S large subunit ribosomal protein L15.E.C12 |
| RAP1 | RRPE | - | J | J4 | - | COG2058 | YOL039W | RPP2A acidic ribosomal protein P2.BETA |
| RAP1 | RRPE | - | J | J4 | - | COG1727 | YOL120C | RPL18A 60S large subunit ribosomal protein S18.E |
| RAP1 | RRPE | - | J | J4 | - | COG1841 | YPL198W | RPL7B 60S large subunit ribosomal protein |
| RAP1 | - | PAC | J | J4 | - | COG2147 | YBL027W | RPL19B 60S large subunit ribosomal protein L19.E |
| RAP1 | - | PAC | J | J4 | - | COG0197 | YLR075W | RPL10 60S large subunit ribosomal protein |
| RAP1 | - | - | J | J4 | - | COG0093 | YBL087C | RPL23A 60S large subunit ribosomal protein L23.E |
| RAP1 | - | - | J | J4 | - | COG2147 | YBR084C-A | RPL19A 60S large subunit ribosomal protein L19.E |
| RAP1 | - | - | J | J4 | - | COG2139 | YBR191W | RPL21A ribosomal protein L21.E |
| RAP1 | - | - | J | J4 | - | COG0255 | YDL191W | RPL35A 60S large subunit ribosomal protein |
| RAP1 | - | - | J | J4 | - | COG0080 | YDR418W | RPL12B 60S large subunit ribosomal protein L12.E |
| RAP1 | - | - | J | J4 | - | COG2126 | YDR500C | RPL37B ribosomal protein L.37.E |
| RAP1 | - | - | J | J4 | - | COG1911 | YGL030W | RPL30 60S large subunit ribosomal protein L30.E |
| RAP1 | - | - | J | J4 | - | COG1841 | YGL076C | RPL7A 60S large subunit ribosomal protein L7.E.A |
| RAP1 | - | - | J | J4 | - | COG0200 | YGL103W | RPL28 60S large subunit ribosomal protein L27A.E |
| RAP1 | - | - | J | J4 | - | COG0094 | YGR085C | RPL11B ribosomal protein |
| RAP1 | - | - | J | J4 | - | COG1358 | YHL033C | RPL8A 60S large subunit ribosomal protein L7A.E.A |
| RAP1 | - | - | J | J4 | - | COG0090 | YIL018W | RPL2B 60S large subunit ribosomal protein L8.E |
| RAP1 | - | - | J | J4 | - | COG2174 | YIL052C | RPL34B ribosomal protein L34.E |
| RAP1 | - | - | J | J4 | - | COG2167 | YJL189W | RPL39 60S large subunit ribosomal protein L39.E |
| RAP1 | - | - | J | J4 | - | COG2163 | YKL006W | RPL14A ribosomal protein |
| RAP1 | - | - | J | J4 | - | COG0244 | YLR340W | RPP0 acidic ribosomal protein L10.E |
| RAP1 | - | - | J | J4 | - | COG0198 | YLR344W | RPL26A 60S large subunit ribosomal protein |
| RAP1 | - | - | J | J4 | - | COG0102 | YNL069C | RPL16B 60S large subunit ribosomal protein |
| RAP1 | - | - | J | J4 | - | COG1631 | YNL162W | RPL42A ribosomal protein L36A.E |
| RAP1 | - | - | J | J4 | - | COG0089 | YOL127W | RPL25 ribosomal protein L23A.E |
| RAP1 | - | - | J | J4 | - | COG2157 | YOR312C | RPL20B 60S large subunit ribosomal protein |
| RAP1 | - | - | J | J4 | - | COG1358 | YOR369C | RPS12 40S small subunit acidic ribosomal protein S12 |
| RAP1 | - | - | J | J4 | - | COG2139 | YPL079W | RPL21B ribosomal protein L21 |
| RAP1 | - | - | J | J4 | - | COG0256 | YPL131W | RPL5 60S large subunit ribosomal protein L5.E |
| RAP1 | - | - | J | J4 | - | COG1997 | YPR043W | RPL43A ribosomal protein L37A.E |
| RAP1 | - | - | J | J4 | - | COG0094 | YPR102C | RPL11A ribosomal protein L11.E |
| rap1 | RRPE | - | J | J4 | - | COG2058 | YDR382W | RPP2B 60S large subunit acidic ribosomal protein |
| rap1 | RRPE | - | J | J4 | - | COG1727 | YNL301C | RPL18B 60S large subunit ribosomal protein L18.E |
| rap1 | RRPE | - | J | J4 | - | COG0087 | YOR063W | RPL3 60S large subunit ribosomal protein L3.E |
| rap1 | - | - | J | J4 | - | COG1717 | YBL092W | RPL32 60S large subunit ribosomal protein L32.E |
| rap1 | - | - | J | J4 | - | COG2097 | YDL075W | RPL31A 60S large subunit ribosomal protein L31.E |
| rap1 | - | - | J | J4 | - | COG0080 | YEL054C | RPL12A 60S large subunit ribosomal protein L12.E |
| rap1 | - | - | J | J4 | - | COG2075 | YGL031C | RPL24A 60S large subunit ribosomal protein L24.E.A |
| rap1 | - | - | J | J4 | - | COG2075 | YGR148C | RPL24B 60S large subunit ribosomal protein L24.E.B |
| rap1 | - | - | J | J4 | - | COG2163 | YHL001W | RPL14B ribosomal protein |
| rap1 | - | - | J | J4 | - | COG0102 | YIL133C | RPL16A 60S large subunit ribosomal protein |
| rap1 | - | - | J | J4 | - | COG1552 | YKR094C | RPL40B ubiquitin |
| rap1 | - | - | J | J4 | - | COG2126 | YLR185W | RPL37A ribosomal protein L37.E |
| RAP1 | RRPE | PAC | K | - | - | COG0724 | YGR159C | NSR1 nuclear localization sequence binding protein |
| RAP1 | RRPE | PAC | K | K1 | - | COG0202 | YPR110C | RPC40 DNA-directed RNA polymerase I, III 40 kD |
| subunit | ||||||||
| Genes not identified experimentally to be regulated by rap1 | ||||||||
| RAP1 | RRPE | - | E | - | - | COG0421 | YLR146C | SPE4 spermine synthase |
| - | RRPE | - | E | - | - | COG0031 | YGR155W | CYS4 cystathionine beta-synthase |
| - | - | PAC | E | - | - | COG0833 | YGR191W | HIP1 histidine permease |
| - | - | - | E | - | - | COG0531 | YGL077C | HNM1 choline permease |
| - | - | - | E | - | - | COG0367 | YGR124W | ASN2 asparagine synthetase |
| - | - | - | E | - | - | COG0367 | YPR145W | ASN1 asparagine synthetase |
| - | RRPE | - | E | E7 | MIT | COG0059 | YLR355C | ILV5 ketol-acid reducto-isomerase |
| - | - | - | E | E7 | MIT | COG0473 | YIL094C | LYS12 homoisocitrate dehydrogenase |
| - | - | - | E | H7 | - | COG0722 | YBR249C | ARO4 2-dehydro-3-deoxyphosphoheptonate aldolase, tyrosine-inhibited |
| - | - | - | E | H7 | - | COG0082 | YGL148W | ARO2 chorismate synthase |
| - | - | - | EH | E7 | - | COG0028 | YGR087C | PDC6 pyruvate decarboxylase 3 |
| - | - | - | EH | E7 | - | COG0028 | YLR134W | PDC5 pyruvate decarboxylase, isozyme 2 |
| - | RRPE | - | F | F2 | - | COG0503 | YML022W | APT1 adenine phosphoribosyltransferase |
| - | - | PAC | F | F2 | - | COG0005 | YLR017W | MEU1 multiple enhancer of UAS2 |
| - | RRPE | PAC | F | F3 | - | COG0504 | YBL039C | URA7 CTP synthase 1 |
| - | RRPE | - | F | F3 | - | COG0167 | YKL216W | URA1 dihydroorotate dehydrogenase |
| - | RRPE | - | F | F3 | - | COG0461 | YML106W | URA5 orotate phosphoribosyltransferase |
| - | - | - | F | F3 | - | COG0563 | YDR226W | ADK1 adenylate kinase, cytosolic |
| - | - | - | F | F3 | - | COG0284 | YEL021W | URA3 orotidine-5'-phosphate decarboxylase |
| - | - | - | F | F3 | - | COG0563 | YKL024C | URA6 uridine-monophosphate kinase |
| - | - | - | F | F3 | - | COG0418 | YLR420W | URA4 dihydroorotase |
| - | RRPE | - | FE | F1 | - | COG0462 | YHL011C | PRS3 ribose-phosphate pyrophosphokinase |
| - | RRPE | - | FE | F1 | - | COG0462 | YKL181W | PRS1 ribose-phosphate pyrophosphokinase |
| - | RRPE | - | G | G1 | - | COG0205 | YGR240C | PFK1 6-phosphofructokinase, alpha subunit |
| - | - | - | G | G1 | - | COG0205 | YMR205C | PFK2 6-phosphofructokinase, beta subunit |
| - | - | - | G | G2 | - | COG0166 | YBR196C | PGI1 glucose-6-phosphate isomerase |
| - | - | - | G | G2 | - | COG0149 | YDR050C | TPI1 triose-phosphate isomerase |
| - | - | - | G | G2 | - | COG0057 | YJL052W | TDH1 glyceraldehyde-3-phosphate dehydrogenase 1 |
| - | - | - | G | G2 | - | COG0191 | YKL060C | FBA1 fructose-bisphosphate aldolase |
| - | - | - | G | G2 | - | COG0158 | YLR377C | FBP1 fructose-1,6-bisphosphatase |
| - | RRPE | PAC | G | G3 | - | COG0120 | YOR095C | RKI1 D-ribose-5-phosphate ketol-isomerase |
| - | RRPE | PAC | G | G3 | - | COG0021 | YPR074C | TKL1 transketolase 1 |
| - | RRPE | - | H | - | - | COG0499 | YER043C | SAH1 S-adenosyl-l-homocysteine hydrolase |
| - | - | - | H | - | - | COG0192 | YDR502C | SAM2 S-adenosylmethionine synthetase 2 |
| - | - | - | H | - | - | COG0192 | YLR180W | SAM1 S-adenosylmethionine synthetase 1 |
| - | RRPE | PAC | J | - | - | COG1889 | YDL014W | NOP1 fibrillarin |
| - | RRPE | PAC | J | - | - | COG1499 | YHR170W | NMD3 nonsense-mediated mRNA decay protein |
| - | RRPE | PAC | J | - | - | COG1498 | YLR197W | SIK1 involved in pre-rRNA processing |
| - | RRPE | PAC | J | - | - | COG0144 | YNL061W | NOP2 nucleolar protein |
| - | RRPE | PAC | J | - | - | COG1498 | YOR310C | NOP58 required for pre-18S rRNA processing |
| - | RRPE | PAC | J | - | - | COG1374 | YPL211W | NIP7 required for efficient 60S ribosome subunit biogenesis |
| - | RRPE | PAC | J | - | - | COG0030 | YPL266W | DIM1 rRNA (adenine-N6, N6-)-dimethyltransferase |
| - | RRPE | - | J | - | - | COG0293 | YCL054W | SPB1 required for ribosome synthesis, putative methylase |
| - | RRPE | - | J | - | - | COG0689 | YGR095C | RRP46 involved in rRNA processing |
| - | RRPE | pac | J | - | - | COG3277 | YHR089C | GAR1 nucleolar rrna processing protein |
| - | RRPE | - | J | - | - | COG2519 | YJL125C | GCD14 translational repressor of GCN4 |
| - | RRPE | - | J | - | - | COG0349 | YOR001W | RRP6 similarity to human nucleolar 100K polymyositis-scleroderma protein |
| - | - | PAC | J | - | - | COG0009 | YGL169W | SUA5 translation initiation protein |
| - | rrpe | PAC | J | - | - | COG1097 | YHR069C | RRP4 3' → 5' exoribonuclease required for 3' end formation of 5.8S rRNA |
| - | - | PAC | J | - | - | COG1736 | YKL191W | DPH2 diphtheria toxin resistance protein |
| - | - | PAC | J | - | - | COG1798 | YLR172C | DPH5 diphthamide methyltransferase |
| - | - | - | J | - | - | COG2123 | YDR280W | RRP45 rRNA processing protein |
| RAP1 | RRPE | - | J | J1 | - | COG2092 | YAL003W | EFB1 translation elongation factor eEF1beta |
| - | RRPE | - | J | J1 | - | COG1503 | YBR143C | SUP45 translational release factor |
| - | RRPE | - | J | J1 | - | COG0480 | YOR133W | EFT1 translation elongation factor EEF2 |
| - | RRPE | - | J | J1 | - | COG1601 | YPL237W | SUI3 translation initiation factor EIF2 beta subunit |
| - | - | - | J | J1 | - | COG0480 | YDR385W | EFT2 translation elongation factor EEF2 |
| - | - | - | J | J1 | - | COG0361 | YMR260C | TIF11 translation initiation factor EIF1A |
| - | RRPE | PAC | J | J2 | - | COG0215 | YNL247W | Cysteinyl-tRNA synthetase |
| - | RRPE | - | J | J2 | - | COG0423 | YBR121C | GRS1 glycine-tRNA ligase |
| - | RRPE | - | J | J2 | - | COG0008 | YGL245W | Strong similarity to glutamine-tRNA ligase |
| - | RRPE | - | J | J2 | - | COG0017 | YHR019C | DED81 asparaginyl-tRNA-Synthetase |
| - | RRPE | - | J | J2 | - | COG0008 | YOR168W | GLN4 glutaminyl-tRNA synthetase |
| - | - | - | J | J2 | - | COG0172 | YDR023W | SES1 seryl-tRNA synthetase, cytosolic |
| RAP1 | - | - | J | J3 | - | COG2051 | YHR021C | RPS27B ribosomal protein S27.E |
| - | RRPE | - | J | J3 | - | COG0199 | YLR388W | RPS29A ribosomal protein S29.E.A |
| - | RRPE | - | J | J3 | - | COG2053 | YOR167C | RPS28A 40S small subunit ribosomal protein S28.E.C15 |
| - | - | - | J | J3 | - | COG1998 | YLR167W | RPS31 ubiquitin/40S small subunit ribosomal protein |
| - | - | - | J | J3 | - | COG2053 | YLR264W | RPS28B 40S small subunit ribosomal protein S28.E.C12 |
| - | - | - | J | J3 | - | COG0096 | YLR367W | RPS22B ribosomal protein S15A.E.C12 |
| - | RRPE | PAC | J | J4 | - | COG0244 | YKL009W | MRT4 mRNA turnover 4 |
| - | RRPE | PAC | J | J4 | - | COG2075 | YLR009W | Similarity to ribosomal protein L24.E.B |
| - | RRPE | - | J | J4 | - | COG0088 | YBR031W | RPL4A ribosomal protein |
| - | RRPE | - | J | J4 | - | COG0088 | YDR012W | RPL4B ribosomal protein L4.E.B |
| - | - | - | J | J4 | - | COG2058 | YDL081C | RPP1A 60S large subunit acidic ribosomal protein A1 |
| - | - | - | J | J4 | - | COG1632 | YMR121C | RPL15B 60S large subunit ribosomal protein L15.E.C13 |
| - | - | - | J | J4 | - | COG2157 | YMR242C | RPL20A 60S large subunit ribosomal protein |
| - | - | - | J | J4 | - | COG0097 | YNL067W | RPL9B ribosomal protein L9.E.C14 |
| - | - | - | J | J4 | - | COG0081 | YPL220W | RPL1A ribosomal protein |
| - | RRPE | - | JE | J1 | - | COG0050 | YDR172W | SUP35 eukaryotic peptide chain release factor GTP-binding subunit |
| - | RRPE | - | JE | J1 | - | COG0050 | YER025W | GCD11 translation initiation factor EIF2 gamma chain |
| - | RRPE | PAC | K | - | - | COG0571 | YMR239C | RNT1 double-stranded ribonuclease |
| - | RRPE | PAC | K | - | - | COG0724 | YPL043W | NOP4 nucleolar protein |
| - | RRPE | - | K | - | - | COG0724 | YER165W | PAB1 mRNA polyadenylate-binding protein |
| - | - | - | K | - | - | COG0724 | YDR429C | TIF35 translation initiation factor EIF3 (P33 subunit) |
| - | - | - | K | - | - | COG0724 | YOR361C | PRT1 translation initiation factor EIF3 subunit |
| - | RRPE | PAC | K | K1 | - | COG2012 | YBR154C | RPB5 DNA-directed RNA polymerase I, II, III 25 kD subunit |
| - | - | PAC | K | K1 | - | COG1761 | YNL113W | RPC19 DNA-directed RNA polymerase I, III 16 kD subunit |
| - | - | - | K | K1 | - | COG0202 | YIL021W | RPB3 DNA-directed RNA-polymerase II, 45 kDA |
| - | - | - | K | K1 | - | COG1644 | YOR210W | RPB10 DNA-directed polymerase I, II, III 8.3 subunit |
| - | - | - | K | K1 | - | COG1758 | YPR187W | RPO26 DNA-directed RNA polymerase I, II, III 18 kD subunit |
| - | RRPE | PAC | L | - | - | COG1643 | YGL120C | PRP43 involved in spliceosome disassembly |
| - | RRPE | PAC | L | - | - | COG1643 | YMR128W | ECM16 similarity to helicases |
| - | RRPE | PAC | LKJ | - | - | COG0513 | YGL078C | DBP3 putative RNA helicase required for pre-rRNA processing |
| - | RRPE | PAC | LKJ | - | - | COG0513 | YGL171W | ROK1 ATP-dependent RNA helicase |
| - | RRPE | PAC | LKJ | - | - | COG0513 | YJL033W | HCA4 can suppress the U14 snoRNA rRNA processing function |
| - | RRPE | PAC | LKJ | - | - | COG0513 | YKR024C | DBP7 RNA helicase required for 60S ribosomal subunit assembly |
| - | RRPE | PAC | LKJ | - | - | COG0513 | YLL008W | DRS1 RNA helicase of the DEAD box family |
| - | RRPE | - | LKJ | - | - | COG0513 | YKR059W | TIF1 translation initiation factor 4A |
| - | - | PAC | LKJ | - | - | COG0513 | YJL138C | TIF2 translation initiation factor EIF4A |
| - | RRPE | - | O | - | - | COG0545 | YLR449W | FPR4 strong similarity to peptidylprolyl isomerase FPR3P |
| - | RRPE | - | O | - | - | COG0443 | YNL209W | SSB2 heat shock protein of HSP70 family, cytosolic |
| - | - | - | O | - | - | COG0443 | YHR064C | PDR13 regulator protein involved in pleiotropic drug resistance |
The first list comprises those genes known to be targeted by Rap1 [18] and the second one those that are not. In the presence of motif columns, '-' means that no motif was identified. Regulatory motifs in uppercase correspond to those found by our computational analysis, those in lowercase correspond to motifs found by others [10,18]. The genes are ordered by their functional category.
The key player in the translation process, the ribosome, is accompanied by its cofactors, RNA polymerases and RNA-handling proteins. These include proteins related to the spliceosome, which was expected as mRNA splicing is mostly devoted to ribosomal proteins in yeast [32]. In addition, a number of other processes may also be necessary for a successful translation. These back-up processes may feed the raw materials necessary for the synthesis of ribosomal/messenger RNA (sugar and nucleotide metabolism) as well as polypeptide chains (amino-acid metabolism). However, the tight association of the translation machinery with sugar metabolism may also reflect the close relation between the amount of nutrients in the environment and cellular growth [32].
The fact that these functional subgroups come together when very different experiments are analyzed supports the hypothesis that their transcription may indeed be co-regulated. Therefore, the upstream regions of the genes encoding these proteins might share common motifs that may serve as binding sites for the same transcription factors. A computational analysis of the upstream regions of this gene subset was carried out to investigate the presence of common local sequences (Materials and methods). Three significant motifs were found: rap1, rrpe and pac. The results are summarized in Table 2 and the sequence consensus for each motif shown in Figure 6.
Figure 6.
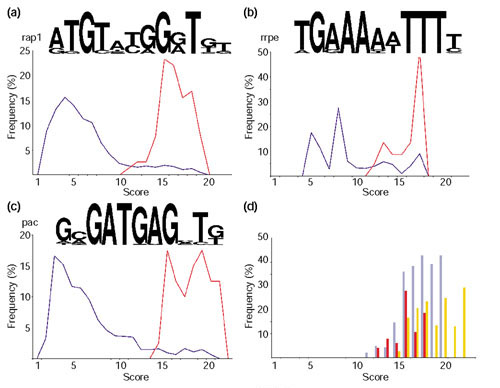
Motifs found in upstream regions of genes involved in protein synthesis. (a-c) The motif logo and the distribution of scores from the matches of these motifs to the upstream regions of all yeast genes (dark blue) or just to those from which the motif was built (red). (a) The motif rap1 presents a periodicity that roughly corresponds to the pitch of a DNA helix and is similar to the sequence repeat found in telomeres, which is also targeted by the protein Rap1 [64]. (b) The motif rrpe contains an A-rich patch followed by a T-rich patch. The lengths of these two patches vary between genes. This motif may be palindromic. (c) The motif pac is made of highly conserved residues (around 100%) at several neighboring positions. (d) Ratio of genes in the cytoplasmic protein translation set and all S. cerevisiae genes matching at a given score to rap1 (purple), rrpe (red) and pac (orange) motifs.
It is well known that repressor-activator P protein 1 (Rap1) targets upstream motifs of a number of ribosomal protein genes (RPG) as well as being central to the cellular economy during rapid growth [33]. Previous experimental studies have uncovered a number of genes regulated by Rap1 including RPGs and genes involved in protein synthesis and sugar metabolism [18]. Approximately half the genes in our set were identified in that study as targets of Rap1. However, the rap1 motif was not found in 25% of these genes, perhaps because they have a degenerate sequence, weaker for computational detection. On the other hand, additional putative rap1 motifs were found in some of the genes that had not been identified previously as Rap1 targets, including genes involved in amino-acid synthesis (SPE4), and genes for a translation elongation factor (EFB1) and a ribosomal protein (RPS27B).
Rrpe is an experimentally uncharacterized motif that has been suggested by computational studies to be specific for rRNA-processing genes [10]. The PAC box (pac motif), which stands for polymerase A and C box, has been found to be conserved in at least 10 genes encoding subunits of RNA polymerases A and C [34], although neither a function nor a trans-acting factor for this motif has been identified. We have identified a number of new genes containing rrpe and pac motifs in their upstream regions.
Genes with either only rap1 (33%) or only rrpe (16%) motifs were frequent, as were genes with a combination of rrpe and pac (around 12%) and rap1 and rrpe (around 13%). The other combinations (pac, rap1/pac and rap1/rrpe/pac) were very rare, that is, less than 4%. None of these motifs was found in a quarter of the analyzed sequences.
To determine the specificity of these motifs for our set of proteins, the motifs were compared to all upstream regions of coding sequences in S. cerevisiae. The distributions of scores for all genes and those in our 'cytoplasmic translation' set are plotted in Figures 6a,6b,6c. As expected, the sequences used to build the motifs matched with higher scores. A more informative plot is shown in Figure 6d, in which the ratios of sequences in our set with respect to all S. cerevisiae genes matching a motif at a given score are depicted, rap1 gives the best ratio, which means that our set contains a good representation of all genes regulated by Rap1, even though it only contains 100 of the approximately 300 genes known to bind Rap1 [18]. The low values for the other two motifs might represent the absence of other functionally related genes containing these motifs in our dataset or the occurrence of these motifs in genes involved in other processes. Although the latter possibility cannot be discarded, previous genes identified to contain rrpe and pac motifs also presented functions related to protein translation [10], which suggests a role for these motifs as regulators of this process.
Genes containing rrpe and pac motifs function as RNA polymerases and helicases, or are involved in RNA processing and the pentose pathway. The rrpe motif may also regulate the expression of some aminoacyl-tRNA synthetases, pyrimidine biosynthetic proteins and the chaperones FPR4 and SSB2. FPR4 is a putative peptidyl-prolyl isomerase, and SSB2 belongs to the Hsp70 family. An SSB2 homolog, SSB1, was shown to be regulated by Rap1 and transcribed at the same time as ribosomal proteins [35]. This suggested an active role of SSB1 in folding of newly synthesized polypeptide chains, as also shown for other SSB proteins [26]. This may also be the case for the chaperones found in the present work. SSB2, an Hsp70 protein, may prevent aggregation of nascent polypeptides, whereas FPR4 may speed up folding by facilitating proline isomerization.
All these genes were repressed under the conditions studied (Figure 7c). It remains to be seen whether the motifs found might control both the repression and activation of genes. In eukaryotes, it is quite common for a protein to serve either as an activator or as a represser, depending on the gene-regulatory proteins present in the cell [36]. In fact, some cases of Rap1 bound to the promoters of inactive genes have been reported, which suggests that other cofactors may determine the transcriptional activity of genes downstream of Rap1 binding [18]. The same mechanism may take place in regulation via pac and rrpe.
Figure 7.
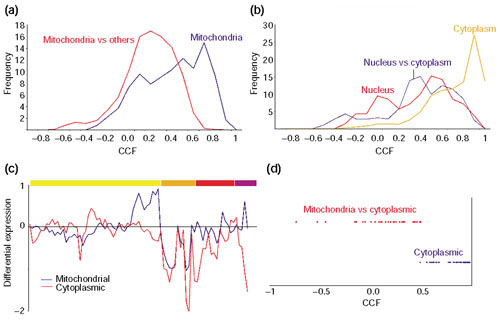
Cytoplasmic versus mitochondrial translation machinery. (a, b) The distribution of CCF values for gene pairs in which both members belong to the COG functional category J, protein translation. (a) The distribution of CCF values for gene pairs in which both members are mitochondrial proteins (dark blue) or one member is mitochondrial and the another one is not (red). (b) The distribution of CCF values for gene pairs in which both members are cytoplasmic (orange), both nuclear (red), or one member is cytoplasmic and the another one is nuclear (dark blue). (c) The averaged expression profiles of the mitochondrial translation machinery (dark blue) and the set of genes involved in protein translation in the cytoplasm, including those involved in the metabolism of sugar, amino acids and nucleotides, as well as RNA processing proteins and polymerases. The color-coding for the experiments is the same as in Figure 4. (d) Distribution of CCF values for pairs of genes that belong to the same COG. The cytoplasmic pairs (dark blue) correspond to paralog-paralog relationships whereas the ones involving cytoplasmic and mitochondrial proteins (red) correspond to orthologs. The expression of duplicated genes can be distinguished from each other because even though two genes can encode two proteins with identical amino-acid sequences, the degree of identity at the DNA level can be low enough to make a selective DNA hybridization onto the chip feasible. This seems to be the case for the genes analyzed in (d).
Analysis of the protein translation class: compartmentalization and homology
The proteins of the COG functional class J, protein translation, are mainly localized in cytoplasm, nucleus and mitochondrion. We observed a clear distinction in the expression of the J genes with respect to their subcellular localization. The correlations of mitochondrial and non-mitochondrial (nuclear and cytoplasmic) gene pairs stopped at 0.7 (Figure 7a). On the other hand, the J genes found in the nucleus and the cytoplasm still correlate at very high thresholds, which implies that their expressions may be coupled (Figure 7b). The nuclear genes, which are mainly involved in the assembly of ribosomal subunits and in the processing and transport of RNAs should, in principle, only be required for cytoplasmic translation.
Functional class J also contains a number of COGs with more than one protein. Some of them are real orthologs and others are paralogs, which are very common in S. cerevisiae as half its genome is duplicated [37]. These paralogs can, in some cases, conserve the function of their ancestors and even replace each other. For example, most ribosomal sub-units are encoded by two genes that yield nearly identical proteins. On the other hand, the 'repeated' orthologs do not complement each other. This is the case for the translational machineries - cytoplasmic and mitochondrial. Even though they have different origins (the mitochondrial one originated by gene transfer from its endosymbiotic bacterial ancestor to the nucleus), they have retained the same function. We observed a poor correlation between cytoplasmic and mitochondrial ribosomal genes, although in some experiments, for example, sporulation, they might seem to be correlated (Figure 7c).
Both cytoplasmic and mitochondrial translation machineries still require the same ingredients for transcription (sugar and nucleotides) and translation (amino acids). However, this metabolic network seems to be coupled only to the cytoplasmic machinery. During the evolution of the endosymbiotic mitochondria, most of the metabolic genes have been passed onto the host nucleus and their function developed in its host cytoplasm [38,39]. The mitochondrion takes in all metabolites and factors by transporters added de novo to its membrane to ensure the delivery of all the necessary primary raw material. However, none of the few mitochondrial transporter genes present in the microarray data was associated with the mitochondrial translation set.
The analysis of the J COGs with more than one protein provides some interesting insight into the behavior of paralogs and orthologs. Plotting the CCF values of paralog-paralog pairs (mostly redundant cytoplasmic ribosomal proteins) and ortholog-ortholog pairs (cytoplasmic and mitochondrial ribosomal partners) shows that there is a spatial and temporal separation of their gene products (Figure 7d). Also, the range of CCF values for cytoplasmic paralogs is somewhat broad, indicating that duplicated genes, even in the case of duplicated ribosomal genes, are not necessarily expressed identically. The reason for keeping duplicated 'redundant' genes is unknown, although it may be related to a selection mechanism for increased level of expression, as many of the over-duplicated gene functional categories include highly expressed genes, for example, heat shock, glucose metabolism and ribosomal proteins [40].
Comparison of the expression of the translation machinery of Escherichia coli and S. cerevisiae
Protein translation is a universal mechanism present in all organisms. In fact, most of the proteins conserved in all organisms are involved either in translation (J) or transcription (K). We wanted to know if the expression of the sets of genes associated with the ribosome is co-regulated across different species in a similar way to our budding yeast cytoplasmic translation set. A comparison against bacterial and archaeal organisms could be carried out by taking advantage of the operon organization of their genomes. An operon is made up of genes that are transcribed as part of a single mRNA molecule. Co-transcribed genes are co-regulated at the transcriptional level and often have related roles, for example involving protein-protein interactions or as part of the same metabolic pathway [41].
The first comparison was against E. coli, as its operons have been best characterized experimentally. All the operons containing ribosomal protein genes as well as others containing genes for proteins that may be involved in translation were selected (see Materials and methods). Table 3 shows all the genes in these operons along with their corresponding COG and whether or not there is a yeast homolog in the microarray dataset. The 'ribosomal operons' contain genes involved in: J (general translation, including translation factors, J1, and ribosomal proteins for the small subunit, J3, and large subunit, J4), K1 (DNA-dependent RNA polymerases), F3 (pyrimidine biosynthesis), EHR (amino-acid transport and metabolism), L (general DNA handling, including basal replication machinery, L1) and a gene with unknown function (R) that may possibly be a nucleic-acid-binding protein. The other operons include genes with other functional categories such as: J2 (aminoacyl-tRNA synthetases), N (protein secretion), H (general coenzyme metabolism, including menaquinone biosynthesis, H7, and pyridoxal phosphate biosynthesis, H9), K2 (basal transcription factors), O (chaperones) and G3 (pentose phosphate pathway).
Table 3.
List of proteins in the E. coli protein translation operons
| Operon name | Eco ORF | Yeast | COG ID | Function | ORF description |
| tRNA synthetase and peptidase | b0026 | Yes* | COG0060 | J2 | Isoleucine tRNA synthetase |
| tRNA synthetase and peptidase | b0027 | No | COG0597 | N | Prolipoprotein signal peptidase |
| tRNA synthetase and peptidase | b0028 | No | COG1047 | O | Probable FKBX-type 16kD peptidyl-prolyl cis-trans isomerase |
| rRNA modification and chaperone | b0049 | Yes* | COG0639 | T | Diadenosine tetraphosphatase |
| rRNA modification and chaperone | b0050 | No | COG2967 | P | Uncharacterized protein affecting Mg2+/Co2+ transport |
| rRNA modification and chaperone | b0051 | Yes** | COG0030 | J | 6-m-2-A methyltransferase; put. 16S rRNA methyltransferase |
| rRNA modification and chaperone | b0052 | No | COG1995 | H9 | Pyridoxine and pyridoxal phosphate biosynthesis |
| rRNA modification and chaperone | b0053 | Yes | COG0760 | O | Peptidyl prolyl isomerase |
| Ribosomal protein 1 | b0169 | Yes** | COG0052 | J3 | Ribosomal protein S2 |
| Ribosomal protein 1 | b0170 | No | COG0264 | J1 | Translation elongation factor EF-Ts |
| Ribosomal protein 1 | b0171 | No | COG0528 | F3 | Uridylate kinase |
| Ribosomal protein 1 | b0172 | Yes* | COG0233 | J1 | Ribosome releasing factor operon? |
| tRNA modification and protein export | b0405 | No | COG0809 | J | S-adenosylmethionine:tRNA ribosyltransferase-isomerase |
| tRNA modification and protein export | b0406 | No | COG0343 | J | Queuine tRNA-ribosyltransferase |
| tRNA modification and protein export | b0407 | No | COG1862 | N | ORF, hypothetical protein |
| tRNA modification and protein export | b0408 | No | COG0342 | N | Protein-export membrane protein SecD |
| tRNA modification and protein export | b0409 | No | COG0341 | N | Protein-export membrane protein SecF |
| Ribosomal protein 2 | b0910 | No | COG0283 | F3 | Cytidine monophosphate kinase |
| Ribosomal protein 2 | b0911 | Yes* | COG0539 | J3 | Ribosomal protein S1 |
| Ribosomal protein 3 | b1088 | No | COG1399 | R | Predicted metal-binding, possibly nucleic acid-binding protein |
| Ribosomal protein 3 | b1089 | Yes | COG0333 | J4 | Ribosomal protein L32 |
| tRNA synthetase and oxidase | b1637 | Yes* | COG0162 | J2 | Transfer RNA-Tyr synthetase |
| tRNA synthetase and oxidase | b1638 | Yes | COG0259 | H9 | Pyridoxamine 5'-phosphate oxidase |
| Phenylalanine tRNA synthetase | b1713 | Yes | COG0073 | R | Phenylalanine tRNA synthetase, beta subunit |
| Phenylalanine tRNA synthetase | b1714 | Yes* | COG0016 | J2 | Phenylalanine tRNA synthetase, alpha subunit |
| Ribosomal protein 4 | b2606 | Yes* | COG0335 | J4 | Ribosomal protein L19 |
| Ribosomal protein 4 | b2607 | No | COG0336 | J | tRNA (m1G) methyltransferae |
| Ribosomal protein 4 | b2608 | No | COG0806 | J | ORF, hypothetical protein |
| Ribosomal protein 4 | b2609 | Yes | COG0228 | J3 | Ribosomal protein S16 |
| Ribosomal protein 5 | b3065 | No | COG0828 | J3 | Ribosomal protein S21 |
| Ribosomal protein 5 | b3066 | No | COG0358 | L1 | DNA primase |
| Ribosomal protein 5 | b3067 | No | COG0568 | K1 | RNA polymerase sigma-subunit |
| Ribosomal protein 6 | b3164 | No | COG1185 | J | Polynucleotide phosphorilase |
| Ribosomal protein 6 | b3165 | Yes** | COG0184 | J3 | Ribosomal protein S15 |
| RNA modification | b3166 | Yes* | COG0130 | J | tRNA pseudouridine 55 synthase; P35 |
| RNA modification | b3167 | No | COG0858 | J | Ribosome-binding factor A; P15B |
| Transcription and translation | b3168 | Yes | COG0532 | J1 | Initiation factor IF2-alpha (infB) |
| Transcription and translation | b3169 | No | COG0195 | K2 | NusA protein |
| Transcription and translation | b3170 | No | COG0779 | S | Hypothetical 16.8 kD protein |
| Ribosomal protein 7 | b3185 | Yes* | COG0211 | J4 | Ribosomal protein L27 |
| Ribosomal protein 7 | b3186 | No | COG0261 | J4 | Ribosomal protein L21 |
| Ribosomal protein 8 | b3230 | Yes** | COG0103 | J3 | 30S ribosomal protein S9 |
| Ribosomal protein 8 | b3231 | Yes** | COG0102 | J4 | 50S ribosomal protein L13 |
| Ribosomal protein 9 | b3258 | Yes* | COG0591 | EHR | Pantothenate permease |
| Ribosomal protein 9 | b3259 | No | COG2264 | J | Ribosomal protein L11 methyltransferase |
| tRNA modification | b3287 | No | COG0242 | J1 | N-formylmethionylaminoacyl-tRNA deformylase |
| tRNA modification | b3288 | Yes | COG0223 | J1 | Methionyl-tRNA formyltransferase |
| tRNA modification | b3289 | Yes** | COG0144 | J | Sun protein (Fmu protein) |
| Ribosomal protein 10 | b3294 | Yes* | COG0203 | J4 | 50S ribosomal protein L17 |
| Ribosomal protein 10 | b3295 | Yes** | COG0202 | K1 | RNA polymerase, alpha subunit |
| Ribosomal protein 10 | b3296 | Yes** | COG0522 | J3 | 30S ribosomal protein S4 |
| Ribosomal protein 10 | b3297 | Yes** | COG0100 | J3 | 30S ribosomal protein Sll |
| Ribosomal protein 10 | b3298 | Yes** | COG0099 | J3 | 30S ribosomal protein S13 |
| Ribosomal protein 11 | b3299 | Yes | COG0257 | J4 | 50S ribosomal protein X |
| Ribosomal protein 11 | b3300 | No | - | - | (secY) membrane protein, protein secretion |
| Ribosomal protein 11 | b3301 | Yes** | COG0200 | J4 | 50S ribosomal protein L15 |
| Ribosomal protein 11 | b3302 | Yes** | COG1841 | J4 | 50S ribosomal protein L30 |
| Ribosomal protein 11 | b3303 | Yes** | COG0098 | J3 | 30S ribosomal protein S5 |
| Ribosomal protein 11 | b3304 | Yes** | COG0256 | J4 | 50S ribosomal protein L18 |
| Ribosomal protein 11 | b3305 | Yes** | COG0097 | J4 | 50S ribosomal protein L6 |
| Ribosomal protein 11 | b3306 | Yes** | COG0096 | J3 | 30S ribosomal protein S8 |
| Ribosomal protein 11 | b3307 | Yes** | COG0199 | J3 | 30S ribosomal protein S14 |
| Ribosomal protein 11 | b3308 | Yes** | COG0094 | J4 | 50S ribosomal protein L5 |
| Ribosomal protein 11 | b3309 | Yes** | COG0198 | J4 | 50S ribosomal protein L24 |
| Ribosomal protein 11 | b3310 | Yes** | COG0093 | J4 | 50S ribosomal protein L14 |
| Ribosomal protein 12 | b3311 | Yes** | COG0186 | J3 | 30S ribosomal protein S17 |
| Ribosomal protein 12 | b3312 | Yes** | COG0255 | J4 | 50S ribosomal protein L29 |
| Ribosomal protein 12 | b3313 | Yes** | COG0197 | J4 | 50S ribosomal protein L16 |
| Ribosomal protein 12 | b3314 | Yes** | COG0092 | J3 | 30S ribosomal protein S3 |
| Ribosomal protein 12 | b3315 | Yes** | COG0091 | J4 | 50S ribosomal protein L22 |
| Ribosomal protein 12 | b3316 | Yes** | COG0185 | J3 | 50S ribosomal protein S19 |
| Ribosomal protein 12 | b3317 | Yes** | COG0090 | J4 | 50S ribosomal protein L2 |
| Ribosomal protein 12 | b3318 | Yes** | COG0089 | J4 | 50S ribosomal protein L23 |
| Ribosomal protein 12 | b3319 | Yes** | COG0088 | J4 | 50S ribosomal protein L4 |
| Ribosomal protein 12 | b3320 | Yes** | COG0087 | J4 | 50S ribosomal protein L3 |
| Ribosomal protein 12 | b3321 | Yes** | COG0051 | J3 | 30S ribosomal protein S10 |
| Ribosomal protein 13 | b3339 | Yes** | COG0050 | J1 | Protein chain elongation factor EF-Tu |
| Ribosomal protein 13 | b3340 | Yes** | COG0480 | J1 | Protein chain elongation factor EF-G |
| Ribosomal protein 13 | b3341 | Yes** | COG0049 | J3 | 30S ribosomal protein S7 |
| Ribosomal protein 13 | b3342 | Yes** | COG0048 | J3 | 30S ribosomal protein S12 |
| Dam superoperon | b3384 | Yes* | COG0180 | J2 | Tryptophanyl-tRNA synthetase |
| Dam superoperon | b3385 | Yes | COG0546 | R | 2-phosphoglycolate phosphatase |
| Dam superoperon | b3386 | Yes | COG0036 | G3 | D-ribulose-5-phosphate epimerase |
| Dam superoperon | b3387 | No | COG0338 | L | Adenine methylase |
| Dam superoperon | b3388 | No | COG3266 | S | Putative membrane protein; interferes with cell division |
| Dam superoperon | b3389 | Yes | COG0337 | H7 | 3-dehydroquinate synthase |
| Dam superoperon | b3390m | Yes | COG0703 | H7 | Shikimic acid kinase I |
| Glycine trna synthetase | b3559 | No | COG0751 | J2 | Glycine tRNA synthetase, beta chain |
| Glycine trna synthetase | b3560 | No | COG0752 | J2 | Glycine tRNA synthetase, alpha chain |
| Ribonuclease and pyrimidine biosynthesis | b3642 | Yes** | COG0461 | F3 | Orotate phosphoribosyltransferase |
| Ribonuclease and pyrimidine biosynthesis | b3643 | Yes** | COG0689 | J | Ribonuclease PH |
| Ribosomal protein 14 | b3703 | Yes | COG0230 | J4 | Ribosomal protein L34 |
| Ribosomal protein 14 | b3704 | No | COG0594 | J | Ribonuclease P protein component |
| Protein export and transcription | b3981 | No | COG0690 | N | Preprotein translocase secE subunit |
| Protein export and transcription | b3982 | Yes* | COG0250 | K2 | Transcription antitermination |
| Ribosomal protein 15 | b3983 | Yes** | COG0080 | J4 | 50S ribosomal protein Lll |
| Ribosomal protein 15 | b3984 | Yes** | COG0081 | J4 | 50S ribosomal protein Ll |
| Ribosomal protein 16 | b3985 | Yes** | COG0244 | J4 | 50S ribosomal protein L10 |
| Ribosomal protein 16 | b3986 | Yes | COG0222 | J4 | 50S ribosomal protein L7/L12 |
| Ribosomal protein 16 | b3987 | Yes* | COG0085 | K1 | DNA-directed RNA polymerase beta chain |
| Ribosomal protein 16 | b3988 | Yes* | COG0086 | K1 | DNA-directed RNA polymerase beta' chain |
| Ribosomal protein 17 | b4200 | Yes* | COG0360 | J3 | 30S ribosomal protein S6 |
| Ribosomal protein 17 | b4201 | No | COG2965 | L | Primosomal replication protein N |
| Ribosomal protein 17 | b4202 | Yes | COG0238 | J3 | 30S ribosomal protein S18 |
| Ribosomal protein 17 | b4203 | Yes | COG0359 | J4 | SOS ribosomal protein L9 |
The Yeast column indicates whether or not there is a yeast homolog of the bacterial gene (Yeast). The notation is as follows: No (genes lacking a yeast homolog), Yes (genes with a yeast homolog that was not present in the microarray data), Yes* (genes with a yeast homolog that, although present in the microarray data, were not part of the 'cytoplasmic translation' cluster) and Yes** (genes with a yeast homolog that are found in the cluster)
The functional categories found in these operons closely resemble those in the yeast cluster deduced from gene-expression experiments, with the exception of the genes involved in protein secretion (N), which do not have yeast homologs. Interestingly, two chaperones were also found in the protein translation operons of E. coli. Both were peptidyl-prolyl cis-trans isomerases (PPI). B0028 is an FKBP-type PPI homologous to the yeast PPI identified above (FPR4). B0053 also has a yeast homolog although it was not present in the microarray dataset. It is well known that in bacteria, in addition to some Hsp70-like chaperones, the trigger factor is associated with the ribosome [42]. The bacterial trigger factor, which lacks a eukaryotic homolog, has PPI and chaperone activity and is thus probably involved in co-translational protein folding. The fact that PPIs are probably co-expressed with ribosomal proteins in two different organisms suggests that there may be a general mechanism to facilitate protein folding by accelerating cis-trans proline conversions during protein translation. The next step was to extend this comparison to other bacterial and archaeal organisms.
Conservation of the expression of the translation machinery across species
Comparison of complete microbial genomes has revealed a large number of conserved gene clusters, that is, sets of adjacent genes that have the same order and orientation in two or more different genomes. A recent study has detected and analyzed these conserved gene pairs to estimate their probability of belonging to the same co-transcribed unit or operon [43]. From this study, we have built a 'translation-machinery' set of probably co-regulated genes for each species by merging all the gene pairs in which one of the members was a ribosomal subunit (see Materials and methods).
Bacteria and archaea were compared separately to the yeast set (Table 4, and see also Additional data files). Most of the genes in these groups have a yeast partner in the COGs (82% for bacteria, and 90% for archaea). However, when ubiquitous genes were not considered, only 55% of the bacterial genes had a yeast homolog, in contrast to 80% for archaea. This suggests that the processes associated with protein translation are more similar between archaea and yeast than between bacteria and yeast. For example, all organisms share a set of key functional classes (J1, J3, J4, F3, K1, K2, N), but some bacteria also have genes involved in DNA replication (L1) as part of the protein translation operon (Table 4). These DNA replication COGs are COG0305 (replicative DNA hell-case), COG0164 (ribonuclease HII) and COG0629 (single-stranded DNA-binding protein). It remains to be seen if this is a special adaptation for some organisms to somehow couple the process of protein translation and cell division, or if the same proteins just facilitate transcription.
Table 4.
Distribution of functional classes in bacterial and archaeal 'translation operons'
| Function | Total | High frequency | Low frequency | |
| Distribution of functional classes in bacterial 'translation operons' | ||||
| No | F3 | 1.57 | 0 | 2.94 |
| H | 0.79 | 0 | 1.47 | |
| I | 0.79 | 0 | 1.47 | |
| I2 | 1.57 | 0 | 2.94 | |
| J | 5.51 | 5.08 | 5.88 | |
| J1 | 1.57 | 3.39 | 0 | |
| J3 | 0.79 | 0 | 1.47 | |
| J4 | 3.15 | 5.08 | 1.47 | |
| K2 | 1.57 | 0 | 2.94 | |
| L | 0.79 | 0 | 1.47 | |
| L1 | 0.79 | 0 | 1.47 | |
| M | 1.57 | 0 | 2.94 | |
| N | 0.79 | 0 | 1.47 | |
| P | 1.57 | 0 | 2.94 | |
| R | 2.36 | 0 | 4.41 | |
| S | 2.36 | 0 | 4.41 | |
| D | 0.79 | 0 | 1.47 | |
| Yes | E | 0.79 | 0 | 1.47 |
| H3 | 0.79 | 0 | 1.47 | |
| H5 | 0.79 | 0 | 1.47 | |
| I | 1.57 | 0 | 2.94 | |
| J | 0.79 | 0 | 1.47 | |
| J1 | 1.57 | 1.69 | 1.47 | |
| J3 | 1.57 | 3.39 | 0 | |
| J4 | 5.51 | 10.17 | 1.47 | |
| L1 | 0.79 | 0 | 1.47 | |
| N | 1.57 | 0 | 2.94 | |
| R | 2.36 | 0 | 4.41 | |
| S | 0.79 | 0 | 1.47 | |
| Yes* | E9 | 0.79 | 0 | 1.47 |
| F5 | 0.79 | 0 | 1.47 | |
| I1 | 0.79 | 0 | 1.47 | |
| J | 1.57 | 0 | 2.94 | |
| J1 | 2.36 | 0 | 4.41 | |
| J2 | 2.36 | 0 | 4.41 | |
| J3 | 1.57 | 1.69 | 1.47 | |
| J4 | 3.15 | 5.08 | 1.47 | |
| K1 | 1.57 | 0 | 2.94 | |
| K2 | 0.79 | 1.69 | 0 | |
| L1 | 0.79 | 0 | 1.47 | |
| N | 1.57 | 1.69 | 1.47 | |
| O | 0.79 | 0 | 1.47 | |
| R | 1.57 | 0 | 2.94 | |
| Yes** | F1 | 0.79 | 0 | 1.47 |
| F3 | 0.79 | 0 | 1.47 | |
| J | 0.79 | 0 | 1.47 | |
| J1 | 2.36 | 3.39 | 1.47 | |
| J3 | 11.81 | 25.42 | 0 | |
| J4 | 14.96 | 30.51 | 1.47 | |
| K1 | 0.79 | 1.69 | 0 | |
| Distribution of functional classes in archaeal 'translation operons' | ||||
| No | E | 0.88 | 0 | 2.78 |
| EM | 0.88 | 0 | 2.78 | |
| F | 0.88 | 1.28 | 0 | |
| F3 | 0.88 | 1.28 | 0 | |
| G2 | 0.88 | 0 | 2.78 | |
| J | 3.51 | 0 | 11.11 | |
| K1 | 0.88 | 1.28 | 0 | |
| K2 | 0.88 | 1.28 | 0 | |
| L | 0.88 | 0 | 2.78 | |
| R | 0.88 | 0 | 2.78 | |
| S | 3.51 | 5.13 | 0 | |
| T | 0.88 | 0 | 2.78 | |
| Yes | J | 0.88 | 0 | 2.78 |
| J1 | 0.88 | 0 | 2.78 | |
| O | 0.88 | 0 | 2.78 | |
| R | 0.88 | 0 | 2.78 | |
| S | 3.51 | 1.28 | 8.33 | |
| Yes* | C | 0.88 | 0 | 2.78 |
| E2 | 0.88 | 0 | 2.78 | |
| F | 0.88 | 0 | 2.78 | |
| F5 | 0.88 | 0 | 2.78 | |
| J | 1.75 | 1.28 | 2.78 | |
| J1 | 2.63 | 2.56 | 2.78 | |
| K | 0.88 | 1.28 | 0 | |
| K1 | 2.63 | 3.85 | 0 | |
| K2 | 0.88 | 1.28 | 0 | |
| L | 0.88 | 0 | 2.78 | |
| N | 1.75 | 1.28 | 2.78 | |
| O | 0.88 | 0 | 2.78 | |
| Yes** | G2 | 0.88 | 1.28 | 0 |
| J | 4.39 | 0 | 13.89 | |
| J1 | 1.75 | 2.56 | 0 | |
| J3 | 19.3 | 26.92 | 2.78 | |
| J4 | 31.58 | 41.03 | 11.11 | |
| K1 | 3.51 | 5.13 | 0 | |
The list of functions is divided into blocks according to the presence or absence of a yeast homolog in the microarray data with the same coding as in Figure3. The last three columns represent the percentage of genes in each functional category with respect to all the genes in the 'operon' (Total), those genes found only in more than (High frequency) or less than 50% (Low frequency) of all the species studied. Functions found with high frequency in different organisms are highlighted in bold. It can be seen that the COG functional categories of the yeast, bacterial and archaeal genes are very similar, suggesting also that proteins with the same function, but not necessarily evolutionarily related, could replace each other in different organisms. More comprehensive tables of the data summarized in Tables 1,2,3,4 are provided as additional data files.
Interestingly, some of the proteins in the archaeal and bacterial groups have been assigned to uncharacterized COGs for which functional information is unavailable. For archaea, these are: COG1325, COG1422, COG1460, COG1500, COG1909, COG2042, COG2106 and COG2118. All the proteins associated to these COGs were exclusively found in archaea, with a few exceptions in which yeast partners were also found. Two of these groups, COG1325 and COG1500, belonged to the functional category S, function unknown, in the earlier version used originally in the analysis. In the recently updated version, however, both are assigned to category J, protein translation, as predicted exosome subunits. Another one, COG2118, has been assigned to class R, general function prediction only, in which one of its members, MTH1615, is a DNA-binding protein. For the rest of the COGs, PSI-BLAST searches were done using as references the genes forming the COG to determine whether homologs from other species absent from the COGs might have been shown to have a role in protein translation or related processes.
No hints on function were found for COG1422 and COG1909. On the other hand, the other three did find characterized partners in other archaea not included in the COGs. COG2106 matched to 024783, a putative ribosomal protein located in a gene cluster coding for ribosomal proteins in Halobacterium marismortui [44]. The other two had similarity to proteins in Sulfolobus solfataricus. COG2042 proteins were similar to Q9UWV6, a RNase P involved in tRNA and 4.5S RNA-processing. COG1460 was similar to q9uxd9 (DNA-directed RNA polymerase, subunit F). Interestingly, the list of hits for COG1460 also contained a DNA-directed RNA polymerase (RPB4_SCHPO) from the fission yeast Schizosaccharomyces pombe with a poor E-score. However, the matching region had the same size as the reference proteins and corresponded to a RPOL4C domain, which is a DNA-directed RNA-polymerase subunit. When this yeast protein was incorporated in the profile, the subsequent iterations also picked a large number of other RNA-polymerases from higher organisms. All of them matched to the profile through their RPOLC domain.
In the bacterial clusters, four COGs of functional category 8 were found, that is, COG0759, COG0779, COG1284 and COG1610. All were bacterial COGs only, with the exception of COG1610, which also included yeast. No additional information could be obtained for COG0779 and COG1284. The proteins of COG0759 were very short and matched to Pfam domain DUF37, whose function is unknown although it is found in a protein from Aeromonas hydrophila that has been shown to have hemolytic activity. In fact, some other members of the hit list were also putative hemolytic proteins. The relevance of this toxin-like protein in the ribosomal cluster is unknown and it may just reflect some particular adaptation of the pathogenic organisms in which it is present (Haemophilus influenzae, Neisseria meningitidis MC58 and Helicobacter pylori J99). Proteins of COG1610 contain the Pfam domain DUF186, which may have a role in tRNA metabolism, specifically as a glutamyl-tRNA aminotransferase.
In general, these uncharacterized proteins seem to have predicted functions in agreement with an active role in protein translation: RNA processing (COG2042), ribosomal subunit (COG2106), RNA polymerase (COG1460) and aminoacyl-tRNA transferase (COG1610). The remaining uncharacterized COGs might also play a part in protein translation.
Discussion
Clustering of genes pregrouped into COG functional categories
The approach presented here has proved useful for a preliminary quick survey of microarray data. General trends can be obtained by analysis of genes with similar functionality without any a priori information about their regulation. For example, the category 'L - -' ('DNA replication, recombination and repair') splits into processes that correspond to some of the characteristic stages of cell division. In this case, it provides an idea about the different cellular states found along a time series. This kind of information could be obtained from any experiment by analyzing one or more relevant functional classes of expected importance under the studied conditions. It may also help to refine sequence annotation by delimiting the temporal distribution of genes within cellular processes with respect to their partners in a given functional class. Two examples have been provided. The 'L - -' group, comprising DNA-handling proteins, has been shown to split into several subgroups that act during cell division (replication control, DNA repair and histones) as well as those helicases involved in transcription during protein synthesis. In these cases, a broad functional group can be described in terms of its more specific subgroups. The second example dealt with the behavior of the expression of genes belonging to the same COG and therefore sharing a high sequence similarity, which precludes finer classification by sequence-similarity approaches. However, the expression of these genes is clearly different and allows finer grouping into subgroups that seem to be more consistent with other biochemical data, as shown for the group of permeases of the major facilitator superfamily.
We have shown that the averaged profiles of genes with the same functional class and a similar expression profile produce meaningful clusters. This may be used for a quick assessment of the quality of the data by investigating the behavior of genes that should always appear together, such as protein complexes [45]. Also, systematic grouping of genes into averages would reduce the number of elements to be handled during the analysis, providing a simple and straightforward way to recognize the processes held in a cluster. Gene averaging also increases the signal-to-noise ratio. Therefore, comparison of averaged genes may work better in those cases for which no duplicated chips are available (which is the case for the data used here). However, this approach will be more powerful with a larger set of genes since then the functional classes could be more or less uniformly represented. Even though genes with unknown function are not initially considered, they could be correlated later to the profiles of the functional classes to get hints about their function.
Cross-talk between functional groups
Clustering of averaged profiles revealed connections between functional classes involved in two highly conserved processes shared by all organisms: protein translation and DNA replication. These two processes are very different in nature. DNA replication occurs once in a cell's life and the proteins involved are tightly regulated in a series of synchronized steps. On the other hand, protein translation is a housekeeping process and its components are constitutively expressed. Both processes require nucleotides and thus they are associated with genes involved in nucleotide synthesis. Interestingly, these sets are different. Protein translation requires a continuously high production of both purines and pyrimidines for RNA synthesis, whereas the DNA replication set is only associated with thymidylate biosynthesis, which is the nucleotide absent from RNA and characteristic of DNA.
The absence of cross-talk between the majority of functional groups could represent the limited number of genes used in our analysis or the adaptability of the cells to the surrounding environment. The latter suggests that only very few processes are tightly associated between them, in this case only protein translation, DNA replication and, to a lesser extent, the TCA cycle (see Materials and methods). The other processes are required together or separately under different conditions, and this may reflect the flexibility of organisms in making a successful response to a range of unpredictable variations in the medium where they live.
Upstream regulatory motifs of genes involved in protein synthesis
Combining microarray experiments is a powerful tool for identification of co-regulated genes that may share upstream regulatory regions. The analysis of the upstream regions of the genes involved in protein translation has revealed three possible key regulatory motifs: pac, rrpe and rap1. Rap1 targets have been well characterized elsewhere [18]. We have identified several new genes containing pac and rrpe motifs. All these genes agree with the functional classes known to contain these motifs, and strengthen their relevance in the regulation of protein translation and transcription. We have also observed some preferential combination between these motifs within genes. Combination of several motifs may be important for regulation [46]. For example, promoters of ribosomal protein genes typically contain a Rap1-binding site adjacent to a T-rich element that also participates in activation by an as-yet unknown mechanism [47]. Also, promoters of genes that encode glycolytic proteins usually contain a Rap1-binding site adjacent to or flanked by multiple binding sites for the GCR1 protein [48]. In the same way, combinations of rap1, rrpe and pac motifs may have a role in protein synthesis regulation. In fact, the tight co-regulated expression of genes containing both pac and rrpe motifs in their upstream regions has recently been reported [49].
Comparison of gene expression across species
The use of the COG information for the analysis of gene-expression data has provided a scaffold for the comparison of expression data between different organisms, revealing a probable link in the expression of peptidyl-prolyl isomerases and ribosomal proteins in two organisms from different phylogenetic kingdoms. It is already known that the cellular information processing systems, at least transcription and translation, are more similar, both at the sequence and presence level, between archaea and eukaryotes when compared to bacteria [50,51]. Furthermore, even though some of the archaeal ribosomal protein genes are organized into 'bacteria-like' operons, the corresponding amino-acid sequences are more similar to those of their eukaryotic, not bacterial, counterparts. Our study suggests that the expression of these genes may also be more similar between archaea and yeast than yeast and bacteria. Interestingly, the protein translation operons of some bacterial organisms also seem to encode proteins involved in other processes, such as cell division and production of toxins. This may be just an adaptation of particular organisms to their niche. Perhaps the indication that the expression of yeast ribosomal genes is associated with sugar metabolism may also represent a yeast adaptation for linking growth rate to carbon source availability, where the organism grows quickly by fermentation in a glucose-rich medium but switches to respiration when the glucose level decreases, accompanied by a drop in the expression of glycolytic enzymes and ribosomal proteins. This tight link between sugar metabolism and ribosomal expression is not observed in the archaeal and bacterial operons analyzed here. However, it cannot be discarded, because operons that do not contain ribosomal proteins may still be co-regulated with 'ribosomal' operons through the same transcription factors.
Conclusions
The expression profiles of genes have been organized into classes defined by their functional annotation and the resulting groups compared with each other to reveal possible interconnections. This approach has proved useful for a preliminary quick survey of microarray data and, in principle, could be used with any type of functional classification. The analysis of yeast genes has revealed a different regulation of the expression of cytoplasmic and mitochondrial proteins involved in translation. It has also identified three main putative regulatory motifs in the upstream regions of the genes in the cytoplasmic set. This set contains not only the ribosomal and RNA-processing proteins but also chaperones and enzymes involved in the synthesis of sugars, nucleotides and amino acids. Homologous genes in bacterial and archaeal organisms showed a potentially similar co-regulation in their expression, including the co-expression of peptidyl-prolyl isomerases and ribosomal proteins. This indicates that the components of the protein translation process are conserved across organisms at the expression level, perhaps with minor specific adaptations.
Materials and methods
COG functional classes of the genes in the expression data
The original dataset [52] contained the expression profiles for 2,467 genes, of which 996 were present in the COGs [53]. This set corresponds to less than half of the S. cerevisiae genes found in the COGs (996 out of 2,175 total genes), but they comprise 559 of the 904 budding yeast COGs. Table 1 shows the distribution of the COG general functional and pathway/system categories of the genes in the microarray data.
Preprocessing of experimental data
A standard filter was applied to prevent the inclusion of genes in which the variation in expression was small (< 2.3-fold) or for which the expression profiles have too many time points missing, and therefore they may yield misleading correlations against other gene profiles. When all the experiments were considered as a whole, all genes but one were differentially expressed, in contrast to the result observed when analyzing individual experiments. The percentage of genes that were differentially expressed in each individual experiment included less than half the genes, except in the sporulation dataset, in which 71% of the genes were regulated. Furthermore, the cell-division experiments, which in principle should involve a very similar set of genes, showed a broad range of values for the different synchronization methods: 9% α-factor, 19% elutriation and 35% cdc-15 strain. The combination of these three resulted in a total of 49%, indicating that some of the genes did not overlap between experiments. This could be due to different synchronization procedures introducing different artifacts [19].
The standard Pearson correlation coefficient was calculated for each gene pair using the profiles of individual experiments (zero was assigned if any of the genes did not pass the cutoff filtering), and all experiments as a whole.
Protein subcellular localization versus gene expression
A list of budding yeast proteins with known localization was retrieved from MIPS [54,55]. Only compartments with more than 50 such proteins were considered: plasma membrane (63), endoplasmic reticulum (63), mitochondrion (136), nucleus (185), cytoplasm (303) and unknown localization (239).
The frequency of gene pairs encoding proteins with the same subcellular localization and a correlation coefficient (when comparing the whole profiles) above/below a given threshold was calculated as:
![]()
where GeneSameloc is the number of distinct genes involved in gene pairs whose products are found in the same compartment, loc; and GeneDiffloc is the number of distinct genes whose products have a different localization to loc but whose expression significantly correlate with at least one of the proteins in loc. The thresholds ranged from 0.5 to 0.8 for correlations, and between -0.5 and -0.8 for anti-correlations.
Furthermore, to estimate the behavior of a random distribution in which there is no relation between expression and localization, the following approach was undertaken. The labels associating genes to compartments were randomly shuffled and the frequencies for the new gene-compartment pairs calculated. This process was repeated 45 times, averaging all the resulting frequencies at the end. The average approximated to a flat line crossing the y-axis at a value proportional to the number of in each class.
'Consistency' and 'comprehensiveness' of predefined functional classes
Functional classes were predefined, as described in the text, by taking into account general function, specific pathway/system (if any) and mitochondrial or non-mitochondrial localization. Two comparisons were carried out to investigate the overall behavior of the classes.
The first one reflects the 'consistency' for a given classification and is the proportion of gene pairs with a CCF higher than a given threshold with respect to the total number of possible pairs for that class. This is calculated as follows:
![]()
where i is the individual group for the classification, p is the total number of groups resulting from the classification, nCCFi is the number of gene pairs for group i at a given CCF, and ci is the total number of unique gene pairs for group i, which corresponds to
![]()
where Ni is the total number of genes for group i.
The second comparison gives an idea of the 'comprehensiveness'. The 'comprehensiveness' of a given classification is the proportion of correlated gene pairs present in that classification with respect to the total number of correlated gene pairs in the whole dataset at a given threshold, and is calculated as follows:
![]()
where i is the individual group for the classification, p is the total number of groups resulting from the classification, nCCFi is the number of correlated gene pairs for group i at a given CCF, and NCCF is the total number of correlated gene pairs (without imposing any a priori classification) at a given CCF.
Subgrouping of genes with the same functional category, pathway/system and subcellular location
Each ORF in the dataset that belonged to the COGs was given an additional string, 'Fn Pn Ln', where F and P are the COG general functional category and pathway/system, if any, respectively; and L is its subcellular location: that is, mitochondrial or non-mitochondrial (Figure 8, step 1). The CCFs, considering the expression profiles as a whole, of gene pairs with the same 'Fn Pn Ln' string were extracted into a matrix (Figure 8, step 2). Then, gene pairs with a CCF lower than a given threshold and whose correlations were inconsistent between individual experiments: that is, both positive and negative values were found in different experiments, were discarded (Figure 8, step 3). Finally, the remaining gene pairs (Figure 8, step 4) were grouped into unique subgroups with non-intersecting elements between them (Figure 8, step 5).
Figure 8.
Procedure for splitting genes of the same functional class into finer subgroups. See text for details.
In general, the resulting subgroups reflected functional (that is, involved in the same, more specific, cellular process) and/or structural (that is, part of the same multi-protein complex) relationships. However, there was no common cutoff value for all the groups, and whereas some genes separated at relatively low CCF (around 0.5), other groups split into finer groups only at high values (> 0.7). At the highest threshold (0.8), usually, only multi-protein complexes remained.
Selection of subgroups
The averaged profiles of selected functional subgroups were compared to investigate how the subgroups obtained relate to each other. The selection of subgroups was the initial issue. As mentioned above, separation into biologically meaningful subgroups was sometimes obtained at different thresholds for different initial classes, and although in principle a more objective approach would be to consider a unique cutoff to select the subgroups, this would have resulted in poorly resolved subgroups at low CCF thresholds, and in a small number of subgroups (that is, poor representation of functions) at high thresholds. Moreover, at continuously increasing thresholds, the number of members in the subgroups decreased. The criterion to select the subgroups was to get a broad representation of functional classes without excluding too many genes but without compromising the quality of the averages by including poorly correlated genes. For that, the subgroups were selected manually (taking advantage of our knowledge of biological processes) and selection may therefore be somewhat subjective. It resulted in a mixture of subgroups obtained at different thresholds. The quality of the chosen subgroups was assessed by correlating the resulting averages against all the individual genes. A subgroup was regarded to be adequate if: the CCF average of all the genes contributing to the averaged profile was higher than 0.7; the CCF of the individual genes was not lower than 0.5 (Figure 9); and the CCF presented a reasonable consistency (data not shown). The list of the chosen subgroups is available as additional data.
Figure 9.
The range of CCF values for the selected classes and their genes. The red dots are the CCFs of the expression profiles of all the genes versus the averaged profile of the class they belong to. The blue points correspond to the means after averaging the CCFs of all the genes within a class.
Cross-talk between functional subgroups
The averaged profiles were correlated and grouped in a similar way to that described in Figure 8. The only difference was that the global CCF was calculated as follows:
![]()
where CCFexp is the correlation coefficient of experiment exp in a set of related experiments (that is, cell cycle comprising α-factor, cdc-15 and elutriation; sporulation; shock experiments comprising heat, temperature and reducing stress; and diauxic shift), Nexp is the number of time points for a given experiment, and Ntotal; is the total number of time points.
The standard correlation is a description of the shape of two profiles without taking into account the intensity. It works well when a single peak is expected in the series. However, when combining several experiments, a number of peaks will be present, and if the intensities in one experiment are much higher than in others, this could introduce a bias in the comparison, resulting in a correlation coefficient that reflects best the similarity for that region of high intensity to the detriment of significant similarities or differences in other regions of lower intensity. The time series of the sporulation data presented extremely high peaks when compared to the data obtained for the other conditions. This is why a weighted correlation was used here.
Initially, a clustering was carried out with a threshold of 0.7. This resulted in a large number of averaged profiles without correlating partners, as well as four sets with more than one of the selected functional subgroups. These sets represented DNA replication, protein translation (both cytoplasmic and mitochondrial) and energy production (TCA cycle). However, not all the averages in the DNA replication group presented the expected periodicity in the cell-cycle region, and this is why the threshold was increased until only averages with a consistent periodicity were grouped together (reached at a cutoff value of 0.71). This periodic behavior of cell-cycle-regulated genes was thus used as a benchmark.
Finding common upstream motifs
AlignACE [10], a program for identifying motifs over-represented in DNA sequences, was used to search for common upstream regions in the complete cluster of genes involved in cytoplasmic protein translation (198 upstream regions) and, subsequently, in two subsets representing the genes that have been already identified to contain a rap1 binding motif (97) and those that may not have it (101). Default values for S. cerevisiae were used, including correction for GC content. AlignACE uses a stochastic algorithm. In general, it will find the strongest motifs in each run, usually with slight differences, although even that is not guaranteed, in particular for the weaker motifs (Jason Hughes, personal communication). Several runs of alignACE yielded slightly different results for our data. Therefore, 20 runs were carried out for each dataset and the results were combined as follows: only motifs found in at least 80% of the runs with a MAP score higher than 10 were considered. The resulting motifs matched to rap1, rrpe and pac motifs with correlation scores higher than 0.8 when compared to known motifs [56] by the compareACE program. The scanACE program was used to find matches for the above motifs in the upstream regions of all yeast genes.
The sequence consensus for each motif was calculated using Weblogo [57,58].
Gathering of E. coli protein translation operons
A collection of experimentally determined E. coli operons was retrieved [59,60]. The selection of operons according to their possible involvement in protein translation was determined both from the operon names and from the genes contained within them. The final selected operons were all those named: 'ribosomal protein', 'RNA modification', 'dam super-operon' (contains tryptophanyl-tRNA synthetase), 'glycine tRNA synthetase', 'phenylalanine tRNA synthetase', 'protein export and transcription', 'rRNA modification and chaper-one', 'ribonuclease and pyrimidine biosynthesis', 'tRNA modification', 'tRNA modification and protein export', 'tRNA synthetase and oxidase', 'tRNA synthetase and peptidase' and 'transcription and translation'.
Each bacterial gene was assigned to its corresponding COG. The occurrence of yeast genes with the same COGs was used to assess whether they may be equivalent representatives when comparing operons against the cluster deduced from yeast expression data. Table 3 summarizes the final list of E. coli protein translation operons used in this work.
Clustering of operon pairs
A previous study has provided a list of pairs of neighboring genes from different organisms with a high probability of being part of the same operon [43,61]. These pairs of genes were merged into groups of pairs with common members. Then, a 'protein-synthesis operon' was obtained by merging all groups containing at least one ribosomal protein, as it was assumed that the stoichiometry of the ribosome has to be maintained and thus the expression of its subunits should be co-regulated. Finally, every gene in each operon was converted to its corresponding COG to allow comparison between organisms through their equivalent orthologs, as described previously.
The table for the bacterial and archaeal operons can be found in the additional data files.
PSI-BLAST of functional uncharacterized COGs
PSI-BLAST is a position-specific identification algorithm that improves the resolution of BLAST for finding distantly related homologs by means of sequence-similarity comparisons weighted by a matrix in which both the mutation frequency of amino acids and the positions of conserved residues are taken into account [62]. PSI-BLAST searches on a non-redundant protein database were conducted using the online facilities provided by the NCBI [63]. For each COG, every protein of the analyzed organisms was used as reference. Thresholds for inclusion of proteins into the profiles was set to 0.005 (default) for archaea, and 0.00001 for bacteria. The stringent cutoff chosen for bacteria ensured that only proteins with a very high similarity to the reference were included. The first BLAST search provided hits to known SMART or Pfam motifs, if any. The iterations were continued until no new sequences were found.
Additional data files
A figure showing diagrams of the F--, FP-, F-L and FPL classes for the COG functional category J, and tables listing: genes of the subgroups shown in Figure 4; genes contained in each chosen subgroup that were used for analysis of crosstalk between functional classes; all COGs found in the 'ribosomal operons' of a number of bacterial organisms; and the archaeal 'ribosomal operons', are available as additional data files with the online version of this paper.
Supplementary Material
A figure showing diagrams of the F--, FP-, F-L and FPL classes for the COG functional category J.
Table listing genes of the subgroups shown in Figure 4.
Table listing genes contained in each chosen subgroup that were used for analysis of crosstalk between functional classes.
Table listing all COGs found in the 'ribosomal operons' of a number of bacterial organisms
Table listing all COGs found in the archaeal 'ribosomal operons'.
Acknowledgments
Acknowledgements
We thank Giampietro Schiavo, Simon Tomlinson, Robert Gilbert, Gavin Kelly and Frederic Meunier for critical reading of the manuscript.
References
- Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Nierman WC, Feldblyum TV, Laub MT, Paulsen IT, Nelson KE, Eisen J, Heidelberg JF, Alley MR, Ohta N, Maddock JR, et al. Complete genome sequence of Caulobacter crescentus. Proc Natl Acad Sci USA. 2001;98:4136–4141. doi: 10.1073/pnas.061029298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenk HP, Clayton RA, Tomb JF, White O, Nelson KE, Ketchum KA, Dodson RJ, Gwinn M, Hickey EK, Peterson JD, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. doi: 10.1038/37052. [DOI] [PubMed] [Google Scholar]
- Ruepp A, Graml W, Santos-Martinez ML, Koretke KK, Volker C, Mewes HW, Frishman D, Stocker S, Lupas AN, Baumeister W. The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature. 2000;407:508–513. doi: 10.1038/35035069. [DOI] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Mewes HW, Albermann K, Bahr M, Frishman D, Gleissner A, Hani J, Heumann K, Kleine K, Maierl A, Oliver SG, et al. Overview of the yeast genome. Nature. 1997;387(Suppl):7–65. doi: 10.1038/42755. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JD, Estep PW, Tavazoie S, Church GM. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Goffeau A. Four years of post-genomic life with 6,000 yeast genes. FEBS Lett. 2000;480:37–41. doi: 10.1016/s0014-5793(00)01775-0. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Brown PO. DNA arrays for analysis of gene expression. Methods Enzymol. 1999;303:179–205. doi: 10.1016/s0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–4868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- Lieb JD, Liu X, Botstein D, Brown PO. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001;28:327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, Umansky L, Drawid A, Jansen R, Liu Y, et al. Subcellular localization of the yeast proteome. Genes Dev. 2002;16:707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamames J, Ouzounis C, Sander C, Valencia A. Genomes with distinct function composition. FEBS Lett. 1996;389:96–101. doi: 10.1016/0014-5793(96)00527-3. [DOI] [PubMed] [Google Scholar]
- Gautschi M, Lilie H, Funfschilling U, Mun A, Ross S, Lithgow T, Rucknagel P, Rospert S. RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc Natl Acad Sci USA. 2001;98:3762–3767. doi: 10.1073/pnas.071057198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfund C, Lopez-Hoyo N, Ziegelhoffer T, Schilke BA, Lopez-Buesa P, Walter WA, Wiedmann M, Craig EA. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosomenascent chain complex. EMBO J. 1998;17:3981–3989. doi: 10.1093/emboj/17.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao SS, Paulsen IT, Saier MH. Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62:1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan S, Johnston M. Function and regulation of yeast hexose transporters. Microbiol Mol Biol Rev. 1999;63:554–569. doi: 10.1128/mmbr.63.3.554-569.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles E, Hollenberg CP. The molecular genetics of hexose transport in yeasts. FEMS Microbiol Rev. 1997;21:85–111. doi: 10.1111/j.1574-6976.1997.tb00346.x. [DOI] [PubMed] [Google Scholar]
- Wieczorke R, Krampe S, Weierstall T, Freidel K, Hollenberg CP, Boles E. Concurrent knock-out of at least 20 transporter genes is required to block uptake of hexoses in Saccharomyces cerevisiae. FEBS Lett. 1999;464:123–128. doi: 10.1016/s0014-5793(99)01698-1. [DOI] [PubMed] [Google Scholar]
- Nourani A, Wesolowski-Louvel M, Delaveau T, Jacq C, Delahodde A. Multiple-drug-resistance phenomenon in the yeast Saccharomyces cerevisiae : involvement of two hexose transporters. Mol Cell Biol. 1997;17:5453–5460. doi: 10.1128/mcb.17.9.5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24:437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- Shore D. RAP1: a protean regulator in yeast. Trends Genet. 1994;10:408–412. doi: 10.1016/0168-9525(94)90058-2. [DOI] [PubMed] [Google Scholar]
- Dequard-Chablat M, Riva M, Carles C, Sentenac A. RPC19, the gene for a subunit common to yeast RNA polymerases A (I) and C (III). J Biol Chem. 1991;266:15300–15307. [PubMed] [Google Scholar]
- Lopez N, Halladay J, Walter W, Craig EA. SSB, encoding a ribosome-associated chaperone, is coordinately regulated with ribosomal protein genes. J Bacteriol. 1999;181:3136–3143. doi: 10.1128/jb.181.10.3136-3143.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AD. The price of repression. Cell. 1995;81:655–658. doi: 10.1016/0092-8674(95)90524-3. [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC. Molecular evidence for an ancient duplication of the entire yeast genome. Nature. 1997;387:708–713. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF. The origin and early evolution of mitochondria. Genome Biol. 2001;2:reviews1018.1–1018.5. doi: 10.1186/gb-2001-2-6-reviews1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283:1476–1481. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- Seoighe C, Wolfe KH. Yeast genome evolution in the post-genome era. Curr Opin Microbiol. 1999;2:548–554. doi: 10.1016/s1369-5274(99)00015-6. [DOI] [PubMed] [Google Scholar]
- Salgado H, Moreno-Hagelsieb G, Smith TF, Collado-Vides J. Operons in Escherichia coli: genomic analyses and predictions. Proc Natl Acad Sci USA. 2000;97:6652–6657. doi: 10.1073/pnas.110147297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer G, Rauch T, Rist W, Vorderwulbecke S, Patzelt H, Schulze-Specking A, Ban N, Deuerling E, Bukau B. L23 protein functions as a chaperone docking site on the ribosome. Nature. 2002;419:171–174. doi: 10.1038/nature01047. [DOI] [PubMed] [Google Scholar]
- Ermolaeva MD, White O, Salzberg SL. Prediction of operons in microbial genomes. Nucleic Acids Res. 2001;29:1216–1221. doi: 10.1093/nar/29.5.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt E, Kromer W, Hatakeyama T. Organization and nucleotide sequence of a gene cluster coding for eight ribosomal proteins in the archaebacterium Halobacterium marismortui. J Biol Chem. 1990;265:3034–3039. [PubMed] [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Wagner A. Genes regulated cooperatively by one or more transcription factors and their identification in whole eukaryotic genomes. Bioinformatics. 1999;15:776–784. doi: 10.1093/bioinformatics/15.10.776. [DOI] [PubMed] [Google Scholar]
- Rotenberg MO, Woolford JL. Tripartite upstream promoter element essential for expression of Saccharomyces cerevisiae ribosomal protein genes. Mol Cell Biol. 1986;6:674–687. doi: 10.1128/mcb.6.2.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker HV. GCR1 of Saccharomyces cerevisiae encodes a DNA binding protein whose binding is abolished by mutations in the CTTCC sequence motif. Proc Natl Acad Sci USA. 1991;88:9443–9447. doi: 10.1073/pnas.88.21.9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilpel Y, Sudarsanam P, Church GM. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat Genet. 2001;29:153–159. doi: 10.1038/ng724. [DOI] [PubMed] [Google Scholar]
- Podani J, Oltvai ZN, Jeong H, Tombor B, Barabasi AL, Szathmary E. Comparable system-level organization of Archaea and Eukaryotes. Nat Genet. 2001;29:54–56. doi: 10.1038/ng708. [DOI] [PubMed] [Google Scholar]
- Olsen GJ, Woese CR. Archaeal genomics: an overview. Cell. 1997;89:991–994. doi: 10.1016/s0092-8674(00)80284-6. [DOI] [PubMed] [Google Scholar]
- Cluster analysis and display of genome-wide expression patterns http://genome-www.stanford.edu/clustering/ [DOI] [PMC free article] [PubMed]
- COGs http://www.ncbi.nlm.nih.gov/COG/
- Mewes HW, Hani J, Pfeiffer F, Frishman D. MIPS: a database for protein sequences and complete genomes. Nucleic Acids Res. 1998;26:33–37. doi: 10.1093/nar/26.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccharomyces cerevisiae subcellular catalogue http://mips.gsf.de/proj/yeast/catalogues/subcell/index.html
- Selected motifs in Saccharomyces cerevisiae http://atlas.med.harvard.edu/cgi-bin/compareace_motifs.pl
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weblogo sequence logo generation form http://www.bio.cam.ac.uk/cgi-bin/seqlogo/logo.cgi
- Itoh T, Takemoto K, Mori H, Gojobori T. Evolutionary instability of operon structures disclosed by sequence comparisons of complete microbial genomes. Mol Biol Evol. 1999;16:332–346. doi: 10.1093/oxfordjournals.molbev.a026114. [DOI] [PubMed] [Google Scholar]
- E.coli operon table http://www.cib.nig.ac.jp/dda/taitoh/ecoli.operon.html
- Predicting operons in microbial genomes http://www.tigr.org/tigr-scripts/operons/operons.cgi
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLAST http://www.ncbi.nlm.nih.gov/BLAST/
- Gilson E, Roberge M, Giraldo R, Rhodes D, Gasser SM. Distortion of the DNA double helix by RAP1 at silencers and multiple telomeric binding sites. J Mol Biol. 1993;231:293–310. doi: 10.1006/jmbi.1993.1283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A figure showing diagrams of the F--, FP-, F-L and FPL classes for the COG functional category J.
Table listing genes of the subgroups shown in Figure 4.
Table listing genes contained in each chosen subgroup that were used for analysis of crosstalk between functional classes.
Table listing all COGs found in the 'ribosomal operons' of a number of bacterial organisms
Table listing all COGs found in the archaeal 'ribosomal operons'.