

Short abstract
MAPPFinder is a tool that creates a global gene-expression profile across all areas of biology by integrating the annotations of the Gene Ontology (GO) Project with the free software package GenMAPP.
Abstract
MAPPFinder is a tool that creates a global gene-expression profile across all areas of biology by integrating the annotations of the Gene Ontology (GO) Project with the free software package GenMAPP http://www.GenMAPP.org. The results are displayed in a searchable browser, allowing the user to rapidly identify GO terms with over-represented numbers of gene-expression changes. Clicking on GO terms generates GenMAPP graphical files where gene relationships can be explored, annotated, and files can be freely exchanged.
Background
DNA microarray experiments simultaneously measure the expression levels of thousands of genes, generating huge amounts of data. The analysis of these data presents a tremendous challenge to biologists and new tools are needed to help gain biological insights from these experiments. Although the data are generated for individual genes, examining a dataset on a gene-by-gene basis is time consuming and difficult to carry out across an entire dataset. One way of accelerating the pace of data analysis is to approach the data from a higher level of organization. This can be done using data-driven methods, such as hierarchical clustering and self-organizing maps [1,2], which identify groups of genes with similar expression patterns. A complementary approach is to view the data at the level of known biological processes or pathways. Identifying those groups of biologically related genes that are showing a large number of gene-expression changes will create an informative description of the biology that is occurring in a particular dataset, making it possible to generate new hypotheses and identify those specific areas of biology that warrant more detailed investigation.
One tool that assists in the identification of important biological processes is GenMAPP (Gene MicroArray Pathway Profiler) [3], a program for viewing and analyzing microarray data on microarray pathway profiles (MAPPs) representing biological pathways or any other functional grouping of genes. When a MAPP is linked to a gene-expression dataset, GenMAPP automatically and dynamically color codes the genes on the MAPP according to criteria supplied by the user. GenMAPP is a useful starting point for pathway-based analysis of gene-expression data, but there are several critical requirements to be met before this tool can be used to identify correlated gene-expression changes across all biology. On a practical level, pathway-based analysis of microarray data needs to be automated, so that all possible pathways can be explored. Identifying correlated gene-expression changes in an individual pathway is often interesting, but it is necessary to know if the gene-expression changes seen on a particular pathway are unique to this pathway or are occurring in many other pathways. Equally important to automation is expanding the pathway information that is digitally represented. GenMAPP currently has over 50 MAPP files depicting various biological pathways and gene families, but this is still only a small fraction of all known biology [3]. Several other pathway programs such as KEGG [4], EcoCyc/MetaCyc [5], Pathway Processor (which uses KEGG) [6] and ViMAc [7] are available for integration with microarray data analysis, but these programs focus on well-defined metabolic pathways, and like GenMAPP, would benefit from a broader base of pathway information.
To address this issue, we have used information available from the Gene Ontology (GO) Consortium [8]. The GO Consortium is creating a defined vocabulary of terms describing the biological processes, cellular components and molecular functions of all genes. The GO is built in a hierarchical manner, with a parent-child relationship existing between GO terms. Curators at the public gene databases are assigning genes to GO terms to provide annotation and a biological context for individual genes. In addition to providing gene annotation, GO also provides a structure for organizing genes into biologically relevant groupings. These groupings can serve as the basis for identifying those areas of biology showing correlated gene-expression changes in a microarray experiment. While GO has been used to annotate microarray data both by hand and by some software packages [9,10,11], there has been no automated way to use it for pathway-based analysis.
We have developed a tool called MAPPFinder that dynamically links gene-expression data to the GO hierarchy. For each of the 11,239 ([12]; as of May 6, 2002]) GO biological process, cellular component and molecular function terms, MAPPFinder calculates the percentage of the genes measured that meet a user-defined criterion. This is done for each specific GO node, and for the cumulative total of the number of genes meeting the criterion in a parent GO term combined with all of its children, giving a complete picture of the number of genes associated with a particular GO term. Using this percentage and a z score (see Materials and methods), the user can rank the GO terms by their relative amounts of gene-expression changes. MAPPFinder therefore generates a gene-expression profile at the level of biological processes, cellular components and molecular functions, rapidly identifying those areas of biology that warrant further study (Figure 1).
Figure 1.
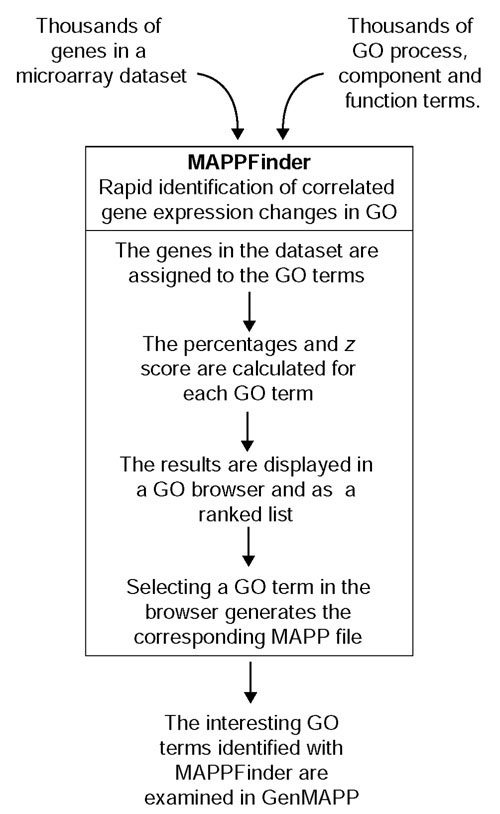
How MAPPFinder works. Microarray data is imported into MAPPFinder as a GenMAPP gene-expression dataset. Using a relational database and the gene-association files from GO, MAPPFinder assigns the thousands of genes in the dataset to the thousands of GO terms. Using a user-defined criterion for a significant gene-expression change, MAPPFinder calculates the percentage of genes meeting the criterion and a statistical score for each GO term. Using the ranked list and GO browser generated by MAPPFinder the user can quickly identify interesting GO terms with high levels of gene-expression changes. The specific genes involved in these GO terms can be examined on automatically generated MAPPs using GenMAPP.
MAPPFinder and GenMAPP are both available free-of-charge at [13].
Results and discussion
To demonstrate the utility of MAPPFinder, we used the program to analyze the publicly available mouse microarray dataset, the FVB benchmark set for cardiac development, maturation and aging [14]. This dataset measures gene-expression levels in the hearts of 12.5-day embryos and adult mice. We have used the 12.5-day embryonic time point to identify those biological processes that show differentially expressed genes between embryonic and adult hearts. We ran the MAPPFinder analysis on this dataset using two criteria, either an increase (fold change > 1.2 and p < 0.05) or decrease (fold change < -1.2 and p < 0.05) in gene expression for the 12.5-day embryo. We chose this dataset for demonstration because of the large number of differences in gene expression observed in the 12.5-day embryo compared to the adult mouse heart tissue.
MAPPFinder linked the 9,946 probe sets measured in this experiment to the 11,239 GO terms [12] in the hierarchy and calculated the percentage of genes meeting the criterion and a z score for each GO term. Table 1 gives an overall summary of the linkages made between the dataset and GO and calculations carried out by MAPPFinder. Nearly half of the 9,946 probe sets measured in the FVB benchmark dataset were connected to a GO term, representing approximately 70% of the mouse genes associated with GO terms [15] and covering a good portion of what is currently known about mouse biology. The proportion of genes in the microarray dataset that link to GO terms will increase as more GO terms and gene associations are added by the Mouse Genome Database (MGD) [16].
Table 1.
Numbers of genes used in the MAPPFinder calculations
| FVB benchmark dataset for development | ||
| Genes measured | 9,946 | |
| Genes linked to MGD directly | 6,267 | |
| Genes linked to MGD via UniGene | 220 | |
| Genes linked to GO terms | 5,120 | |
| Unique genes linked to GO | 4,574 | |
| Genes measured/associated in GO process | 3,544/4,962 (71.4%) | |
| Genes measured/associated in GO component | 3,238/4,691 (69.0%) | |
| Genes measured/associated in GO function | 3,999/5,846 (68.4%) | |
| 12.5-day embryo | ||
| Increased | Decreased | |
| Genes changed | 2,219 | 1,775 |
| Genes linked to GO process | 806 | 711 |
| Genes linked to GO component | 726 | 657 |
| Genes linked to GO function | 885 | 783 |
Of the 9,946 genes measured by this array, 6,267 were linked to the MGD database via the GenBank accession numbers referenced by MGD. An additional 220 genes were linked to MGD using UniGene as an intermediate step (see Materials and methods). Of these 6,487 genes, 5,120 were found in the mouse GO gene-association files. Once duplicate probes were removed, 4,574 unique genes were used for the MAPPFinder analysis. This dataset comprised 71.4% of the 4,962 genes associated with GO process terms, 69% of the 4,691 genes associated with GO component terms, and 68.4% of the 5,846 genes associated with GO function terms [15]. In the 12.5-day embryo, 2,219 genes met the criterion for increased gene expression, 806 having process annotation, 726 having component annotation, and 885 having function annotation. The decreased criterion found 1,775 genes, 711 in process, 657 in component, and 783 in function.
After MAPPFinder assigns the genes in the microarray dataset to the GO structure, it calculates for each GO term the percentage and z score (see Materials and methods) for the genes that meet the user's criterion. These two values can be used to identify GO terms with an over- (or under-) represented number of gene-expression changes. The MAPPFinder results are displayed in two forms. The first is a GO browser that graphically displays the MAPPFinder results in the structure of the GO hierarchy (Figures 2a,3a). The second is a text file listing all the GO terms measured, ranked by the z score. The number of genes meeting the criterion, the number of genes measured in the experiment, and the number of genes assigned to each GO term by MGD are given, along with the respective percentages and z score, in the text file and GO browser (Figure 2b). Table 2 shows the list of process, component and function terms with a z score greater than 2 for the significantly increased and decreased criteria at the 12.5-day embryonic time point. GO terms that had fewer than 5 or more than 100 genes changed were removed from the list because these terms were either too specific or too general for our data analysis. This filter identified the top 108 (8.0%) GO terms for the significantly increased criterion and the top 63 (4.8%) GO terms for the significantly decreased criterion. The stringency of this filter can be increased or decreased by raising or lowering the z score cutoff, or by including terms with larger or smaller numbers of genes. The filtered list was then pruned by hand for related GO terms to remove any over-represented branches of the GO hierarchy (for the complete results, see Additional data files). When both a parent and a child term were present in the list, the parent term was removed if its presence was due entirely to genes meeting the criterion for the child term. The remaining terms on the list still have a large degree of interrelatedness, but have been retained here for completeness.
Figure 2.
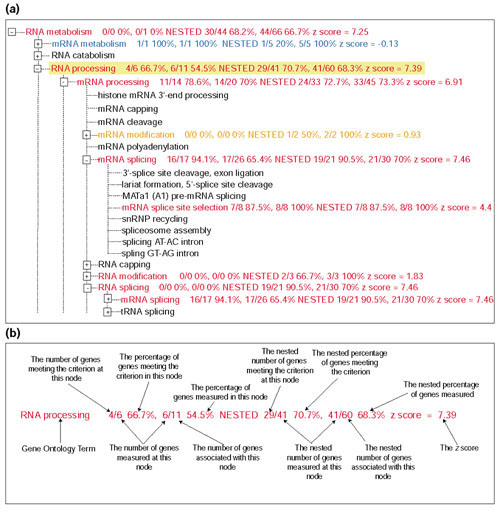
The MAPPFinder browser. (a) The branch of the GO hierarchy rooted at the biological process term 'RNA processing' is shown. The terms are colored with the MAPPFinder results for genes significantly increased in the 12.5-day embryo versus the adult mice. Terms with 0-5% of genes changed are colored black, 5-15% purple, 15-25% dark blue, 25-35% light blue, 35-45% green, 45-55% orange, and greater than 55% red. The term RNA processing is highlighted in yellow, indicating that it meets the search or filter requirements. (b) The MAPPFinder results. The term RNA processing is shown with the various MAPPFinder results labeled. The percentage of genes meeting the criterion and the percentage of genes in GO measured in this experiment are calculated. The results are calculated for both this node individually and in combination with all of its child nodes (that is, nested results). The z score indicates whether the number of genes meeting the criterion is higher or lower than expected. A positive score indicates that more genes are changed than expected; a negative score means fewer genes are changed than expected, and a score near 0 indicates that the number of changes approximates to the expected value for that GO term.
Figure 3.
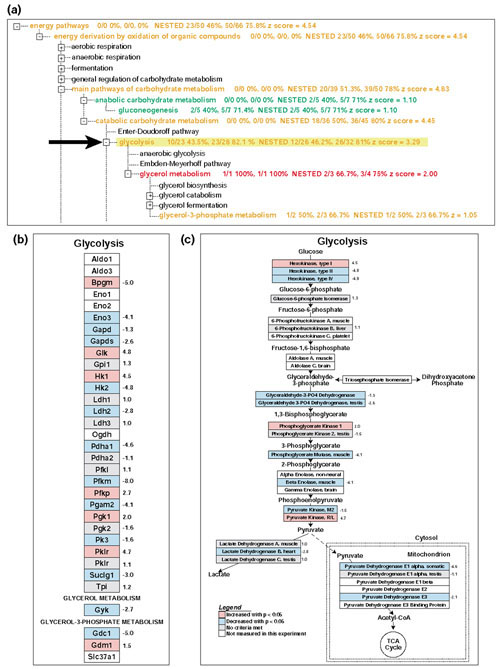
Linking MAPPFinder to GenMAPP. (a) The MAPPFinder browser displaying the 12.5-day embryo increased results for the GO process term 'glycolysis'. Color-coding of GO terms is the same as in Figure 2. (b) Clicking on the GO term glycolysis in the MAPPFinder browser builds the corresponding GenMAPP MAPP file. This MAPP file contains a list of genes associated with this term and all of its children. (c) Genes in the GO list were rearranged with the tools in GenMAPP to depict the glycolysis pathway with the metabolic intermediates and cellular compartments. Color-coding of genes for (b) and (c) is as follows: Red, fold change >1.2 and p < 0.05 in the 12.5-day embryo versus adult mice. Blue, fold change < -1.2 and p < 0.05. Gray, neither of the above criteria met. White, gene not found on the array.
Table 2.
MAPPFinder results for genes significantly increased and significantly decreased in 12.5-day embryos versus adult mice
| GO name | Number changed | Number measured | Number in GO | % Changed | % Present | z score |
| Significantly increased | ||||||
| Process | ||||||
| Mitotic cell cycle | 44 | 70 | 89 | 62.9 | 78.7 | 8.1631 |
| DNA metabolism | 67 | 135 | 163 | 49.6 | 82.8 | 7.6807 |
| mRNA splicing | 19 | 21 | 30 | 90.5 | 70 | 7.4868 |
| RNA processing | 29 | 41 | 60 | 70.7 | 68.3 | 7.4411 |
| RNA metabolism | 30 | 44 | 66 | 68.2 | 66.7 | 7.3038 |
| Cell cycle | 98 | 240 | 291 | 40.8 | 82.5 | 7.0096 |
| mRNA processing | 24 | 33 | 45 | 72.7 | 73.3 | 6.9456 |
| Protein biosynthesis | 52 | 104 | 152 | 50 | 68.4 | 6.8095 |
| Macromolecule biosynthesis | 57 | 121 | 172 | 47.1 | 70.3 | 6.5863 |
| DNA replication | 28 | 46 | 55 | 60.9 | 83.6 | 6.2752 |
| DNA replication and chromosome cycle | 29 | 49 | 62 | 59.2 | 79 | 6.1944 |
| Ribosome biogenesis | 19 | 28 | 37 | 67.9 | 75.7 | 5.7749 |
| Biosynthesis | 89 | 242 | 334 | 36.8 | 72.5 | 5.4866 |
| DNA dependent DNA replication | 13 | 18 | 22 | 72.2 | 81.8 | 5.0697 |
| Mitosis | 13 | 18 | 24 | 72.2 | 75 | 5.0697 |
| Nuclear division | 14 | 21 | 30 | 66.7 | 70 | 4.8663 |
| DNA packaging | 20 | 36 | 46 | 55.6 | 78.3 | 4.7782 |
| Cell organization and biogenesis | 74 | 207 | 294 | 35.7 | 70.4 | 4.6913 |
| M phase | 15 | 25 | 36 | 60 | 69.4 | 4.5110 |
| mRNA splice site selection | 7 | 8 | 8 | 87.5 | 100 | 4.4125 |
| DNA replication initiation | 6 | 7 | 7 | 85.7 | 100 | 4.0138 |
| Chromosome organization and biogenesis (sensu Eukarya) | 18 | 37 | 51 | 48.6 | 72.5 | 3.8338 |
| DNA repair | 21 | 46 | 53 | 45.7 | 86.8 | 3.7895 |
| Protein folding | 12 | 22 | 31 | 54.5 | 71 | 3.6157 |
| Cytoplasm organization and biogenesis | 56 | 169 | 241 | 33.1 | 70.1 | 3.3912 |
| Establishment and/or maintenance of chromatin architecture | 13 | 27 | 35 | 48.1 | 77.1 | 3.2089 |
| Protein synthesis elongation | 6 | 9 | 37 | 66.7 | 24.3 | 3.1815 |
| Chromatin assembly/disassembly | 10 | 20 | 25 | 50 | 80 | 2.9585 |
| Biological process unknown | 34 | 98 | 250 | 34.7 | 39.2 | 2.9354 |
| Protein-ligand dependent protein degradation | 17 | 43 | 58 | 39.5 | 74.1 | 2.6968 |
| Ubiquitin-dependent protein degradation | 16 | 42 | 57 | 38.1 | 73.7 | 2.4404 |
| Protein-nucleus import | 5 | 9 | 10 | 55.6 | 90 | 2.3820 |
| Ubiquitin cycle | 6 | 12 | 16 | 50 | 75 | 2.2896 |
| Nucleocytoplasmic transport | 6 | 12 | 17 | 50 | 70.6 | 2.2896 |
| Actin cytoskeleton organization and biogenesis | 6 | 12 | 19 | 50 | 63.2 | 2.2896 |
| Transmembrane receptor protein Ser/Thr kinase signaling pathway | 10 | 25 | 31 | 40 | 80.6 | 2.1081 |
| Induction of apoptosis | 7 | 16 | 24 | 43.8 | 66.7 | 2.0449 |
| Component | ||||||
| Spliceosome | 17 | 20 | 42 | 85 | 47.6 | 6.7175 |
| Cytosolic ribosome (sensu Eukarya) | 19 | 26 | 33 | 73.1 | 78.8 | 6.2032 |
| Cytosol | 40 | 85 | 112 | 47.1 | 75.9 | 5.4872 |
| Ribosome | 35 | 71 | 93 | 49.3 | 76.3 | 5.4624 |
| Chromosome | 19 | 36 | 55 | 52.8 | 65.5 | 4.3772 |
| Nuclear envelope-endoplasmic reticulum network | 9 | 12 | 17 | 75 | 70.6 | 4.3676 |
| Adherens junction | 6 | 7 | 14 | 85.7 | 50 | 4.0138 |
| Endoplasmic reticulum membrane | 7 | 9 | 13 | 77.8 | 69.2 | 3.9811 |
| Chromatin | 15 | 28 | 41 | 53.6 | 68.3 | 3.9579 |
| Cellular component unknown | 41 | 117 | 291 | 35 | 40.2 | 3.3057 |
| Nucleolus | 10 | 19 | 34 | 52.6 | 55.9 | 3.1587 |
| 26S proteasome | 11 | 22 | 23 | 50 | 95.7 | 3.1036 |
| Endoplasmic reticulum | 39 | 117 | 141 | 33.3 | 83 | 2.8569 |
| 20S core proteasome | 9 | 19 | 19 | 47.4 | 100 | 2.6078 |
| Nuclear membrane | 6 | 11 | 18 | 54.5 | 61.1 | 2.5536 |
| Cytoskeleton | 64 | 223 | 306 | 28.7 | 72.9 | 2.2918 |
| Collagen | 10 | 25 | 31 | 40 | 80.6 | 2.1081 |
| Golgi membrane | 7 | 16 | 18 | 43.8 | 88.9 | 2.0449 |
| Actin cytoskeleton | 16 | 46 | 63 | 34.8 | 73 | 2.0140 |
| Function | ||||||
| RNA binding | 51 | 113 | 155 | 45.1 | 72.9 | 5.8498 |
| Cyclin-dependent protein kinase | 17 | 24 | 33 | 70.8 | 72.7 | 5.6944 |
| Structural constituent of ribosome | 39 | 83 | 101 | 47 | 82.2 | 5.4055 |
| Cyclin-dependent protein kinase, regulator | 12 | 17 | 24 | 70.6 | 70.8 | 4.7646 |
| Structural molecule | 77 | 223 | 278 | 34.5 | 80.2 | 4.4306 |
| Pre-mRNA splicing factor | 7 | 8 | 12 | 87.5 | 66.7 | 4.4125 |
| mRNA binding | 10 | 14 | 19 | 71.4 | 73.7 | 4.3979 |
| Protein serine/threonine kinase | 62 | 181 | 243 | 34.3 | 74.5 | 3.8821 |
| Actin binding | 25 | 58 | 83 | 43.1 | 69.9 | 3.7927 |
| Proteasome endopeptidase | 11 | 19 | 19 | 57.9 | 100 | 3.7096 |
| DNA-directed DNA polymerase | 7 | 10 | 15 | 70 | 66.7 | 3.6069 |
| RHO small monomeric GTPase | 7 | 10 | 10 | 70 | 100 | 3.6069 |
| Nucleotidyltransferase | 16 | 33 | 41 | 48.5 | 80.5 | 3.5964 |
| Kinase regulator | 15 | 33 | 42 | 45.5 | 78.6 | 3.1777 |
| DNA dependent adenosinetriphosphatase | 8 | 14 | 16 | 57.1 | 87.5 | 3.1151 |
| Cytoskeletal protein binding | 33 | 93 | 144 | 35.5 | 64.6 | 3.0423 |
| DNA repair protein | 11 | 23 | 27 | 47.8 | 85.2 | 2.9232 |
| Translation factor, nucleic acid binding | 14 | 32 | 43 | 43.8 | 74.4 | 2.8970 |
| Transcription co-activator | 6 | 10 | 14 | 60 | 71.4 | 2.8483 |
| Chromatin binding | 5 | 8 | 11 | 62.5 | 72.7 | 2.7166 |
| Kinase | 89 | 311 | 394 | 28.6 | 78.9 | 2.6983 |
| Phosphotransferase, alcohol group as acceptor | 87 | 305 | 386 | 28.5 | 79 | 2.6301 |
| Protein kinase | 76 | 263 | 336 | 28.9 | 78.3 | 2.5796 |
| Exonuclease | 6 | 11 | 15 | 54.5 | 73.3 | 2.5536 |
| Small monomeric GTPase | 15 | 38 | 46 | 39.5 | 82.6 | 2.5247 |
| GTP binding | 43 | 141 | 201 | 30.5 | 70.1 | 2.3248 |
| Peptidylprolyl cis-trans isomerase | 6 | 12 | 16 | 50 | 75 | 2.2896 |
| Translation elongation factor | 6 | 12 | 16 | 50 | 75 | 2.2896 |
| Transcription factor binding | 11 | 27 | 43 | 40.7 | 62.8 | 2.2838 |
| Guanyl nucleotide binding | 46 | 155 | 219 | 29.7 | 70.8 | 2.1927 |
| Adenosinetriphosphatase | 12 | 31 | 38 | 38.7 | 81.6 | 2.1763 |
| Molecular_function unknown | 29 | 91 | 230 | 31.9 | 39.6 | 2.1739 |
| Protein binding | 99 | 368 | 539 | 26.9 | 68.3 | 2.1328 |
| Chaperone | 16 | 45 | 62 | 35.6 | 72.6 | 2.1166 |
| Extracellular matrix structural constituent conferring tensile strength | 10 | 25 | 31 | 40 | 80.6 | 2.1081 |
| DNA-directed RNA polymerase | 5 | 10 | 11 | 50 | 90.9 | 2.0897 |
| Structural constituent of cytoskeleton | 21 | 63 | 79 | 33.3 | 79.7 | 2.0838 |
| Transferase, transferring one-carbon groups | 8 | 19 | 29 | 42.1 | 65.5 | 2.0570 |
| GTPase | 25 | 78 | 95 | 32.1 | 82.1 | 2.0488 |
| Isomerase | 12 | 32 | 42 | 37.5 | 76.2 | 2.0468 |
| Significantly decreased | ||||||
| Process | ||||||
| Fatty acid metabolism | 19 | 30 | 41 | 63.3 | 73.2 | 5.9082 |
| Main pathways of carbohydrate metabolism | 20 | 39 | 50 | 51.3 | 78 | 4.8600 |
| Energy derivation by oxidation of organic compounds | 23 | 50 | 66 | 46 | 75.8 | 4.5739 |
| Catabolic carbohydrate metabolism | 18 | 36 | 45 | 50 | 80 | 4.4754 |
| Tricarboxylic acid cycle | 6 | 8 | 10 | 75 | 80 | 3.8664 |
| Hexose metabolism | 18 | 41 | 49 | 43.9 | 83.7 | 3.8016 |
| Lipid metabolism | 42 | 127 | 167 | 33.1 | 76 | 3.6708 |
| Lipid transport | 5 | 7 | 11 | 71.4 | 63.6 | 3.3807 |
| Glycolysis | 12 | 26 | 32 | 46.2 | 81.2 | 3.3091 |
| Peroxisome organization and biogenesis | 7 | 12 | 15 | 58.3 | 80 | 3.2972 |
| Glucose metabolism | 15 | 36 | 42 | 41.7 | 85.7 | 3.2247 |
| Lymph gland development | 8 | 15 | 17 | 53.3 | 88.2 | 3.2043 |
| Cell proliferation | 10 | 21 | 34 | 47.6 | 61.8 | 3.1400 |
| Humoral immune response | 15 | 37 | 79 | 40.5 | 46.8 | 3.0982 |
| Carbohydrate metabolism | 31 | 95 | 135 | 32.6 | 70.4 | 3.0557 |
| Regulation of cell proliferation | 5 | 8 | 15 | 62.5 | 53.3 | 2.9848 |
| Muscle contraction | 9 | 20 | 28 | 45 | 71.4 | 2.7716 |
| Muscle development | 13 | 34 | 43 | 38.2 | 79.1 | 2.6328 |
| Mesoderm development | 28 | 90 | 111 | 31.1 | 81.1 | 2.6096 |
| Potassium transport | 17 | 49 | 60 | 34.7 | 81.7 | 2.5450 |
| Metal ion transport | 24 | 77 | 100 | 31.2 | 77 | 2.4230 |
| Monovalent inorganic cation transport | 21 | 67 | 88 | 31.3 | 76.1 | 2.2935 |
| Complement activation | 8 | 20 | 23 | 40 | 87 | 2.2132 |
| Cation transport | 28 | 98 | 135 | 28.6 | 72.6 | 2.0923 |
| Electron transport | 25 | 87 | 113 | 28.7 | 77 | 2.0075 |
| Component | ||||||
| Mitochondrion | 88 | 187 | 293 | 47.1 | 63.8 | 9.3508 |
| Peroxisome | 18 | 29 | 42 | 62.1 | 69 | 5.6381 |
| Mitochondrial inner membrane | 19 | 36 | 60 | 52.8 | 60 | 4.8922 |
| Mitochondrial electron transport chain complex | 10 | 14 | 32 | 71.4 | 43.8 | 4.7848 |
| Mitochondrial membrane | 20 | 40 | 72 | 50 | 55.6 | 4.7195 |
| Cytochrome C oxidase | 6 | 8 | 16 | 75 | 50 | 3.8664 |
| Mitochondrial matrix | 9 | 22 | 33 | 40.9 | 66.7 | 2.4283 |
| Basement lamina | 5 | 11 | 11 | 45.5 | 100 | 2.0910 |
| Cytoskeleton | 57 | 223 | 306 | 25.6 | 72.9 | 2.0527 |
| Function | ||||||
| Hydrogen ion transporter | 11 | 15 | 33 | 73.3 | 45.5 | 5.1373 |
| Primary active transporter | 27 | 64 | 107 | 42.2 | 59.8 | 4.4175 |
| Cation transporter | 17 | 36 | 61 | 47.2 | 59 | 4.0585 |
| Ion transporter | 19 | 43 | 79 | 44.2 | 54.4 | 3.9406 |
| Cytochrome c oxidase | 6 | 8 | 16 | 75 | 50 | 3.8664 |
| Oxidoreductase | 48 | 149 | 207 | 32.2 | 72 | 3.7213 |
| Major histocompatibility complex antigen | 13 | 30 | 54 | 43.3 | 55.6 | 3.1700 |
| Oxidoreductase, acting on the aldehyde or oxo group of donors | 7 | 13 | 16 | 53.8 | 81.2 | 3.0285 |
| Carrier | 40 | 131 | 196 | 30.5 | 66.8 | 2.9960 |
| Complement component | 8 | 16 | 19 | 50 | 84.2 | 2.9770 |
| P-type ATPase | 5 | 9 | 11 | 55.6 | 81.8 | 2.6467 |
| Hydrolase, acting on acid anhydrides, catalyzing transmembrane movement of substances | 15 | 42 | 67 | 35.7 | 62.7 | 2.5199 |
| Nucleobase, nucleoside, nucleotide kinase | 7 | 16 | 19 | 43.8 | 84.2 | 2.3531 |
| Phosphotransferase, phosphate group as acceptor | 5 | 10 | 13 | 50 | 76.9 | 2.3520 |
| Glutathione transferase | 5 | 10 | 13 | 50 | 76.9 | 2.3520 |
| P-P-bond-hydrolysis-driven transporter | 17 | 52 | 78 | 32.7 | 66.7 | 2.2609 |
| ATP-binding cassette (ABC) transporter | 11 | 30 | 50 | 36.7 | 60 | 2.2573 |
| Potassium channel | 15 | 45 | 56 | 33.3 | 80.4 | 2.2093 |
| Carbon-carbon lyase | 5 | 11 | 18 | 45.5 | 61.1 | 2.0910 |
All terms with a z score of 2 and at least 5, but less than 100 genes meeting the criterion are shown.
The MAPPFinder results present a global picture of the biological processes, cellular components and molecular functions that are increased and decreased in the 12.5-day embryo compared with the adult mouse (Table 2). Using the criterion for a significantly increased gene-expression change, MAPPFinder primarily identified GO terms involved in cell division and growth. Notable GO terms include the processes 'mitotic cell cycle' (62.9% of 70 genes, z score of 8.1), 'mRNA splicing' (90.5% of 21 genes, z score of 7.5), and 'protein biosynthesis' (50% of 104 genes, z score of 6.8). The top-ranked component and function terms reflected the same biological processes. For example, the component term 'spliceosome' shows that 17 out of 20 genes (85%, z score of 6.7) were upregulated. The upregulation of these processes is consistent with the fact that cardiomyocytes remain mitotically active throughout embryonic development [17]. Apart from processes involved in cell division and growth, the MAPPFinder results indicate that the processes 'transmembrane receptor protein serine/threonine kinase signaling pathway' and 'induction of apoptosis' are upregulated, with a z score of approximately 2. The presence of the term 'transmembrane receptor protein serine/threonine kinase signaling pathway' is due to the upregulation of genes involved in transforming growth factor-β (TGFβ) receptor signaling, which is thought to regulate the induction of apoptosis required for morphogenesis during heart development [18,19].
Genes involved in energy metabolism showed the highest levels of downregulation in the 12.5-day embryo heart versus the adult heart. In particular, the process terms 'fatty acid metabolism' (63.3% of 30 genes, z score of 5.9) and 'main pathways of carbohydrate metabolism' (51.3% of 39 genes, z score 4.8), which is the parent of the terms 'glycolysis' and 'tricarboxylic acid cycle', indicate that metabolic genes as a whole are downregulated in an embryo when compared to an adult mouse. In addition, the component term 'mitochondrion' shows that 88 out of 187 genes (47.1%, z score of 9.1) are downregulated. The downregulation of genes involved in fatty-acid metabolism is consistent with research that has shown that the developing heart, unlike the adult heart, does not derive its energy from fatty acids [20].
Overall, the MAPPFinder results provide a global perspective of the processes that are up- and down-regulated in the 12.5-day embryonic heart compared to an adult heart. The results confirmed what was expected: when compared to the adult heart, the embryonic heart is undergoing increased cell division and growth and has decreased energy metabolism. In addition, the global gene-expression profile presented by MAPPFinder allows the gene-expression changes observed for cell division and growth and energy metabolism to be put in the context of other regulatory and developmental processes such as TGFβ signaling and apoptosis.
The MAPPFinder browser
Viewing the MAPPFinder results as a ranked list is informative, but it does not take full advantage of the fact that GO is arranged in a hierarchy. MAPPFinder also presents the results in the context of the GO hierarchy (Figures 2a,3a) showing the entire hierarchy, color-coded by the percentage of genes changed. Users can step through the hierarchy, expanding those branches of the tree that are showing gene expression changes, moving from broad terms to more specific categories. Often the ranked list of terms will show many interrelated terms, and it is necessary to view the results in the hierarchy to identify the relationships among them. For example, the terms 'RNA metabolism', 'RNA processing', 'mRNA processing', and 'mRNA splicing' appear as upregulated in Table 2. However, the tree view (Figure 2a) clearly shows that mRNA splicing is a child term of both RNA splicing and mRNA processing, which are in turn child terms of RNA metabolism. Similarly, the terms 'main pathways of carbohydrate metabolism', 'catabolic carbohydrate metabolism', and 'glycolysis' also appear as downregulated in Table 2. The MAPPFinder browser (Figure 3a) shows that 'glycolysis' is related to 'main pathways of carboyhydrate metabolism' through the hierarchical relationship between these terms.
The MAPPFinder browser also provides three search and navigation functions. First, the user can search by a keyword or an exact GO term name. Second, the user can search by a gene identifier to find which GO term(s) the gene is associated with. For example, searching for the gene alpha-myosin heavy chain using its SWISS-PROT identifier MYH6_MOUSE or its MGD identifier MGI:97255 finds the GO process terms 'striated muscle contraction', 'cytoskeleton organization and biogenesis', 'protein modification', and 'muscle development'. Third, the user can expand the GO tree automatically to show all nodes with a minimum number of genes or minimum percentage of genes meeting the criterion or with a minimum z score. The terms meeting the filter are highlighted in yellow to clearly indicate the results of the search.
Once the GO terms of interest have been identified with MAPPFinder, the user will want to know exactly which genes are associated with these terms and exactly which genes are being differentially expressed. This can be accomplished using GenMAPP. Selecting a GO term in the MAPPFinder browser automatically builds a MAPP containing the genes associated with that GO term and all of its children, and opens this MAPP in GenMAPP. Figure 3b shows the MAPP generated by selecting the GO term 'glycolysis' in the MAPPFinder browser. The genes on the MAPP are color-coded with the same criteria used to calculate the MAPPFinder results, significantly increased and decreased at the 12.5-day embryo time point. Clicking on a gene on the MAPP opens a 'back page' containing annotations, gene-expression data and hyperlinks to that gene's page in the public databases. By integrating GenMAPP and MAPPFinder, it is possible to seamlessly move from a global gene-expression profile at the level of all biological processes, components and functions to a detailed description of the gene-expression levels for the specific genes involved. For example, a closer examination of the glycolysis MAPP indicates that hexokinase I is upregulated in the 12.5-day embryo and isoforms II and IV are downregulated, as compared with the adult heart. This is consistent with hexokinase I being the predominant isoform in the embryonic heart [21].
Expanding MAPPFinder beyond GO
GO is a good starting point for analyzing microarray data in the context of biological pathways, but this is by no means the only way to group related genes. Instead of representing each GO process as an alphabetical list on a MAPP, it would be more useful to represent the relationships between these genes as a fully delineated pathway. As a start in this direction, GenMAPP.org [13] has created over 50 MAPPs depicting metabolic pathways, signaling pathways and gene families. MAPPFinder can incorporate any MAPP file into its analysis to augment the GO hierarchy. For the FVB benchmark developmental dataset, we have run MAPPFinder on an archive of 54 mouse MAPPs available from [13] (see Additional data files for the complete results). These results for the 12.5-day embryonic time point agree with the GO results, showing that the expression of genes involved in the metabolic pathways 'tricarboxylic acid cycle' (83.3% of 12 genes measured, z score of 5.91) and 'fatty acid degradation' (69.2% of 13 genes measured, z score 4.82) is significantly decreased. In addition, the significantly increased criterion identified genes encoding ribosomal proteins (71.1% of 45 genes, z score 6.75) and genes involved in the cell cycle (53.3% of 15 genes, z score 2.4).
The archive of MAPPs provided by GenMAPP is in no way comprehensive. The growth of this archive depends on assistance from the entire biological community. Our hope is that, as MAPPFinder users see the added utility of viewing the GO biological processes as fully delineated pathways, they will use GenMAPP to organize the gene lists into more descriptive biological pathways. Figure 3c gives an example of how the genes from the GO term 'glycolysis' can be rearranged using the tools in GenMAPP to depict the full pathway showing the direction of the enzymatic cascade, metabolic intermediates and cellular compartments. GenMAPP.org is currently accepting submissions of new MAPP files. MAPPs contributed by the community will be included in the downloadable MAPP archive.
MAPPFinder is a necessary complement to current analysis tools
By approaching large datasets from a higher level or organization, MAPPFinder helps to ease the data analysis and shorten the time necessary to gain a biological understanding of the microarray data. MAPPFinder has greatly expanded current pathway-based tools by using the large amount of annotations available from the GO. This broad analysis will help identify biological processes that have not yet been implicated in a particular experimental condition and begin to make connections between biological processes previously thought to be unrelated.
MAPPFinder is available for yeast, mouse and human data. We plan to extend the program to many of the other species that are in GO and updates will be available at [13].
Materials and methods
Gene-expression data
The publicly available mouse microarray dataset, the FVB benchmark set for cardiac development, maturation and aging, was obtained from the CardioGenomics Program for Genomics Applications [14]. These data compare healthy mouse hearts at different time points during development, using male and female FVB/N mice. Specifically, this dataset examines heart tissue from 12.5-day embryos, 1-day neonatal mice, 1-week mice, 4-week mice, and adult mice at 5 months and 1 year. Our analysis focused on the 12.5-day embryonic time point and the control adult mice. Three Affymetrix U74A version 1 arrays were used for each time point. For the embryonic time point, three hearts were pooled for each array because of their small size. To improve the statistical power in our analysis, the 5-month and the 1-year mice were combined into a single group of normal adult mice. Signal intensity values were obtained with Affymetrix MAS 5.0 software. Signal values less than 20 were raised to 20 and the log base 2 was taken. Log folds were determined from the average of each time point when compared with the average of the combined control group. P values were calculated with a permutation t test. The statistical analysis was done using the multest package of the R statistical programming language [22]. These data were imported into GenMAPP, and the resulting GenMAPP Expression Dataset file (.gex) was exported to MAPPFinder.
MAPPFinder requires a user-defined criterion for a meaningful gene-expression change. In this case we combined a fold change with a statistical filter to determine significance. We are using a fold change of greater than 1.2 with a p value of less than 0.05 to define a significant gene-expression increase, and a fold change of less than -1.2 with a p-value of less than 0.05 to define a significant gene-expression decrease. To determine the overall number of gene-expression changes in each GO term, an additional criterion of a fold change greater than 1.2 or less than -1.2 and a p value of less than 0.05 is used (data not shown).
It is important to note that while we have used gene-expression data generated from Affymetrix GeneChips, data from other microarray platforms and other techniques such as SAGE (serial analysis of gene expression) can be used equally easily.
Linking the expression data to Gene Ontology
MAPPFinder builds a local copy of the GO hierarchy using the three ontology files (Process, Component and Function) available from GO [12]. The directed acyclic graph (DAG) structure of GO [23] allows a node to be a child of multiple parents. This makes the navigation, visualization and computation of the MAPPFinder results more difficult than if the GO were stored in a classical tree structure. To ease the programming necessary to implement the MAPPFinder algorithm, the DAG structure was converted to a classical tree. For each node of the DAG that contained multiple parents, multiple copies were inserted into the tree representation of the GO using local identifiers to handle duplicate GO terms. This tree structure maintains the 'true path' rule enforced in the GO DAG structure. MAPPFinder handles this conversion internally, and to the user the GO hierarchy seen in the MAPPFinder browser will be identical to that seen in other GO browsers.
The links between the GO terms and the genes in the expression dataset are made with the gene-association files [15]. These associations are taken from the European Bioinformatics Institute [24] for human genes, the Mouse Genome Database (MGD) [16] for mouse genes, and the Saccharomyces Genome Database (SGD) [25] for yeast genes. Currently, the genes in the input data must be identified with GenBank, SWISS-PROT or SGD identifiers.
MAPPFinder uses a relational database to link the expression dataset to the gene-association files. The MAPPFinder database relates gene-expression data to the appropriate gene-identifrcation systems for each species (Figure 1). For human data, the gene-association files use SWISS-PROT identifiers, requiring a SWISS-PROT-to-GenBank relational table to link datasets using GenBank accession numbers to the GO annotations. For yeast data, the gene-association files use SGD identifiers. A SWISS-PROT-to-SGD relational table is also included for expression datasets using SWISS-PROT identifiers. For mouse data, the GO gene-association files use MGD identifiers, requiring a GenBank-to-MGD relational table, and a SWISS-PROT-to-MGD relational table. MAPPFinder takes advantage of the fact that MGD is also related to UniGene, allowing additional ESTs that are not in the MGD-GenBank relational table to be used as gene identifiers. With this intermediate step, many more GenBank identifiers can be linked to GO annotations. Currently, there is no direct relationship between SWISS-PROT and UniGene, so a similar intermediate step was not used for human data.
Calculating the MAPPFinder results
MAPPFinder calculates the percentage of genes measured within each GO term that meet a user-defined criterion, and this measurement is known as the 'percent changed'. MAPPFinder also calculates the percentage of the genes associated with a GO term that are measured in the experiment, and this measurement is known as the 'percent present'. Calculating the percent present is necessary to determine how well represented a GO term is in the dataset.
The GO gene-association files [17] are potentially problematic, because they treat each GO term independently, removing the implicit parent-child relationship. As a result, looking at the GO terms individually is often uninformative because the number of genes associated with any one term is smaller than the actual number of genes involved in that process, component, or function. To address this issue, we calculate the nested percentage for a parent term with all its children below it in the hierarchy. By combining the child terms with their parent, the results incorporate genes associated with the entire branch of the hierarchy, providing a much more accurate representation of the number of genes involved in that process, component or function. As more specific branches of the GO are examined, the denominator of the two equations will become smaller and the user can find their desired level of specificity. One complication that arises from this method is that in some cases a gene is associated with both the parent and child terms or multiple child terms. When the percentages are calculated for the sub-tree, we ensure that each gene is only counted once, so that genes with multiple annotations are not weighted more heavily.
Another complication that arises while calculating the MAPPFinder results is the issue of multiple probes of the same gene on the array. In this case, the features or duplicate genes are clustered to one unique gene. If any of the instances of this gene on the array meet the user-defined criterion, then that gene meets the user-defined criterion. The number of unique genes is also used to calculate the z score, meaning that the statistics are based only on a single occurrence of each gene in the dataset.
A statistical rating of the relative gene-expression activity in each MAPP and GO term is also provided. It is a standardized difference score (z score) using the expected value and standard deviation of the number of genes meeting the criterion on a GO term under a hypergeometric distribution. The z score is useful for ranking GO terms by their relative amounts of gene expression changes. Positive z scores indicate GO terms with a greater number of genes meeting the criterion than is expected by chance. Negative z scores indicate GO terms with fewer genes meeting the criterion than expected by chance. A z score near zero indicates that the number of genes meeting the criterion approximates the expected number. Extreme positive scores suggest GO terms with the greatest confidence that the correlation between the expression changes of the genes in this grouping are not occurring by chance alone. P values are not assigned to the GO terms or MAPPs because, while such a standardized difference score could approximate a normal z score for an individual MAPP, the lack of independence between GO terms and the multiple testing occurring among them most certainly makes the normal p value for such a z score unreliable. As a result, p values are not assigned to the GO terms and MAPPs.
The z score is calculated by subtracting the observed number of genes in a GO term (or MAPP) meeting the criterion from the expected number of genes, and dividing by the standard deviation of the observed number of genes. The equation used is
![]()
or
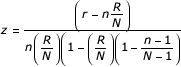
where N is the total number of genes measured, R is the total number of genes meeting the criterion, n is the total number of genes in this specific MAPP, and r is the number of genes meeting the criterion in this specific MAPP.
Therefore, if two GO terms contain the same number of genes, the term with the greater number of genes meeting the criterion will receive a higher score. Dividing by the standard deviation adjusts for the size of the GO term, ranking a GO term (or MAPP) with a large number of genes meeting the criterion higher than a GO term (or MAPP) with the same percentage of genes changed, but fewer total genes.
The MAPPFinder results are generated in the GO browser for analysis in the context of the GO hierarchy and as tab-delimited text files that can be used for sorting and filtering the data in a spreadsheet program.
Additional data files
The following additional data files are available:
The FVBN developmental data in the form of a GenMAPP expression dataset file (.gex). It contains the microarray dataset and the criteria used to define increased and decreased gene-expression change. It can be opened for editing in GenMAPP and is the appropriate data type for use with MAPPFinder.
The FVBN developmental data as a database file generated by MAPPFinder (.gdb). It contains the relationships between the genes in the dataset and the GO hierarchy. The file can be opened for viewing in Microsoft Access. This file must be present to build GenMAPP MAPPs from existing MAPPFinder results.
The MAPPFinder results for the 12.5-day embryos versus the adult mice are contained in the files: 12.5-day Embryo - significantly increased - Gene Ontology results, 12.5-day Embryo - significantly increased - Local results, 12.5-day Embryo - significantly decreased - Gene Ontology results, 12.5-day Embryo - significantly decreased - Local results, 12.5-day Embryo - All Changes - Gene Ontology results, 12.5-day Embryo - All Changes - Local Results. These text files contain the MAPPFinder results for both criteria and both the GO hierarchy and the GenMAPP.org MAPPs. These files can be loaded into MAPPFinder for view in the MAPPFinder GO browser. These files are tab-delimited and can also be viewed as tables in Microsoft Excel. The 'All Changes' files contain the results for a criteria looking for either increased or decreased gene expression changes. A zip file containing all aditional data files is available.
Supplementary Material
The FVBN developmental data in the form of a GenMAPP expression dataset file (.gex). It contains the microarray dataset and the criteria used to define increased and decreased gene-expression change. It can be opened for editing in GenMAPP and is the appropriate data type for use with MAPPFinder.
The FVBN developmental data as a database file generated by MAPPFinder (.gdb). It contains the relationships between the genes in the dataset and the GO hierarchy. The file can be opened for viewing in Microsoft Access. This file must be present to build GenMAPP MAPPs from existing MAPPFinder results.
12.5-day Embryo - significantly increased - Gene Ontology results
12.5-day Embryo - significantly increased - Local results
12.5-day Embryo - significantly decreased - Gene Ontology results
12.5-day Embryo - significantly decreased - Local results
12.5-day Embryo - All Changes - Gene Ontology results
12.5-day Embryo - All Changes - Local results
A zip file containing all aditional data files.
Acknowledgments
Acknowledgements
We thank A. Zambon, W. Tingley, T. Speed, P. Bacchetti and J. Myers for helpful conversations about the design and implementation of MAPPFinder, B. Taylor for help with the preparation of this manuscript, and S. Izumo and CardioGenomics for making the microarray dataset publicly available. This work is supported by the J. David Gladstone Institutes, the San Francisco General Hospital General Clinical Research Center, the National Heart, Lung, and Blood Institute, San Francisco General Hospital General Clinical Research Center MO1RR00083 (B.R.C.) and the NHLBI Programs for Genomic Applications (BayGenomics).
References
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Dmitrovsky E, Lander ES, Golub TR. Interpreting gene expression with self-organizing maps: methods and application to hematopoeitic differentiation. Proc Natl Acad Sci USA. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. doi: 10.1038/ng0502-19. [DOI] [PubMed] [Google Scholar]
- Nakao M, Bono H, Kawashima S, Kamiya T, Sato K, Goto S, Kanehisa M. Genome-scale gene expression analysis and pathway reconstruction in KEGG. Genome Inform Ser WorkShop Genome Inform. 1999;10:94–103. [PubMed] [Google Scholar]
- Karp PD, Riley M, Paley SM, Pellegrini-Toole A. The MetaCyc Database. Nucleic Acids Res. 2002;30:59–61. doi: 10.1093/nar/30.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosu P, Townsend J, Hartl D, Cavalieri D. Pathway processor: a tool for integrating whole-genome expression results into metabolic networks. Genome Res. 2002;12:1121–1126. doi: 10.1101/gr.226602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyf A, de Gast J, van Kampen A. Visualizing metabolic activity on a genome-wide scale. Bioinformatics. 2002;18:813–818. doi: 10.1093/bioinformatics/18.6.813. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletcher SD, MacDonald SJ, Marguerie R, Certa U, Stearns SC, Goldstein DB, Partridge L. Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr Biol. 2002;12:712–723. doi: 10.1016/S0960-9822(02)00808-4. [DOI] [PubMed] [Google Scholar]
- Silcon Genetics Products: Gene Spring http://www.silicongenetics.com/cgi/SiG.cgi/Products/GeneSpring/index.smf
- Affymetrix - NetAffx Analysis Center http://www.affymetrix.com/analysis/index.affx
- The Gene Ontology Project Ontology Files ftp://ftp.geneontology.org/go/ontology/
- GenMAPP http://www.GenMAPP.org
- CardioGenomics: FVB benchmark data set for cardiac development, maturation, and aging http://www.cardiogenomics.org:1550/groups/proj1/pages/fvb_home.html
- The Gene Ontology Project Gene Association Files ftp://ftp.geneontology.org/go/gene-associations
- Blake JA, Richardson JE, Bult CJ, Kadin JA, Eppig JT, the Mouse Genome Database Group The Mouse Genome Database (MGD): the model organism database for the laboratory mouse. Nucleic Acids Res. 2002;30:113–115. doi: 10.1093/nar/30.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H. Myocardial cellular development and morphogenesis. In: Langer GA, editor. In The Myocardium. San Diego, CA: Academic Press; 1997. pp. 33–80. [Google Scholar]
- Poelmann R, Molin D, Wisse L, Gittenberger-de Groot A. Apoptosis in cardiac development. Cell Tissue Res. 2000;301:43–52. doi: 10.1007/s004410000227. [DOI] [PubMed] [Google Scholar]
- Kubalak S, Hutson D, Scott K, Shannon R. Elevated transforming growth factor β2 enhances apoptosis and contributes to abnormal outflow tract and aortic sac development in retinoic X receptor α knockout embryos. Development. 2002;129:733–746. doi: 10.1242/dev.129.3.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopaschuk G, Collins-Nakai R, Itoi T. Developmental changes in energy substrate use by the heart. Cardiovasc Res. 1992;26:1172–1180. doi: 10.1093/cvr/26.12.1172. [DOI] [PubMed] [Google Scholar]
- Fritz H, Smoak I, Branch S. Hexokinase I expression and activity in embryonic mouse heart during early and late organogenesis. Histochem Cell Biol. 1999;112:359–365. doi: 10.1007/s004180050417. [DOI] [PubMed] [Google Scholar]
- Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments http://www.stat.berkeley.edu/users/terry/zarray/TechReport/578.pdf
- The Gene Ontology Consortium Creating the Gene Ontology resource: design and implementation. Genome Res. 2001;11:1425–1433. doi: 10.1101/gr.180801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMBL-EBI: GOA project online http://www.ebi.ac.uk/GOA/project.html
- Dwight SS, Harris MA, Dolinski K, Ball CA, Binkley G, Christie KR, Fisk DG, Issel-Tarver L, Schroeder M, Sherlock G, et al. Saccharomyces Genome Database (SGD) provides secondary gene annotation using the Gene Ontology (GO). Nucleic Acids Res. 2002;30:69–72. doi: 10.1093/nar/30.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The FVBN developmental data in the form of a GenMAPP expression dataset file (.gex). It contains the microarray dataset and the criteria used to define increased and decreased gene-expression change. It can be opened for editing in GenMAPP and is the appropriate data type for use with MAPPFinder.
The FVBN developmental data as a database file generated by MAPPFinder (.gdb). It contains the relationships between the genes in the dataset and the GO hierarchy. The file can be opened for viewing in Microsoft Access. This file must be present to build GenMAPP MAPPs from existing MAPPFinder results.
12.5-day Embryo - significantly increased - Gene Ontology results
12.5-day Embryo - significantly increased - Local results
12.5-day Embryo - significantly decreased - Gene Ontology results
12.5-day Embryo - significantly decreased - Local results
12.5-day Embryo - All Changes - Gene Ontology results
12.5-day Embryo - All Changes - Local results
A zip file containing all aditional data files.