

Abstract
Bone development is extremely sensitive to alterations in thyroid status. Recently, we analyzed the skeletal phenotypes of mice with the dominant negative resistance to thyroid hormone (RTH) mutation PV targeted to either the thyroid hormone receptor (TR) α1 or β gene. This perspective summarizes our findings to date and explores the wider implications for thyroid status and T3 target gene expression in individual tissues.
Thyroid hormone and the skeleton
Thyroid hormone (T3) is a major regulator of skeletal development. T3-actions are mainly mediated via TRα and TRβ nuclear receptors, which are alternatively spliced to produce multiple isoforms [Cheng, 2000]. The inception and timing of TR expression is variable and occurs in tissue-specific patterns in utero, throughout development and in adulthood. Thus, TR expression is regulated in a temporo-spatial manner and, as a consequence, the ratios of expressed TR isoforms vary between individual tissues [Cheng, 2000; Forrest et al., 1990]. In the skeleton, TRα1 mRNA is expressed at 12-fold higher concentrations than TRβ1 [O'Shea et al., 2003]. TRα1, TRα2 and TRβ1 mRNAs and proteins have been identified in growth plate chondrocytes and osteoblasts at sites of endochondral and intramembranous ossification [Ballock et al., 1999; Robson et al., 2000; Williams et al., 1994]. However, it is not known whether TRα and β isoforms are co-expressed in individual bone cells or whether discrete actions will eventually be ascribed to each isoform.
Resistance to thyroid hormone and mouse models
RTH is an autosomal dominant condition characterized by reduced tissue sensitivity to thyroid hormones and disruption in the hypothalamic-pituitary-thyroid (HPT) axis leading to elevated levels of T3 and T4 together with inappropriately raised TSH concentrations. Affected individuals possess dominant-negative mutant TRβ proteins that interfere with the actions of wild-type TRs and disrupt T3-regulated gene transcription. The clinical features of RTH are variable and phenotypic differences occur between families with different mutations, between families that harbor an identical mutation, and between individuals of one family with an identical mutation [Weiss and Refetoff, 2000]. An example of the phenotypic variability is the skeleton where a wide variety of abnormalities have been described [Kvistad et al., 2004; Weiss and Refetoff, 1996; Weiss and Refetoff, 2000].
We previously analyzed skeletal development in mice with a PV mutation targeted to the TRβ gene [O'Shea et al., 2003]. The PV mutation, derived from a patient with severe RTH and an affected skeletal phenotype [Parrilla et al., 1991], is a C-insertion at codon 448, which produces a frameshift of the 14 amino acids at the carboxyl-terminal of the TRβ gene [Kaneshige et al., 2000]. The mutant protein does not bind T3, has no transactivation activity and is a potent dominant-negative antagonist. Heterozygous TRβPV/+ mice mirror human RTH with circulating concentrations of T3, T4 and TSH increased 2-, 2.5- and 2.1-fold, respectively. Homozygous TRβPV/PV mice have very high levels of circulating T3, T4 and TSH, elevated 9-, 15- and 412-fold, respectively [Kaneshige et al., 2000]. We demonstrated that homozygous TRβPV/PV mice displayed advanced endochondral and intramembranous ossification [O'Shea et al., 2003]. The expression of fibroblast growth factor receptor-1 (FGFR1), previously identified as a skeletal T3-target gene [Stevens et al., 2003], was elevated in TRβPV mice indicating increased T3 signaling in the mutant skeleton. Heterozygous TRβPV/+ mice demonstrated an intermediate phenotype. Taken together, the results suggested a phenotype of skeletal thyrotoxicosis, consistent with the hypothesis that elevated circulating thyroid hormone levels drive the thyrotoxic phenotype via increased stimulation of an intact TRα1 signaling pathway in bone.
To investigate this hypothesis and determine whether TRα1 is the predominant TR isoform in bone, we studied mice carrying the identical mutation targeted to TRα1 [O'Shea P et al., 2005]. The mutant TRα1PV protein is also a potent dominant-negative antagonist capable of interfering with the activities of wild-type TRα1 and TRβ. The homozygous mutation was lethal and heterozygous TRα1PV/+ mice displayed only mild thyroid failure, with circulating T3 and T4 levels in the euthyroid range. The observed minor increase in T3 (1.15 fold) and no change in T4 [Kaneshige et al., 2001] was consistent with other mice harboring dominant-negative RTH mutations in TRα1 [Liu et al., 2003; Tinnikov et al., 2002]. Heterozygous TRα1PV/+ mice were dwarfs, exhibiting severely delayed endochondral and intramembranous ossification and postnatal growth retardation. In contrast to TRβPV mice, FGFR1 expression was reduced in TRα1PV/+ mutants, demonstrating impaired skeletal T3 signaling and skeletal hypothyroidism in TRα1PV/+ mice, consistent with significant levels of expressed mutant TRα1PV in bone that impairs the activities of wild-type TRα1 and β proteins. Our studies also indicated that the skeletal consequences of the PV mutation, when affecting either TRα1 or TRβ, result from dysregulated local GH/IGF-1 signaling in the growth plate, suggesting this signaling pathway lies downstream of TRα1 during bone development.
Thus, TRα1PV/+ mice display skeletal hypothyroidism despite the presence of biochemical euthyroidism [O'Shea P et al., 2005] and, in contrast, TRβPV mice have severe RTH but a phenotype of skeletal thyrotoxicosis [O'Shea et al., 2003]. Our studies indicate this paradox results from the differing effects of the PV mutations at the level of the HPT axis and in the skeleton (Figure 1). In the hypothalamus and pituitary, TRβ is the predominant isoform. Mutation of TRβ has been shown to produce RTH with elevated thyroid hormone concentrations and impaired feedback regulation of TRH and TSH. Thus, TRβ is critical for the determination of both the set-point of the HPT axis and the levels of circulating thyroid hormones [Abel et al., 2001; Abel et al., 2003; Dupre et al., 2004; Forrest et al., 1996; Gothe et al., 1999; Guissouma et al., 1998; Kaneshige et al., 2000]. In TRα0/0β-/- mice, TSH elevation is more severe than in TRβ-/- animals suggesting the TRα gene may also play a role in set-point regulation [Gauthier et al., 2001]. However, this role is likely to be compensatory and, in the physiological setting, TRα1 is much less important in HPT axis regulation. Indeed, mutation of the TRα locus has been shown to amplify the regulation of TRH in the hypothalamus but not interfere with TSH feedback regulation, circulating T4 and T3 levels, or affect pituitary function [Dupre et al., 2004; Gauthier et al., 2001; Kaneshige et al., 2001; Liu et al., 2003; Tinnikov et al., 2002]. Thus, in TRβPV mice, the pituitary displays tissue hypothyroidism and in TRα1PV/+ animals severe growth retardation occurs despite normal pituitary function. In contrast, our studies establish that TRα1 is the predominant isoform in bone [O'Shea P et al., 2005; O'Shea et al., 2003; Stevens et al., 2003]. Thus, in PV mutant mice, the hyperthyroid TRβPV/PV skeleton results from increased TRα1 activity stimulated by thyrotoxic circulating hormone levels resulting from HPT axis dysregulation. The phenotype is less severe in TRβPV/+ heterozygous animals because peripheral hormone concentrations are less markedly elevated. In the TRα1PV/+ skeleton, impaired TRα1 function determines the hypothyroid skeletal phenotype observed (Figure 2).
Figure 1. Hypothalamic-pituitary-thyroid (HPT) axis, tissue distribution of TRs and predominant TR-isoform responsiveness of selective T3-target tissues.
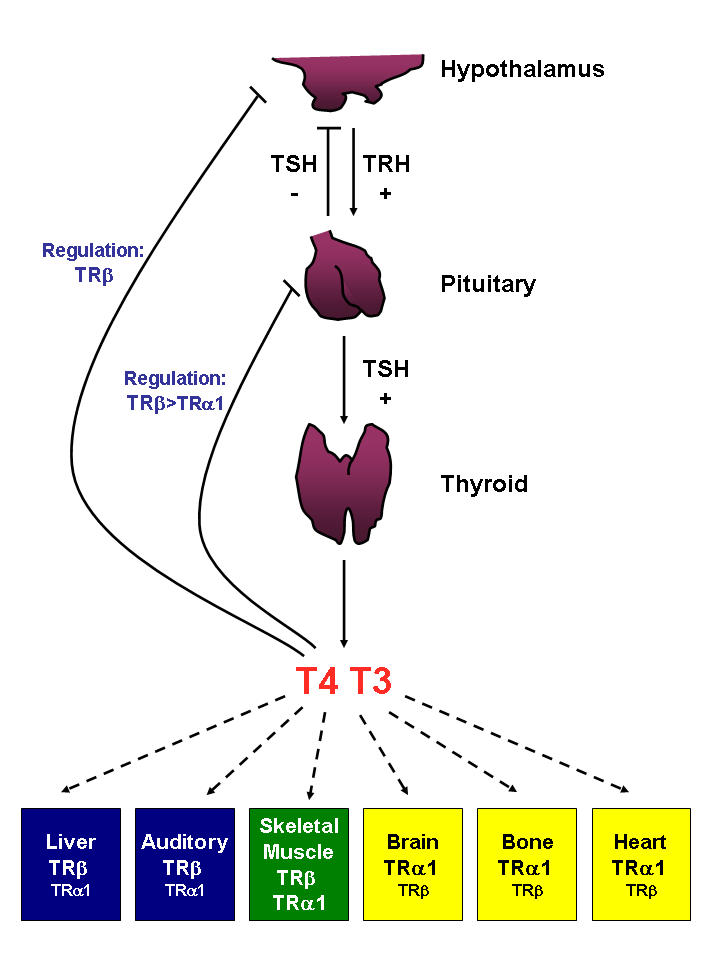
Serum concentrations of the thyroid hormones (T4 and T3) are maintained by a negative feedback system involving the hypothalamus, the pituitary and the thyroid gland. The circulating T3 and T4 exert direct negative feedback control on both TRH and TSH secretion, predominantly via regulation by TRβ. TSH itself inhibits the secretion of TRH. Relative TR distribution in individual tissues is highlighted; blue, TRβ predominant tissue; green, mixed; yellow, TRα1 predominant tissue.
Figure 2. Proposed mechanism for TRα1PV/+ and mutant TRβPV mutant mice.
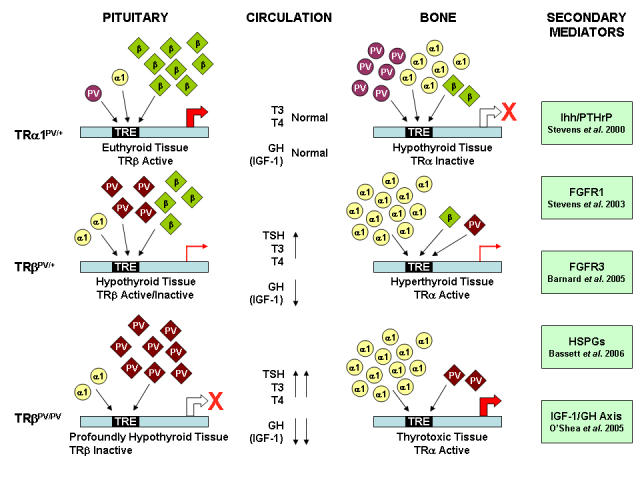
TRα1PV/+: In a TRβ predominant tissue, e.g. pituitary, the low levels of PV mutant receptor are unable to interfere with the actions of wild-type TRβ. In contrast, in a TRα1 predominant tissue, e.g. bone, the increased levels of dominant negative mutant receptor interfere with the actions of the wild-type receptors and impair the expression of T3-target genes. As a consequence, in TRα1PV/+ mutant mice, TRβ predominant tissues exhibit a euthyroid phenotype and TRα1 predominant tissues display a hypothyroid phenotype. TRβPV/+ and TRβPV/PV: In the TRβ predominant pituitary, there is a high level of TRβ expression relative to TRα1. Consequently, there are increasing levels of mutant and either low levels or no wild type TRβ in TRβPV/+ and TRβPV/PV tissues, respectively. In TRα1 predominant bone, the situation differs because levels of TRβ are low compared to TRα1. In both TRβPV/+ and TRβPV/PV mice, the low levels of mutant receptor are unable to interfere with the action of TRα1. Thus, in TRβPV mice, where the TRβ mutant acts as a rheostat and disrupts HPT axis regulation, TRβ predominant tissues display a hypothyroid phenotype with impaired expression of T3-target genes. TRα1 predominant tissues appear hyperthyroid in response to increased TRα1 activity that is stimulated by thyrotoxic circulating hormone levels resulting from impaired HPT axis regulation. The phenotype is less severe in TRβPV/+ heterozygous animals because peripheral hormone concentrations are less markedly elevated. A range of secondary mediators are included that have recently been shown to be targets of T3 action [Barnard et al., 2005; Bassett et al., 2006; O'Shea P et al., 2005; Stevens et al., 2003; Stevens et al., 2000].
Implications of skeletal observations
Phenotypic variability is a characteristic of human RTH and it is possible this may result from a range of individual RTH mutations that interact with genetic background factors leading to the production of mutant TRβ proteins with unique properties. Each mutation may affect T3 binding affinity or cofactor interactions in a specific manner resulting in variable dysregulation of the HPT axis and a spectrum of dominant negative activity. In the physiological setting, the TRβ mutation may act as a rheostat to determine the magnitude of resistance and the relationship of TSH to T4 and T3 levels. For instance, if an RTH mutation had a minimal effect on T3 binding affinity, resistance to negative feedback regulation of TSH may be overcome easily resulting in a minor change in circulating thyroid hormone levels. In contrast, if a mutation severely impaired T3 binding, as in the case of PV, TSH negative feedback regulation would be abolished and TSH levels profoundly increased irrespective of T4 and T3 levels. It is also likely that the dominant negative properties of each RTH mutation in each target tissue, combined with the differing TR isoform expression ratios in individual tissues, modifies the phenotype and provides a potential explanation for the heterogeneity observed in RTH.
Our analysis of TRα1PV/+ and TRβPV mutant mice has provided a new understanding of the complex relationship between central pituitary thyroid status and peripheral skeletal thyroid status that arises because the pituitary gland is a TRβ target tissue, whereas bone is a TRα1 target organ. Numerous groups have demonstrated differential TR isoform specific actions in individual tissues suggesting the hypothesis that tissue-specific responses to thyroid hormones result from differential TR expression patterns. Thus, the liver, ear and retina have also been identified as TRβ target tissues whilst the heart, like bone, is a TRα1 target organ [Brent, 2000; Flamant and Samarut, 2003]. Our studies in liver and heart from TRβPV mice further indicated that tissue-specific expression of TR isoforms dictates the dominant negative activity of the mutant receptor [Zhang et al., 2002]. The new studies in bone refine the hypothesis further and suggest the model can be extended to analyze the relationship between central and peripheral thyroid status in any T3-target tissue, depending on whether the peripheral tissue in question is predominantly responsive to TRα1 or TRβ.
Evidence from genetically modified mice supports this contention. For example, in the heart, in which TRα1 predominates, TRβ-/- mice exhibit features of thyrotoxicosis including tachycardia and increased expression of the cyclic nucleotide-gated channels HCN2 and HCN4, which are cardiac T3-target genes [Gloss et al., 2001]. In contrast, in TRα1-/- mice, features of hypothyroidism have been reported including bradycardia and reduced body temperature [Wikstrom et al., 1998]. Heart glucose utilization was reduced in TRα1PV/+ mice and increased in TRβPV/PV animals and these opposite effects on cardiac energy metabolism are consistent with bradycardia associated with hypothyroidism and tachycardia observed in hyperthyroidism and RTH [Esaki et al., 2004]. In the reproductive system, sertoli cell development is predominantly mediated through TRα1. TRα-null mice exhibited hypothyroid features including increased sertoli cell number and testis weight while TRβ-/- animals maintained normal sertoli cell responsiveness to T3 [Holsberger et al., 2005]. In the liver, a TRβ-predominant tissue, T3 influences cholesterol metabolism via regulation of cholesterol 7α-hydroxylase (CYP7A). TRβ-/- mice exhibit features of hypothyroidism with loss of hepatic T3-responsiveness, whereas normal CYP7A activity is observed in TRα1 deficient mice [Gullberg et al., 2000]. These insights into TR isoform-specific actions of T3 have important clinical implications. Thus, development of TR isoform-specific agonists and antagonists is likely to provide new therapeutic options for the treatment of diseases such as hypercholesterolemia, cardiac arrhythmias, growth disorders and osteoporosis [Baxter et al., 2001].
Acknowledgments
The skeletal phenotype characterization of TRα1PV and TRβPV mice was supported by a Medical Research Council (MRC) PhD Studentship (to PJO’S), MRC Clinician Scientist Fellowship (to JHDB) and MRC Career Establishment Grant (G9803002) (to GRW).
Abbreviations
- CYP7A
Cholesterol 7α-hydroxylase
- FGFR1
Fibroblast growth factor receptor-1
- GH
Growth hormone
- HPT
Hypothalamic-pituitary-thyroid
- HSPG
Heparan sulfate proteoglycan
- IGF-1
Insulin-like growth factor-1
- RTH
Resistance to thyroid hormone
- T3
Thyroid hormone
- TR
Thyroid hormone receptor
- TRH
Thyrotropin-releasing hormone
- TSH
Thyroid-stimulating hormone, thyrotropin
References
- Abel E. D., Ahima R. S., Boers M. E., Elmquist J. K., Wondisford F. E. Critical role for thyroid hormone receptor beta2 in the regulation of paraventricular thyrotropin-releasing hormone neurons. J Clin Invest. 2001;107:1017–23. doi: 10.1172/JCI10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel E. D., Moura E. G., Ahima R. S., Campos-Barros A., Pazos-Moura C. C., Boers M. E., Kaulbach H. C., Forrest D., Wondisford F. E. Dominant inhibition of thyroid hormone action selectively in the pituitary of thyroid hormone receptor-β null mice abolishes the regulation of thyrotropin by thyroid hormone. Mol Endocrinol. 2003;17:1767–76. doi: 10.1210/me.2003-0109. [DOI] [PubMed] [Google Scholar]
- Ballock R., Mita B. C., Zhou X., Chen D. H., Mink L. M. Expression of thyroid hormone receptor isoforms in rat growth plate cartilage in vivo. J Bone Miner Res. 1999;14:1550–6. doi: 10.1359/jbmr.1999.14.9.1550. [DOI] [PubMed] [Google Scholar]
- Baxter J. D., Dillmann W. H., West B. L., Huber R., Furlow J. D., Fletterick R. J., Webb P., Apriletti J. W., Scanlan T. S. Selective modulation of thyroid hormone receptor action. J Steroid Biochem Mol Biol. 2001;76:31–42. doi: 10.1016/s0960-0760(01)00052-8. [DOI] [PubMed] [Google Scholar]
- Brent G. A. Tissue-specific actions of thyroid hormone: insights from animal models. Rev Endocr Metab Disord. 2000;1:27–33. doi: 10.1023/a:1010056202122. [DOI] [PubMed] [Google Scholar]
- Cheng S. Y. Multiple mechanisms for regulation of the transcriptional activity of thyroid hormone receptors. Rev Endocr Metab Disord. 2000;1:9–18. doi: 10.1023/a:1010052101214. [DOI] [PubMed] [Google Scholar]
- Dupre S. M., Guissouma H., Flamant F., Seugnet I., Scanlan T. S., Baxter J. D., Samarut J., Demeneix B. A., Becker N. Both thyroid hormone receptor (TR)β 1 and TR β 2 isoforms contribute to the regulation of hypothalamic thyrotropin-releasing hormone. Endocrinology. 2004;145:2337–45. doi: 10.1210/en.2003-1209. [DOI] [PubMed] [Google Scholar]
- Esaki T., Suzuki H., Cook M., Shimoji K., Cheng S. Y., Sokoloff L., Nunez J. Cardiac glucose utilization in mice with mutated α- and β-thyroid hormone receptors. Am J Physiol Endocrinol Metab. 2004;287:E1149–53. doi: 10.1152/ajpendo.00078.2004. [DOI] [PubMed] [Google Scholar]
- Flamant F., Samarut J. Thyroid hormone receptors: lessons from knockout and knock-in mutant mice. Trends Endocrinol Metab. 2003;14:85–90. doi: 10.1016/s1043-2760(02)00043-7. [DOI] [PubMed] [Google Scholar]
- Forrest D., Sjoberg M., Vennstrom B. Contrasting developmental and tissue-specific expression of α and β thyroid hormone receptor genes. Embo J. 1990;9:1519–28. doi: 10.1002/j.1460-2075.1990.tb08270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest D., Hanebuth E., Smeyne R. J., Everds N., Stewart C. L., Wehner J. M., Curran T. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor β: evidence for tissue-specific modulation of receptor function. Embo J. 1996;15:3006–15. [PMC free article] [PubMed] [Google Scholar]
- Gauthier K., Plateroti M., Harvey C. B., Williams G. R., Weiss R. E., Refetoff S., Willott J. F., Sundin V., Roux J. P., Malaval L., Hara M., Samarut J., Chassande O. Genetic analysis reveals different functions for the products of the thyroid hormone receptor α locus. Mol Cell Biol. 2001;21:4748–60. doi: 10.1128/MCB.21.14.4748-4760.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloss B., Trost S., Bluhm W., Swanson E., Clark R., Winkfein R., Janzen K., Giles W., Chassande O., Samarut J., Dillmann W. Cardiac ion channel expression and contractile function in mice with deletion of thyroid hormone receptor α or β. Endocrinology. 2001;142:544–50. doi: 10.1210/endo.142.2.7935. [DOI] [PubMed] [Google Scholar]
- Gothe S., Wang Z., Ng L., Kindblom J. M., Barros A. C., Ohlsson C., Vennstrom B., Forrest D. Mice devoid of all known thyroid hormone receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes Dev. 1999;13:1329–41. doi: 10.1101/gad.13.10.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guissouma H., Ghorbel M. T., Seugnet I., Ouatas T., Demeneix B. A. Physiological regulation of hypothalamic TRH transcription in vivo is T3 receptor isoform specific. Faseb J. 1998;12:1755–64. doi: 10.1096/fasebj.12.15.1755. [DOI] [PubMed] [Google Scholar]
- Gullberg H., Rudling M., Forrest D., Angelin B., Vennstrom B. Thyroid hormone receptor β-deficient mice show complete loss of the normal cholesterol 7alpha-hydroxylase (CYP7A) response to thyroid hormone but display enhanced resistance to dietary cholesterol. Mol Endocrinol. 2000;14:1739–49. doi: 10.1210/mend.14.11.0548. [DOI] [PubMed] [Google Scholar]
- Holsberger D. R., Kiesewetter S. E., Cooke P. S. Regulation of neonatal Sertoli cell development by thyroid hormone receptor alpha1. Biol Reprod. 2005;73:396–403. doi: 10.1095/biolreprod.105.041426. [DOI] [PubMed] [Google Scholar]
- Kaneshige M., Suzuki H., Kaneshige K., Cheng J., Wimbrow H., Barlow C., Willingham M. C., Cheng S. A targeted dominant negative mutation of the thyroid hormone α 1 receptor causes increased mortality, infertility, and dwarfism in mice. Proc Natl Acad Sci U S A. 2001;98:15095–100. doi: 10.1073/pnas.261565798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneshige M., Kaneshige K., Zhu X., Dace A., Garrett L., Carter T. A., Kazlauskaite R., Pankratz D. G., Wynshaw-Boris A., Refetoff S., Weintraub B., Willingham M. C., Barlow C., Cheng S. Mice with a targeted mutation in the thyroid hormone β receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc Natl Acad Sci U S A. 2000;97:13209–14. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvistad P. H., Lovas K., Boman H., Myking O. L. Retarded bone growth in thyroid hormone resistance. A clinical study of a large family with a novel thyroid hormone receptor mutation. Eur J Endocrinol. 2004;150:425–30. doi: 10.1530/eje.0.1500425. [DOI] [PubMed] [Google Scholar]
- Liu Y. Y., Schultz J. J., Brent G. A. A thyroid hormone receptor α gene mutation (P398H) is associated with visceral adiposity and impaired catecholamine-stimulated lipolysis in mice. J Biol Chem. 2003;278:38913–20. doi: 10.1074/jbc.M306120200. [DOI] [PubMed] [Google Scholar]
- O'Shea P J., Bassett J. H., Sriskantharajah S., Ying H., Cheng S. Y., Williams G. R. Contrasting skeletal phenotypes in mice with an identical mutation targeted to thyroid hormone receptor {α}1 or {β} Mol Endocrinol. 2005 doi: 10.1210/me.2005-0224. [DOI] [PubMed] [Google Scholar]
- O'Shea P. J., Harvey C. B., Suzuki H., Kaneshige M., Kaneshige K., Cheng S. Y., Williams G. R. A thyrotoxic skeletal phenotype of advanced bone formation in mice with resistance to thyroid hormone. Mol Endocrinol. 2003;17:1410–24. doi: 10.1210/me.2002-0296. [DOI] [PubMed] [Google Scholar]
- Parrilla R., Mixson A. J., McPherson J. A., McClaskey J. H., Weintraub B. D. Characterization of seven novel mutations of the c-erbA β gene in unrelated kindreds with generalized thyroid hormone resistance. Evidence for two "hot spot" regions of the ligand binding domain. J Clin Invest. 1991;88:2123–30. doi: 10.1172/JCI115542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson H., Siebler T., Stevens D. A., Shalet S. M., Williams G. R. Thyroid hormone acts directly on growth plate chondrocytes to promote hypertrophic differentiation and inhibit clonal expansion and cell proliferation. Endocrinology. 2000;141:3887–97. doi: 10.1210/endo.141.10.7733. [DOI] [PubMed] [Google Scholar]
- Stevens D. A., Harvey C. B., Scott A. J., O'Shea P. J., Barnard J. C., Williams A. J., Brady G., Samarut J., Chassande O., Williams G. R. Thyroid hormone activates fibroblast growth factor receptor-1 in bone. Mol Endocrinol. 2003;17:1751–66. doi: 10.1210/me.2003-0137. [DOI] [PubMed] [Google Scholar]
- Tinnikov A., Nordstrom K., Thoren P., Kindblom J. M., Malin S., Rozell B., Adams M., Rajanayagam O., Pettersson S., Ohlsson C., Chatterjee K., Vennstrom B. Retardation of post-natal development caused by a negatively acting thyroid hormone receptor alpha1. Embo J. 2002;21:5079–87. doi: 10.1093/emboj/cdf523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R. E., Refetoff S. Effect of thyroid hormone on growth. Lessons from the syndrome of resistance to thyroid hormone. Endocrinol Metab Clin North Am. 1996;25:719–30. doi: 10.1016/s0889-8529(05)70349-2. [DOI] [PubMed] [Google Scholar]
- Weiss R. E., Refetoff S. Resistance to thyroid hormone. Rev Endocr Metab Disord. 2000;1:97–108. doi: 10.1023/a:1010072605757. [DOI] [PubMed] [Google Scholar]
- Wikstrom L., Johansson C., Salto C., Barlow C., Campos Barros A., Baas F., Forrest D., Thoren P., Vennstrom B. Abnormal heart rate and body temperature in mice lacking thyroid hormone receptor α 1. Embo J. 1998;17:455–61. doi: 10.1093/emboj/17.2.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G. R., Bland R., Sheppard M. C. Characterization of thyroid hormone (T3) receptors in three osteosarcoma cell lines of distinct osteoblast phenotype: interactions among T3, vitamin D3, and retinoid signaling. Endocrinology. 1994;135:2375–85. doi: 10.1210/endo.135.6.7988420. [DOI] [PubMed] [Google Scholar]
- Zhang X. Y., Kaneshige M., Kamiya Y., Kaneshige K., McPhie P., Cheng S. Y. Differential expression of thyroid hormone receptor isoforms dictates the dominant negative activity of mutant β receptor. Mol Endocrinol. 2002;16:2077–92. doi: 10.1210/me.2002-0080. [DOI] [PubMed] [Google Scholar]