

Short abstract
DNA microarrays based on long oligonucleotides are powerful tools for the functional annotation of the Plasmodium falciparum genome. Expression profiling of trophozoites and schizonts revealed genes associated with stage-specific processes and may serve as the basis for future drug targets and vaccine development.
Abstract
Background
The worldwide persistence of drug-resistant Plasmodium falciparum, the most lethal variety of human malaria, is a global health concern. The P. falciparum sequencing project has brought new opportunities for identifying molecular targets for antimalarial drug and vaccine development.
Results
We developed a software package, ArrayOligoSelector, to design an open reading frame (ORF)-specific DNA microarray using the publicly available P. falciparum genome sequence. Each gene was represented by one or more long 70 mer oligonucleotides selected on the basis of uniqueness within the genome, exclusion of low-complexity sequence, balanced base composition and proximity to the 3' end. A first-generation microarray representing approximately 6,000 ORFs of the P. falciparum genome was constructed. Array performance was evaluated through the use of control oligonucleotide sets with increasing levels of introduced mutations, as well as traditional northern blotting. Using this array, we extensively characterized the gene-expression profile of the intraerythrocytic trophozoite and schizont stages of P. falciparum. The results revealed extensive transcriptional regulation of genes specialized for processes specific to these two stages.
Conclusions
DNA microarrays based on long oligonucleotides are powerful tools for the functional annotation and exploration of the P. falciparum genome. Expression profiling of trophozoites and schizonts revealed genes associated with stage-specific processes and may serve as the basis for future drug targets and vaccine development.
Background
Plasmodium falciparum, a parasitic protozoan, is the causative agent of the most lethal form of human malaria. It is responsible for 300-500 million infections per year in some 90 countries and regions throughout the tropical and subtropical world. Of these clinical cases, approximately 2.1 million result in death annually [1]. In areas where mosquito abatement has failed, chemotherapy, consisting of a limited selection of antimalarial agents, is the only defense against this disease. The increase in drug resistance throughout the malaria endemic regions is cause for great concern and calls for the development of new antimalarial measures, which would involve a larger variety of drug targets as well as a wider array of vaccine strategies (reviewed in [2,3]).
The study of malaria will be greatly helped by the publicly available complete genome sequence of P. falciparum. The sequencing project, driven by the Sanger Centre, the Institute for Genomic Research (TIGR), and Stanford University is essentially complete [4]. The sequence of the completed chromosomes are available for download from each sequencing center and from the Plasmodium Genome Resource, PlasmoDB [5,6]. Preliminary analysis of the 23 megabase-pair (Mbp) P. falciparum genome indicates the presence of approximately 5,400 genes spread across 14 chromosomes, a circular plastid genome and a mitochondrial genome. Strikingly, more than 60% of the predicted open reading frames (ORFs) lack orthologs in other genomes [4]. This fact underscores the need to elucidate gene function, yet many of the tools that have propelled the study of model organisms remain inefficient or nonexistent in Plasmodium. Despite recent improvements in P. falciparum transformation techniques, [7] the efficiency of stable transfection under a direct drug selection remains approximately 10-6, making knockout and gene replacement experiments difficult, and genetic complementation strategies nearly impossible. Genome-wide expression profiling by microarray technology provides an easy alternative for the functional genomic exploration of P. falciparum.
In organisms ranging from bacteria to humans, expression profiling has proved a powerful tool. Profiling has been used to gain important insights into processes such as development, responses to environmental perturbations, gene mutation, pathogen and host response, and cancer [8,9,10,11,12,13,14]. Expression profiling has already been successfully applied to the partial genome sequence of P. falciparum, and has been used to characterize the role of previously unannotated genes [15,16,17].
Here we present the design and assembly of a long-oligonucleotide P. falciparum gene-specific microarray using the currently available genomic sequence generated by the Malaria Genome Consortium [18,19,20]. During the course of this work, we have developed software, improved by experimental data and an open-source policy, for rapidly selecting unique sequences from predicted ORFs of any genome. Subsequently, we constructed a long-oligonucleotide-based P. falciparum microarray, which we used to evaluate changes in the global expression profile between two distinct stages of P. falciparum erythrocytic-stage asexual development - mid-trophozoite and mid-schizont. The large number of differentially expressed genes detected in this analysis suggests that extensive transcriptional regulation has a major role in the functional specialization of parasite development.
Results and discussion
P. falciparum ORF predictions
At the outset of these studies, a total of 27.6 Mbp of P. falciparum genomic sequence was obtained from the publicly available sources presented by the Malaria Genome Consortium [18,19,20] in October 2000. The sequence comprised two completely assembled chromosomes, the complete mitochondrial and plastid genomes, and the sum of all the partial contigs from the remaining chromosomes. ORF predictions were carried out using GlimmerM, a gene-finding tool trained with P. falciparum specific sequences [21,22]. Using default parameters, GlimmerM frequently yielded a large number of overlapping predictions (competing gene models) and thus additional filtering of the initial prediction output was required. As slight overprediction of ORFs is generally desirable for the purpose of expression array building, the post-prediction filtering of the GlimmerM output was modified with respect to the process used by the Malaria Genome Consortium [21]. Briefly, individual predictions that overlapped and were on opposite strands or in different reading frames were retained. For competing predictions within a given GlimmerM gene model, ORFs that were extended downstream by at least 300 bp and were within 300 bp of the total size compared to the size of the largest prediction were chosen. In all other cases, the largest predicted ORF was selected. This selection method resulted in 290 ORF predictions for chromosome 2, whereas the Malaria Genome Consortium selected 210 for the same chromosome [21].
The first round of predictions, carried out on the publicly available genomic sequence as of August 2000, yielded 8,008 putative ORFs. The predicted ORFs are available as additional data with the online version of this paper (see Additional data files) and from [23]. As a first step to annotation, the translation of all predicted ORFs were used to search the Astral, SwissProt, and non-redundant (NR) databases for sequence similarities using the Smith-Waterman algorithm [24]. In addition, all ORF predictions were linked to their counterparts in PlasmoDB [5,6].
ArrayOligoSelector: array element design
To construct a gene-specific microarray of the P. falciparum genome, we designed 70 mer oligonucleotide array elements. We chose this length for a number of reasons. Long oligonucleotides are a highly sensitive alternative to PCR products and provide a means to readily distinguish between genes with high degrees of sequence similarity [25]. In addition, the presence of various types of repetitive sequences and highly homologous gene families in the AT-rich P. falciparum genome contributes to a high rate of PCR failure ([17] and J.L.D., unpublished results). A software program, ArrayOligoSelector, was developed specifically for the purpose of systematically selecting gene-specific long oligonucleotide probes for entire genomes. The latest version and complete source code for ArrayOligoSelector is freely available at [26]. For each ORF, the program optimizes the oligonucleotide selection on the basis of several parameters, including uniqueness in the genome, sequence complexity, lack of self-binding, and GC content (Figure 1). Similar approaches to oligonucleotide design have previously been described, but the exact algorithms, source code, and/or accompanying hybridization data are not available [25,27,28].
Figure 1.
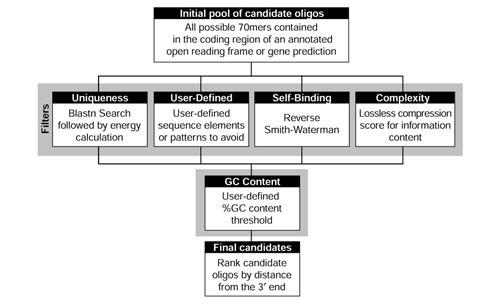
Schematic of the ArrayOligoSelector oligonucleotide-selection algorithm. Oligonucleotide selection begins with the collection of all possible 70 mer oligonucleotides from a given ORF. Four filters are executed in parallel: selection for uniqueness within the genome, an optional user-defined pattern filter, avoidance of significant secondary structure (self-binding), and avoidance of low-complexity sequence. The intersection of the set of all oligonucleotides passing these filters are then further selected for a desired base composition and then ranked by proximity to the 3' end of the ORF.
ArrayOligoSelector helps ensure complete genome coverage and optimal array hybridization while avoiding several potential problems originating from the peculiar characteristics of the P. falciparum genome. The algorithm attempts to minimize cross-hybridization between the oligonucleotide and other regions of the genome. To evaluate the potential for cross-hybridization, early versions of ArrayOligoSelector used a simple BLASTN alignment identity [29]. Although this method prevents the selection of troublesome sequences, it does not take into account the effect of mismatch distribution or base composition. Subsequent versions of ArrayOligoSelector were improved by calculating a theoretical energy of binding between the oligonucleotide and its most probable cross-hybridization target in the genome ('second best target'). The binding energy (kcal/mol) is calculated using a nearest-neighbor model using established thermodynamic parameters [30,31,32,33,34,35]. Thus, a sequence with high cross-hybridization potential will have a more stable binding energy with a larger absolute value. In contrast, a sequence unique in the genome will yield a smaller absolute value for the binding energy. A representative plot of the calculated binding energies for all possible 70 bp oligonucleotides from a putative var gene (PlasmoDB v4.0 annotated gene ID PF08_0140) is shown in Figure 2a.
Figure 2.
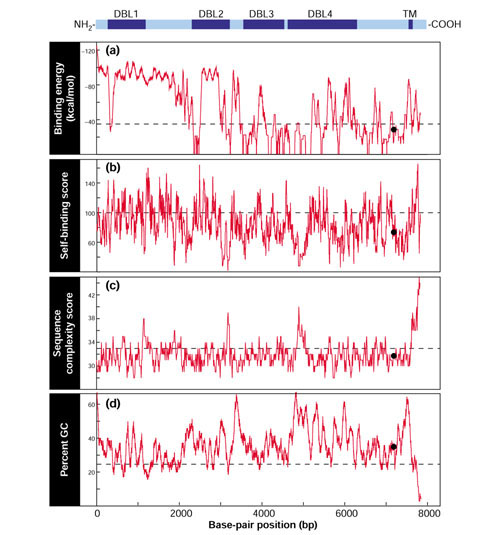
Example of sequence parameters measured by ArrayOligoSelector for a putative member of the var gene family (PlasmoDB v4.0 annotated gene ID PF08_0140). A schematic of the target gene is shown above plots for each filter indicating the positions of four Duffy binding-like domains (DBL) and a putative transmembrane domain (TM). For each filter, a 70 bp window was measured for all possible positions within the gene. The dashed line represents the average value obtained for each filter where 6,000 random 70 bp sequences were chosen from the total collection of P. falciparum predicted ORFs. The black circle denotes the position of the final candidate oligonucleotide (oligo ID F44871_2) chosen for the array.
An important aspect of oligonucleotide design for microarray hybridization is avoiding secondary structures within the oligonucleotide, as these are likely to be detrimental to hybridization performance. To avoid selecting oligonucleotides with secondary complex structure, ArrayOligoSelector uses the Smith-Waterman algorithm with the PAM47 DNA matrix to calculate the optimal alignment score between the candidate oligonucleotide sequence and the reverse complement of that sequence [24]. A high Smith-Waterman score indicates the potential to create secondary structures (Figure 2b).
The presence of low-complexity sequence could also result in significant nonspecific cross-hybridization. For example, the P. falciparum genome contains a large number of low-complexity sequence elements as a result of a high frequency of continuous stretches of A and T nucleotides in both the non-coding and the coding regions. ArrayOligoSelector automatically detects such sequences by subjecting candidate oligonucleotide sequences to a lossless compression [36]. The compression score, calculated as the difference in bytes between the original sequence and the compressed version, is inversely proportional to complexity (Figure 2c). Using this score, repeats of essentially any nature are detected in a computationally efficient manner.
In addition, in order to avoid specific sequence features, ArrayOligoSelector supports filtering based on user-defined patterns. This feature can be used to implement filtering rules based on empirically derived data. Finally, the melting temperature of an oligonucleotide is largely determined by its GC content. As is the case with most ORFs, there exists a large range of %GC values (< 10 to > 60%) over a 70 bp window (Figure 2d). For this reason, a user-defined %GC target range is used by ArrayOligoSelector such that the majority of the array elements will share a similar base composition and hybridization properties across the array.
Given the above parameters, ArrayOligoSelector evaluates every 70 mer sequence within an ORF and chooses an optimal set on the following criteria. The uniqueness-filter requires oligonucleotides to satisfy two simultaneous threshold criteria based on the calculation of the binding energy to their second-best target (the best target is itself). First, the oligonucleotide must rank among the top 5% of the unique or almost unique 70 mers in the entire ORF. Second, its binding energy must be within 5 kcal/mol of the best candidate for the ORF. In addition, an optional user-defined energy threshold can operate in conjunction with the default threshold. Initial settings for the low-complexity and the self-binding terms allow the top-scoring 33% of 70 mers to pass to the next selection step. Finally, an optional 'user-defined sequence filter' simply eliminates the 70 mer candidates containing the defined sequences. These four filters operate on the entire set of 70 mer candidates for a particular ORF and generate four independent output sets. The intersection of the four outputs is then subjected to the final selection. If no common oligonucleotide is identified in the first intersection, the self-binding and complexity filters are incrementally relaxed until an intersection becomes available. The final selection of candidate oligonucleotides depends upon the %GC filter and 3'-end proximity ranking. Initially, oligonucleotides are allowed to pass if they meet the user-specified %GC. If no oligonucleotide with the desired GC content is found, the target %GC range is relaxed by one percentage point in each direction until one or more oligonucleotides pass. As a final step, a single candidate, closest to the 3' end of the gene is chosen. Finally, ArrayOligoSelector generates an output file containing the oligonucleotide selections for each putative ORF.
From our initial set of predictions, a total of 6,272 70 mer oligonucleotides were selected and synthesized. For our first pass of malaria oligonucleotide selections, the earlier version of ArrayOligoSelector utilizing the BLASTN-based identity threshold was used. The identity cutoff was adjusted to a very conservative value of < 30 bp of identity. The initial setting of the GC content filter was set to 28% GC (73°C Tm. Subsequently, with the release of additional sequence information, a new set of predictions was generated in April 2002 and an additional 1,025 oligonucleotides were selected using the upgraded version of ArrayOligoSelector. In this selection, the user-defined uniqueness threshold was set at -35 kcal/mol, the value at which cross-hybridization is essentially eliminated (Figure 3). The GC content target was set at 28%. The sequence and location of each oligonucleotide is available online [23]. The experiments described in the following section were conducted with the first set of predictions only. As additional annotations become available for the whole genome sequence, additional oligonucleotides will be selected and added to the existing collection. We expect the final set to contain approximately 8,500 oligonucleotides.
Figure 3.
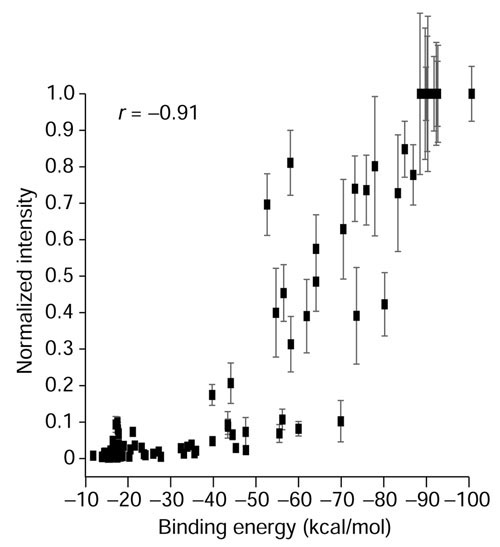
Relationship of calculated binding energy to relative hybridization intensity. For each of the oligonucleotides shown in Figure 4, a binding energy (kcal/mol) to the perfect match sequence was calculated using ArrayOligoSelector and plotted against relative hybridization intensity. The Pearson correlation coefficient |r| = 0.91 indicates a strong correlation between the calculated binding energy and hybridization performance.
Oligonucleotide performance
Hughes et al. [25] showed that 60 mer oligonucleotides make highly sensitive specific microarray elements for expression profiling of Saccharomyces cerevisiae [25]. The oligonucleotides used in that study were synthesized in situ using ink-jet technology whereas the oligonucleotides used in our study were commercially synthesized and subsequently printed using mechanical deposition. Similarly to the experiments of Hughes et al., we wished to test experimentally the effect of mismatches on sensitivity and specificity of 70 mer oligonucleotides in the context of a complex hybridization mixture (P. falciparum total RNA). Ten separate malaria ORF predictions were arbitrarily selected for analysis and for each of these ORFs a set of ten oligonucleotides were synthesized. The first oligonucleotide in each set represents the original 70 mer selection from ArrayOligoSelector. Each successive oligonucleotide within a set contains an increasing number of mutations made in increments of 10%. Thus, the second oligonucleotide in each set had seven bases (10%) altered, while the last oligonucleotide had 63 bases (90%) mutated. For the first set of five ORFs (Figure 4a,4b,4c,4d,4e), which is referred to as the 'distributed set', both the position and the identity of each mutation was random. For the second set of five ORFs (Figure 4g,4h,4i,4j,4k), referred to as the 'anchored set', the mutations in each oligonucleotide were limited to the ends of the sequence. In this manner, a contiguous stretch of perfectly matched bases was always preserved in the center of each oligonucleotide.
Figure 4.
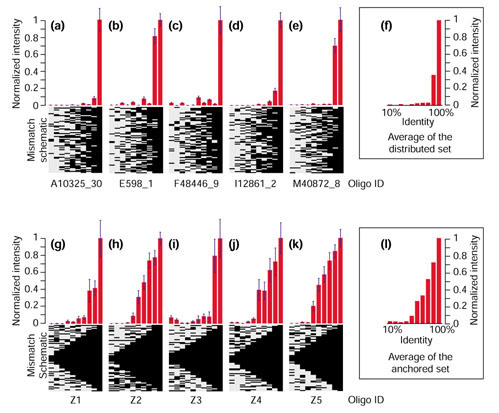
The hybridization performance of long oligonucleotides in relation to internal versus terminal mismatches. Ten putative genes were chosen arbitrarily. Each gene is represented by a series of ten oligonucleotides. The first (rightmost) oligonucleotide in each series represents a perfect match to the target sequence. Mismatches were introduced in increasing numbers (10% steps) into each subsequent oligonucleotide in each series. The pattern of matches and mismatches within each oligonucleotide is shown below each graph, where a black bar indicates a perfect match and a gray bar indicates a mismatch. (a-e) Mismatch positions were randomized ('distributed set'). (g-k) Mismatch positions were chosen such that a contiguous stretch of perfect-match sequence remained in the middle of the oligonucleotide ('anchored set'). The average hybridization performance, measured as the normalized total intensity for 16 independent experiments, is plotted for each oligonucleotide. (f) The average plot for a-e; (l) the average plot for g-k.
Figure 4 summarizes normalized hybridization intensities of control oligonucleotides obtained from the global gene-expression comparisons between trophozoite and schizont stages. The results originate from the six microarray hybridizations presented in Figure 5 and 10 additional hybridizations available as additional data files [23]. The resulting hybridization intensity measurements for each oligonucleotide were averaged across all hybridizations and scaled as a fraction of the average intensity of the perfect-match oligonucleotide for each set. As is evident from Figure 4a,4b,4c,4d,4e, the presence of internal mismatches (bubbles and bulges) had a large effect on hybridization performance: oligonucleotides with 10% mismatches (7 bases) suffered an average reduction of 64% in hybridization intensity when compared to the perfect match, while oligonucleotides with 20% (14 bases) or more mismatches were reduced by an average of 97% (Figure 4f). For the anchored set (Figure 44g,4h,4i,4j,4k), a more gradual hybridization trend was observed. Mutating the terminal 14 bases (7 bases at each end) resulted in an average loss of 49% of the maximal hybridization intensity. Not until 42 bases had been mutated (21 bases at each end) did the relative intensity of hybridization drop by an average of 97.5% (Figure 4l). In agreement with the findings of Hughes et al. [25], the data from the anchored set of oligonucleotides reveal a strong relationship between the length of contiguous match (the equivalent of oligonucleotide length) and overall hybridization performance.
Figure 5.
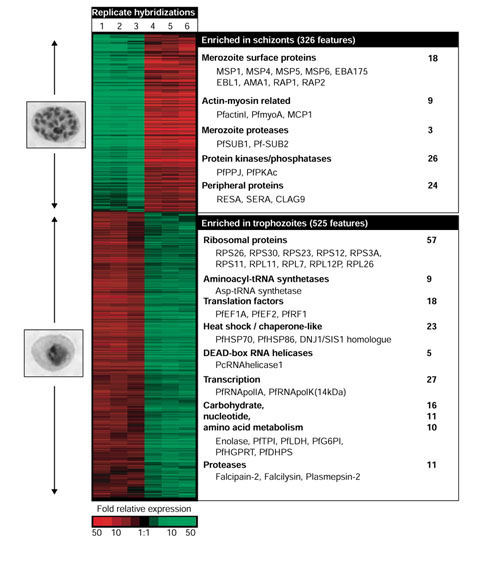
Comparison of trophozoite and schizont stages of P. falciparum. Hierarchical cluster analysis of six replicate microarray hybridizations is shown for the 854 genes that yielded at least a twofold expression difference in at least four experiments. In hybridizations 1-3, the schizont RNA was labeled with Cy3 (green signal) and trophozoite RNA was labeled with Cy5 (red signal) and in analyses 4-6, the fluorophore order was reversed. Examples of major functional gene groups enriched in either studied stage, number of corresponding ORFs and several previously characterized representatives of each group are indicated. All gene expression data are available at PlasmoDB and the DeRisi Lab website [23].
To measure the extent to which the energy calculation implemented in ArrayOligoSelector matches reality, we have plotted in Figure 3 the calculated energy of the 100 control oligos shown in Figure 4 and their relative intensities of hybridization. The calculated energy and relative intensity of hybridization correlate at |r| = 0.91. For comparison, the relative intensity of hybridization and number of nucleotide identities correlate at |r| = 0.72. This indicates that a calculated binding energy approach may be used to estimate the potential for cross-hybridization for any sequence relative to the rest of the genome. The specificity for each oligonucleotide is uniquely and computationally determined and expressed as a binding energy (kcal/mol).
To further address the question of specificity of oligonucleotide hybridization to their targets in a complex sample we introduced a set of probes targeting a set of 19 non-repetitive sequences from S. cerevisiae to the microarray. To control for the nucleotide bias of the malaria genome relative to yeast, the selection criteria for this set were identical to selection of the plasmodial microarray elements. The average GC content of the S. cerevisiae oligonucleotides was 31.5%, whereas the average GC content of plasmodial oligonucleotides is 32.5%. The average signal-to-background ratios across all hybridizations for these negative control spots was less than twofold, which is well below the conservative fivefold signal-to-background threshold used to filter data (see Materials and methods). In addition, a series of 10 hybridizations was carried out where total RNA from an asynchronous parasite culture was hybridized against PCR products corresponding to the negative control S. cerevisiae sequences. In these hybridizations the yeast PCR fragment hybridized strictly to its cognate sequence, while the average signal to background value for plasmodial elements in the same channel was 1.17 ± 0.05. In no individual case did a plasmodial element yield a signal greater than 2.3% of the target hydridization signal intensity. The results of these microarray hybridizations are available as additional data files [23].
To assess whether separate oligonucleotides designed to represent the same target gene perform in a similar manner, we examined three distinct situations: elements dispersed over a long single exon ORF (Figure 6a), overlapping oligonucleotides (Figure 6b), and oligonucleotides representing multiple exons of a single gene (Figure 6c). In each case we observed consistent oligonucleotide performance.
Figure 6.

Hybridization performance of multiple oligonucleotides representing single ORFs. The average expression ratios were calculated for each oligonucleotide overlapping distinct regions of three ORFs: (a) PFD0985w encoding a predicted hypothetical protein; (b) PFE0040c encoding PfEMP2; and (c) PFA0110w encoding RESA1. The arrows indicate the location of oligonucleotide elements within the coding sequence (thick line), and the ratios express fold enrichment in either trophozoite stage versus schizont stage (t) or schizont stage versus trophozoite stage (s).
Gene-expression profiling of trophozoites and schizonts
We chose a direct comparison of the trophozoite and schizont stages of the P. falciparum asexual intraerythrocytic life cycle as a first step toward comprehensively profiling all life-cycle stages of this parasite. The trophozoite and schizont represent two distinct developmental stages within the 48-hour plasmodial erythrocytic life cycle. These stages vary greatly in morphology, biochemical properties, and transcriptional activity (reviewed in [15,37]). The mid-trophozoite stage, 18-24 hours post-invasion, contains a highly transcriptionally active nucleus with abundant euchromatin. In addition, trophozoites are characterized by massive hemoglobin ingestion, intake of nutrients from the surrounding medium, increasing concentration of cytoplasmic ribosomes and rapid formation of organelles. In contrast, the mid-schizonts, at 36-42 hours post-invasion, are characterized by DNA replication (16-32 copies) and compaction into newly formed nuclei. In addition, maturation of merozoite cells begins at the schizont stage and is characterized by the appearance of merozoite organelles such as the rhoptry and dense granules. The several trophozoite- and schizont-specific genes identified previously provide an excellent source of positive controls for the experiments described below.
For microarray hybridization, total RNA was prepared from synchronized in vitro P. falciparum cultures representing the trophozoite stage and the schizont stage (see Materials and methods). Six independent hybridizations were carried out; in three, the trophozoite-derived cDNA was labeled with Cy3 and the schizont-derived cDNA with Cy5. In the other three hybridizations, the fluorophore assignment was reversed. Of the genes assayed, 854 features displayed a differential expression greater than twofold (Figure 5): 525 showed higher relative transcript abundance in trophozoites than in schizonts, whereas 326 had greater relative transcript abundance in schizonts. Linear regression ratios were calculated for each possible pair of microarray hybridizations using the filtered dataset. The correlation between hybridizations with the same Cy3/Cy5 assignment was r = 0.94 ± 0.02, while correlation of hybridizations with the opposite Cy3/Cy5 order was r = 0.89 ± 0.03.
Northern blot hybridizations
To confirm the microarray results, we examined six genes by northern blot analysis. In the microarray hybridization, the expression levels of two of the selected genes were unchanged (< 2-fold) while four additional genes showed a differential expression between the trophozoite and schizont stage (> 2-fold). An equal mass of total RNA from both the trophozoite and schizont stages was hybridized with PCR-generated DNA probes corresponding to the selected genes. Subsequently, each northern blot was stripped and rehybridized with a probe specific for the 41 kD antigen (p41), fructose-bisphosphate aldolase (PfALDO; PlasmoDB v4.0 ID PF14_0425; Oligo ID M11919_1) [38], as a loading control. While the relative amount of PfALDO transcript differs by more than twofold between trophozoite and schizont stages when equal masses of total RNA are blotted, we found that the relative amount of PfALDO to be essentially equivalent when equal masses of poly(A)+ RNA were used for the northern blot (Figure 7a). The discrepancy between northern blots with total RNA and poly(A)+ mRNA are probably due to changes in the relative amounts of mRNA and ribosomal RNA during the intraerythrocytic life cycle. The poly(A)+ northern blot measurements agree well with the replicate array hybridizations, in which PfALDO was consistently less than 1.5-fold differentially expressed (Figure 7b). To make northern blot measurements comparable to the normalized expression array ratios, the ratio between the two stages was measured using a phosphoimager and divided by the ratio obtained for the PfALDO control in each case. The normalized ratios of the radiolabel signal were highly consistent with the averaged ratios from the six microarray hybridizations (Figure 7b).
Figure 7.
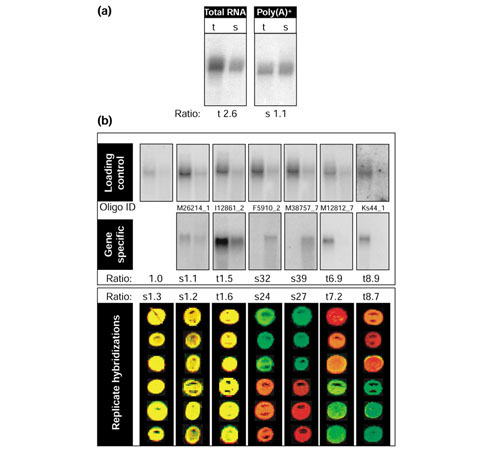
Northern blot validation of the microarray results. (a) Comparison of total RNA and poly(A)+ RNA for northern analysis. 12 μg total RNA and 2 μg poly(A)+ mRNA from both the schizont (s) and trophozoite (t) stages of P. falciparum were blotted and probed with a PCR fragment overlapping the genomic sequence corresponding to oligonucleotide ID M11919_1, which represents the 41 kD antigen (p41), fructose-bisphosphate aldolase (PlasmoDB v4.0 annotated gene ID PF14_0425). (b) Total RNA northern blots were probed with PCR probes overlapping the corresponding oligonucleotide elements ('gene specific') for each gene. The selected gene set included a probable calcium-transporting ATPase (PlasmoDB MAL13P1.61), plasmodial heat-shock protein (PlasmoDB PFI0875w), a member of a family of conserved hypothetical proteins (PlasmoDB PFI1445w) and an ORF with sequence similarity to a sodium-and chloride-dependent taurine transporter (PlasmoDB MAL13P1.130), lactate dehydrogenase (PlasmoDB PF13_0141), and a heat-shock protein homolog of Hsp70-3 (PlasmoDB PF11_0351). The northern blot membranes were stripped and reprobed with the fructose bisphosphate aldolase control probe. Northern blot expression levels were measured by a phosphorimager. For each gene-specific probe, the measured ratio of trophozoite to schizont was divided by the ratio measured for aldolase on the same membrane. The average ratio from six replicate hybridizations on the microarray, including fluorophore reversal, is shown in the lower panel. The ratios express fold enrichment in either the trophozoite stage versus schizont stage (t) or schizont stage versus trophozoite stage (s).
Biological significance of the gene-expression results
The genome-wide expression data summarized by hierarchical cluster analysis (Figure 5) resulted in two main gene categories, corresponding to genes differentially expressed between trophozoite and schizont stages. Serving as internal positive controls, a number of previously well-characterized plasmodial genes were detected in both categories. In addition, an evaluation of the homology-based gene identities within these categories revealed several functional gene groups. All data from these experiments are available at PlasmoDB and the DeRisi Lab website [23].
Trophozoite genes
The predominant group of features with elevated expression in the trophozoite stage comprised genes encoding various components of the eukaryotic translation machinery. This group contained 24 and 33 proteins of the 40S and 60S ribosomal subunits (RPS and RPL), respectively. In addition, nine orthologs of aminoacyl-tRNA synthetases, and 10 initiation and seven elongation translation factors were detected among trophozoite-specific genes. Several previously identified plasmodial genes were present in this group, including Asp-tRNA synthetase, two plasmodial elongation factors (PfEF1A and PfEF2) and one ribosome-releasing factor, PfRF1 [21,39]. Consistent with our findings, PfEF-1A has been previously shown to have peak expression during the trophozoite stage [40]. Two additional gene groups whose functions are linked to the process of protein synthesis were present among the trophozoite genes: five DEAD-box RNA helicases, including a close homolog of P. cynomolgi RNA helicases-1 [41] and 23 molecular chaperone-like molecules, including two P. falciparum heat-shock proteins such as PfHSP70 (GenBank accession number M19753) and PfHSP86 (accession number L34028), and a homolog of a DnaJ-domain-containing protein family, DNJ1/SIS1 homolog [42]. These data agree with previous studies that found a group of DEAD-box RNA helicases to be overexpressed during the trophozoite stage in P. cynomolgi [41]. Along with the genes for the translation machinery a number of genes involved in various steps of RNA synthesis and processing were located among the 'trophozoite genes', including 16 ORFs belonging to various RNA polymerase complexes and 11 splicing factors (Figure 5). Two previously identified plasmodial RNA polymerase components were found in this group, including the largest subunit of P. falciparum RNA polymerase II, PfRNApolIIA (M73770), and a homolog RNApolK (14 kD) [39]. The expression characteristics revealed are also consistent with several previous studies that suggested that the plasmodial transcription and translation machinery is active through the late ring and early trophozoite stage before decaying during the late schizont stage [15,40].
Another functional group of genes that encode enzymes of cellular biosynthetic pathways was distinguished within the trophozoite category. This gene set includes 16 enzymes of carbohydrate metabolism, 10 ORFs likely to be involved in nucleotide metabolism, and 11 ORFs involved in the biosynthetic pathways of several amino acids. Several well-characterized plasmodial genes were identified in this metabolic collection, including P. falciparum lactate dehydrogenase (PfLDH; 027743), enolase (U00152), triose-phosphate isomerase (PfTPI; L01654), glucose-6-phosphate isomerase (PfG6PI; J05544), hypoxanthine-guanine phosphoribosyl-transferase (PfHGPRT; X16279) and dihydropteroate synthetase (PfDHPS; U07706). In addition, a group of 11 proteolytic enzymes potentially involved in hemoglobin degradation was detected among the trophozoite genes; these include a cysteine protease, falcipain-2 (AF251193), a metalloprotease falcilysine, (AF123458), and a member of an aspartic protease family, plasmepsin-2 (L10740). Falcipain-2 and plasmepsin-2 have been the targets of recent drug discovery research [43,44].
Overall, the emergent gene clusters suggest that the trophozoite stage, a central phase of plasmodial intraerythrocytic development, is characterized by the activation of general cellular growth functions such as transcription, translation and hemoglobin degradation and biosynthesis of basic metabolites.
Schizont genes
A large number of ORFs found in the schizont-expressed category correspond to genes previously associated with the various steps by which newly released merozoites invade new host cells. The initial step of this process, adhesion of the merozoite to the surface of an erythrocyte, is facilitated by several classes of proteins exposed on the surface of the parasite. Eighteen ORFs, identical or homologous to proteins associated with the merozoite surface, were present among the schizont-enriched genes. This group included four merozoite surface proteins (MSP): MSP1 (M19753), MSP4, MSP5, (AF033037) and MSP6 (AY007721). Additional members of this group include two ORFs containing Duffy-like binding domains, erythrocyte-binding antigen, EBA 175 (L07755), a putative erythrocyte-binding protein, EBL1 (AF131999), and proteins known to be delivered to the surface from apical organelles, including apical membrane antigen, AMA1 (U65407), and finally two rhoptry-associated proteins (RAP1 (U20985) and RAP2).
Initial attachment of the merozoite is followed by reorientation of the parasite cell with its apical part toward the erythrocyte membrane followed by invagination of the membrane. Previous studies suggested that both steps are facilitated by the action of actomyosin, which requires ATP hydrolysis [45]. Consistent with these findings, we found five proteins previously associated with this process, pf-actinI (M19146), pf-myoA (AF255909), and merozoite cap protein-1 (U14189), and two subtilisin-like proteases (PfSUB1 and PfSUB2 (AJ132422)) differentially enriched in schizonts. Interestingly, one additional homolog of PfSUB1 was identified among the schizont genes. Moreover, the expression levels of a set of plasmodial protein kinases were previously found to be augmented during the late stages of the malarial erythrocytic life cycle [15]. Our findings confirm and extend this report: 26 unique ORFs sharing a high to medium level of homology with protein kinases and phosphorylases had elevated mRNA levels during the schizont stage (Figure 5). Two previously identified representatives were present in this set: a cAMP-dependent protein kinase, PfPKAc (AF126719), and a plasmodial serine/threonine protein phosphatase, PfPPJ (AF126719).
A second functional group of genes with increased expression in schizonts encodes proteins that are thought to function on the periphery of a newly infected erythrocyte at the early stages of asexual development. Representatives include: the genes for ring-infected erythrocyte surface antigen (RESA) (X04572) and several close RESA homologs, CLAG9 (AF055476) the related gene CLAG3.1, and two members of the serine-repeat rich protein (SERA) family [21]. In addition to these well-characterized surface proteins, the schizont-enriched set of transcripts contained a number of ORFs identical or homologous to proteins recognized by antibodies present in plasmodium immune sera obtained either from model organisms [46] or from acute and/or convalescent patients [47]. In summary, the schizont stage of plasmodial development featured genes predominantly occupied with the process of merozoite function as well as the advance synthesis of transcripts for proteins that facilitate parasite establishment within the newly infected erythrocyte.
Taken together, these results suggest that the parasite cell in the trophozoite stage is dedicated to cell growth, and the predominant function of the mid-late schizont stage is maturation of the next generation of merozoites. Of particular interest is the large number of ORFs within both categories (39% in trophozoite and 61% in schizonts) with no putative functions assigned. These ORFs have little to no homology to any other known genes and may possibly represent highly specialized functions not likely to be shared outside this family of parasites.
Conclusions
In this study, we present a P. falciparum ORF-specific microarray utilizing 70 mer oligonucleotides as individual microarray elements. This approach helped to overcome potential problems originating from low PCR amplification and allowed us to select probes with a high specificity, thereby minimizing potential cross-hybridization. Moreover, the oligonucleotide-selection algorithm allowed a balanced GC content (around 28%) across the entire microarray set, which is significantly higher than the plasmodial genome average, which is 19.4% with 23.7% in coding regions [4].
Application of the ArrayOligoSelector is not restricted to the P. falciparum genome, but is broadly useful for the automated selection of hybridization probes for a range of species. The flexibility of the selection parameters controlling stringency of uniqueness, self-binding, complexity, user-defined filters and GC content, allows the selection of oligonucleotides appropriate for any genome.
Evaluation of results from derivative control oligonucleotides showed that long oligonucleotides could tolerate 10% mismatches; however, alteration of the target sequence by more then 20% eliminated most of the hybridization signal. Therefore, small sequencing errors and natural variation among isolates are not likely to impact on sensitivity. These performance characteristics imply that the array design for this effort can accommodate the study of essentially any P. falciparum strain with a high degree of specificity.
At present, the P. falciparum microarray used in this study consists of approximately 6,000 gene-specific elements corresponding to the majority of the total coding content predicted for the P. falciparum genome. As new sequence and improved gene predictions arise, additional elements will be added to this evolving platform. Moreover, the present oligonucleotide representation could be further extended for investigation of several unusual P. falciparum genetic and transcriptional phenomena, including antisense mRNA transcription [48] and alternative splicing and/or transcriptional initiation [49,50]. This may be achieved by designing exon-specific array features, as well as antisense oligonucleotides. The oligonucleotide collection could also be expanded by sequences corresponding to intergenic genomic regions. Inclusion of such elements was found to be extremely useful for identifying protein-binding DNA regions by chromatin-immunoprecipitation as well as genes not detected by automated gene-prediction algorithms [51].
Within both the trophozoite and schizont categories, large numbers of genes belong to functionally related processes. These include genes encoding ribosomal subunits, multiple factors for transcription and translation, enzymes of biosynthetic and catabolic pathways, or merozoite adherence and invasion machinery. These results are consistent with predictions that a large number of plasmodial genes undergo strict stage-specific transcriptional regulation, and that such (co-)regulation is shared among functionally related genes [15,52]. Naturally, a 'fine-resolution' global gene-expression profile including the different steps of the plasmodial life cycle for multiple divergent strains will be necessary to characterize fully the intraerythrocytic life of the parasite. At present, our laboratory is analyzing a global gene-expression profile of the 48-hour erythrocytic life cycle with 1-hour resolution for three strains of P. falciparum.
In a number of model organisms, high-resolution gene-expression maps have served as extremely powerful tools for discovery and characterization of novel genes as well as exploration of multiple cellular functions [9,11]. The gene-expression maps typically comprise genome-wide expression profiles at a number of different stages of cellular development, profiles of multiple strains and genetic variants, and global expression responses to number of growth perturbations and growth-inhibitory drugs. Following a similar approach in P. falciparum is most likely to provide substantial information about the many ORFs that lack functional annotation. Further understanding of cellular physiology of this parasite including basic metabolic functions and the intricate interactions between the parasite cell and human host immune system will be a key step in uncovering new targets for antimalarial drug discoveries and vaccine development.
Materials and methods
Microarray fabrication
The 70-bp oligonucleotides were synthesized (Operon Technologies, CA), resuspended in 3 × SSC to a final concentration of 60 pmol/μl, and spotted onto poly-L-lysine-coated microscopic slides, as previously described [53]. All oligo sequences are available at [23].
Cell cultures
P. falciparum parasite cells (W2 strain) were cultured as described [54] with slight modifications: 2% suspension of purified human red blood cells in RPMI1640 media supplemented with 0.25% AlbumaxI (GIBCO/Invitrogen, San Diego, CA), 2 g/l sodium bicarbonate, 0.1 mM hypoxanthine, 25 mM HEPES pH 7.4, and 50 μg/I gentamycin. Cells were synchronized by two consecutive sorbitol treatments on two consecutive cell cycles (a total of four treatments) and harvested at the subsequent trophozoite stage (18-24 h post-invasion) and schizont stage (36-42 h post-invasion). For the trophozoite stage collection, visual inspection of the Giemsa stains show a nearly pure trophozoite population with less than 1% schizonts. For the schizont stage collection, we estimate the amount of ring contamination to be around 3%. The cells were harvested in prewarmed PBS at 37°C, and spun at 1,500 g for 5 min. Cell pellets were rapidly frozen in liquid nitrogen and stored at -80°C.
RNA preparation and microarray hybridization
Total RNA was prepared directly from the frozen pellets of parasitized erythrocytes, where approximately 1 ml of cell pellet was lysed in 7.5 ml Trizol (GIBCO) and RNA was extracted according to the manufacturer's instructions. mRNA was isolated from total RNA preparations using the Oligotex mRNA Mini Kit (Qiagen, Valencia, CA). For the hybridization experiments, 12 μg total RNA was used for first-strand cDNA synthesis as follows: RNA was mixed with a mixture of random hexamer (pdN6) oligonucelotides and oligo-(dT20) at final concentration 125 μg/μl for each oligonucleotide. The mixture was heated to 70°C for 10 min and then incubated on ice for 10 min. Reverse transcription was started by adding dNTPs to a final concentration of 1 mM dATP and 500 μM each: dCTP, dGTP, dTTP and 5-(3-aminoallyl)-2'-deoxyuridine-5'-triophosphate, (aa-dUTP) (Sigma), with 150 units of StrataScript (Stratagene, La Jolla, CA). The reaction was carried out at 42°C for 120 min and the residual RNA was hydrolyzed with 0.1 mM EDTA and 0.2 M NaOH at 65°C for 15 min. The resulting aa-dUTP-containing cDNA was coupled to CyScribe Cy3 or Cy5 (Amersham, Piscataway, NJ) monofunctional dye in the presence of 0.1 M NaHCO3 pH 9.0. Coupling reactions were incubated for a minimum of 1 h at room temperature. The labeled product was purified using QIAquick PCR purification system (Qiagen). Hybridizations and final washing procedures were carried out as described [9] with slight modifications. Briefly, the hybridization medium contained 3 × SSC, 1.5 μg/μl poly(A) DNA (Pharmacia Biotech, Uppsala), and 0.5% SDS. Hybridizations were incubated at 65°C for 8-16 h. Arrays were washed in 2 × SSC/0.2% SDS and then 0.1 × SSC at room temperature. The microarrays were scanned with a GenePix 4000B scanner and the images analyzed using GenePix Pro 3.0 software (Axon Instruments, Union City, CA). Subsequently, the data were normalized using the AMAD microarray database and subjected to the cluster analysis using the CLUSTER and TREEVIEW software, as described [53]. For the CLUSTER analysis, low-quality features and features with a signal level less than fivefold the background were filtered from the initial raw data set, yielding 4,737 elements. Subsequently, features with an arbitrary twofold fluorescence signal difference in at least four experiments were considered. All programs and microarray-related protocols are available online [55].
Probe preparation and northern blot analysis
The northern blot probes were generated by PCR using the following oligonucleotide sequences:
FWD-M11919_1: 5'-TAGAAAACAGAGCTAGCTACAGAG; REV-M11919_1: 5'-AGTTGGTTTTCCTTTGGCTGTGTG; FWD-M1282_7: 5'-CTGTAGGTGGTATCCCTTTACAAG; REV-M12812_7: 5'-GACAAATAATAATGCCATACCAGG; FWD-I12861_2: 5'-AAATGCAGTTGTTACTGTCCCTG; REV-I12861_2: 5'-GCTCTTTTGTCAGTTCTTAAATCG; FWD-F5910_2: 5'-ACAACCAGTTTGCTCTGCTTATC; REV-F5910_2: 5'-GGCCGACATTAATTGCTTATATGC; FWD-M38757_7: 5'-TAGAAGTATATCATTCCGAAGGTG; REV-M38757_7: 5'-GTAGAAGCTTCAATATCAAGCTC; FWD-M1282_7: 5'-CTGTAGGTGGTATCCCTTTACAAG; REV-M12812_7: 5'-GCTAATGCCTTCATTCTCTTAGTT; FWD-Ks44_1: 5'-GGCAAGCTATAACAAATCCTGAGA; REV-Ks44_1: 5'-GCTAAAGCGGCAGCAGTTGGTTCA.
Total RNA (10 μg) or poly(A)+ RNA (0.4 μg) was resolved on a denaturing 1% agarose gel, transferred to nitrocellulose membrane and hybridized with a radiolabeled probe as described [56]. The blots were analyzed using ImageQuant v1.2 (Molecular Dynamics, Sunnyvale, CA).
Additional data files
The predicted ORFs and GenePix results (GPR) files containing raw data for Figure 5 and from 10 additional hybridizations are available as additional data files with the online version of this paper and from [23]. Data for Figure 5: three hybridizations (1, 2,3) with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5; Three hybridizations (4,5,6) with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3. Additional hybridizations: Six hybridizations (7,8,9,10,11,12) with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5; four hybridizations (14,15,15,16) with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3.
ORF predictions of August 2000 were predicted from contig sequences available in August 2000, using GlimmerM software. These predictions were used to design the first set of 70 mer oligonucleotides and includes genes from the plastid genome. ORF predictions of October 2000 were predicted from contig sequences available in October 2000, using GlimmerM software. These predictions also include genes from the plastid and mitochondrial genomes.
Supplementary Material
Figure 5: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Figure 5: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Figure 5: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Figure 5: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Figure 5: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Figure 5: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Additional hybridization: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Additional hybridization: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Additional hybridization: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Acknowledgments
Acknowledgements
We thank Pradip Rathod and his lab for ongoing discussion and assistance. We also thank the Howard Hughes Medical Institute, Anita Sil, Phil Rosenthal, David Roos and Patrick O. Brown. Z.B., M.P.J., and J.L.D. are supported by an award from the Searle Foundation and the Burroughs Wellcome Fund.
References
- Sachs J, Malaney P. The economic and social burden of malaria. Nature. 2002;415:680–685. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- Ridley RG. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature. 2002;415:686–693. doi: 10.1038/415686a. [DOI] [PubMed] [Google Scholar]
- Richie TL, Saul A. Progress and challenges for malaria vaccines. Nature. 2002;415:694–701. doi: 10.1038/415694a. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berrlman M, Hyman R, Carlton JM, Pain A, Nelson K, Bowman S, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PlasmoDB: The Plasmodium Genome Resource http://plasmodb.org
- Bahl A, Brunk B, Coppel RL, Crabtree J, Diskin SJ, Fraunholz MJ, Grant GR, Gupta D, Huestis RL, Kissinger JC, et al. PlasmoDB: the Plasmodium genome resource. An integrated database providing tools for accessing, analyzing and mapping expression and sequence data (both finished and unfinished). Nucleic Acids Res. 2002;30:87–90. doi: 10.1093/nar/30.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitsch K, Driskill C, Wellems T. Transformation of malaria parasites by the spontaneous uptake and expression of DNA from human erythrocytes. Nucleic Acids Res. 2001;29:850–853. doi: 10.1093/nar/29.3.850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet. 1996;14:457–460. doi: 10.1038/ng1296-457. [DOI] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Marton MJ, DeRisi JL, Bennett HA, Iyer VR, Meyer MR, Roberts CJ, Stoughton R, Burchard J, Slade D, Dai H, et al. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat Med. 1998;4:1293–1301. doi: 10.1038/3282. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Avalos SV, Blader IJ, Fisher M, Boothroyd JC, Burleigh BA. Immediate/early response to Trypanosoma cruzi infection involves minimal modulation of host cell transcription. J Biol Chem. 2002;277:639–644. doi: 10.1074/jbc.M109037200. [DOI] [PubMed] [Google Scholar]
- Ben Mamoun C, Gluzman IY, Hott C, MacMillan SK, Amarakone AS, Anderson DL, Carlton JM, Dame JB, Chakrabarti D, Martin RK, et al. Co-ordinated programme of gene expression during asexual intraerythrocytic development of the human malaria parasite Plasmodium falciparum revealed by microarray analysis. Mol Microbiol. 2001;39:26–36. doi: 10.1046/j.1365-2958.2001.02222.x. [DOI] [PubMed] [Google Scholar]
- Hayward RE. Plasmodium falciparum phosphoenolpyruvate carboxykinase is developmentally regulated in gametocytes. Mol Biochem Parasitol. 2000;107:227–240. doi: 10.1016/S0166-6851(00)00191-2. [DOI] [PubMed] [Google Scholar]
- Hayward RE, Derisi JL, Alfadhli S, Kaslow DC, Brown PO, Rathod PK. Shotgun DNA microarrays and stage-specific gene expression in Plasmodium falciparum malaria. Mol Microbiol. 2000;35:6–14. doi: 10.1046/j.1365-2958.2000.01730.x. [DOI] [PubMed] [Google Scholar]
- The Sanger Centre Plasmodium falciparum Genome Project http://www.sanger.ac.uk/Projects/P_falciparum
- Stanford Genome Technology Center Malaria Genome Project http://sequence-www.stanford.edu/group/malaria/index.html
- TIGR Plasmodium falciparum Genome Database (PFDB) http://www.tigr.org/tdb/edb2/pfa1/htmls
- Gardner MJ, Tettelin H, Carucci DJ, Cummings LM, Aravind L, Koonin EV, Shallom S, Mason T, Yu K, Fujii C, et al. Chromosome 2 sequence of the human malaria parasite Plasmodium falciparum. Science. 1998;282:1126–1132. doi: 10.1126/science.282.5391.1126. [DOI] [PubMed] [Google Scholar]
- Salzberg SL, Pertea M, Delcher AL, Gardner MJ, Tettelin H. Interpolated Markov models for eukaryotic gene finding. Genomics. 1999;59:24–31. doi: 10.1006/geno.1999.5854. [DOI] [PubMed] [Google Scholar]
- Joseph DeRisi lab: web supplement http://derisilab.ucsf.edu/falciparum
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Hughes TR, Mao M, Jones AR, Burchard J, Marton MJ, Shannon KW, Lefkowitz SM, Ziman M, Schelter JM, Meyer MR, et al. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat Biotechnol. 2001;19:342–347. doi: 10.1038/86730. [DOI] [PubMed] [Google Scholar]
- ArrayOligoSelector http://arrayoligosel.sourceforge.net
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Rouillard J-M, Herbert CJ, Zuker M. OligoArray: genome-scale oligonucleotide design for microarrays. Bioinformatics. 2002;18:486–487. doi: 10.1093/bioinformatics/18.3.486. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- Jaeger J, Turner DH, Zuker M. Improved predictions of secondary structures for RNA. Proc Natl Acad Sci USA. 1989;86:7706–7710. doi: 10.1073/pnas.86.20.7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allawi HT, SantaLucia J., Jr Nearest neighbor thermodynamic parameters for internal G.A mismatches in DNA. Biochemistry. 1998;37:2170–2179. doi: 10.1021/bi9724873. [DOI] [PubMed] [Google Scholar]
- Lyngso RB, Zuker M, Pedersen CN. Fast evaluation of internal loops in RNA secondary structure prediction. Bioinformatics. 1999;15:440–445. doi: 10.1093/bioinformatics/15.6.440. [DOI] [PubMed] [Google Scholar]
- Peritz AE, Kierzek R, Sugimoto N, Turner DH. Thermodynamic study of internal loops in oligoribonucleotides: symmetric loops are more stable than asymmetric loops. Biochemistry. 1991;30:6428–6436. doi: 10.1021/bi00240a013. [DOI] [PubMed] [Google Scholar]
- Peyret N, Seneviratne PA, Allawi HT, Santa Lucia J., Jr Nearest-neighbor thermodynamics and NMR of DNA sequences with internal A.A, C.C, G.G, and T.T mismatches. Biochemistry. 1999;38:3468–3477. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Nakano S, Yoneyama M, Honda K. Improved thermodynamic parameters and helix initiation factor to predict stability of DNA duplexes. Nucleic Acids Res. 1996;24:4501–4505. doi: 10.1093/nar/24.22.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv J, Lempel A. A universal algorithm for sequential data compression. IEEE Trans Inf Theory. 1977;23:337–343. [Google Scholar]
- Kumar VP, Datta S. Use of variability in the stage-specific transcription levels of Plasmodium falciparum in the selection of target genes. Parasitol Int. 2001;50:165–173. doi: 10.1016/S1383-5769(01)00075-7. [DOI] [PubMed] [Google Scholar]
- Certa U, Ghersa P, Dobeli H, Matile H, Kocher HP, Shrivastava IK, Shaw AR, Perrin LH. Aldolase activity of a Plasmodium falciparum protein with protective properties. Science. 1988;240:1036–1038. doi: 10.1126/science.3285469. [DOI] [PubMed] [Google Scholar]
- Bowman S, Lawson D, Basham D, Brown D, Chillingworth T, Churcher CM, Craig A, Davies RM, Devlin K, Feltwell T, et al. The complete nucleotide sequence of chromosome 3 of Plasmodium falciparum. Nature. 1999;400:532–538. doi: 10.1038/22964. [DOI] [PubMed] [Google Scholar]
- Vinkenoog R, Speranca MA, van Breemen O, Ramesar J, Williamson DH, Ross-MacDonald PB, Thomas AW, Janse CJ, del Portillo HA, Waters AP. Malaria parasites contain two identical copies of an elongation factor 1 alpha gene. Mol Biochem Parasitol. 1998;94:1–12. doi: 10.1016/S0166-6851(98)00035-8. [DOI] [PubMed] [Google Scholar]
- Song P, Malhotra P, Tuteja N, Chauhan VS. RNA helicase-related genes of Plasmodium falciparum and Plasmodium cynomolgi. Biochem Biophys Res Commun. 1999;255:312–316. doi: 10.1006/bbrc.1999.0204. [DOI] [PubMed] [Google Scholar]
- Watanabe J. Cloning and characterization of heat shock protein DnaJ homologues from Plasmodium falciparum and comparison with ring infected erythrocyte surface antigen. Mol Biochem Parasitol. 1997;88:253–258. doi: 10.1016/S0166-6851(97)00073-X. [DOI] [PubMed] [Google Scholar]
- Joachimiak MP, Chang C, Rosenthal PJ, Cohen FE. The impact of whole genome sequence data on drug discovery - a malaria case study. Mol Med. 2001;7:698–710. [PMC free article] [PubMed] [Google Scholar]
- Coombs GH, Goldberg DE, Klemba M, Berry C, Kay J, Mottram JC. Aspartic proteases of Plasmodium falciparum and other parasitic protozoa as drug targets. Trends Parasitol. 2001;17:532–537. doi: 10.1016/S1471-4922(01)02037-2. [DOI] [PubMed] [Google Scholar]
- Pinder J, Fowler R, Bannister L, Dluzewski A, Mitchell GH. Motile systems in malaria merozoites: how is the red blood cell invaded? Parasitol Today. 2000;16:240–245. doi: 10.1016/S0169-4758(00)01664-1. [DOI] [PubMed] [Google Scholar]
- McColl DJ, Silva A, Foley M, Kun JF, Favaloro JM, Thompson JK, Marshall VM, Coppel RL, Kemp DJ, Anders RF. Molecular variation in a novel polymorphic antigen associated with Plasmodium falciparum merozoites. Mol Biochem Parasitol. 1994;68:53–67. doi: 10.1016/0166-6851(94)00149-9. [DOI] [PubMed] [Google Scholar]
- de Stricker K, Vuust J, Jepsen S, Oeuvray C, Theisen M. Conservation and heterogeneity of the glutamate-rich protein (GLURP) among field isolates and laboratory lines of Plasmodium falciparum. Mol Biochem Parasitol. 2000;111:123–130. doi: 10.1016/S0166-6851(00)00304-2. [DOI] [PubMed] [Google Scholar]
- Patankar S, Munasinghe A, Shoaibi A, Cummings LM, Wirth DF. Serial analysis of gene expression in Plasmodium falciparum reveals the global expression profile of erythrocytic stages and the presence of antisense transcripts in the malarial parasite. Mol Biol Cell. 2001;12:3114–3125. doi: 10.1091/mbc.12.10.3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Lin LH, Pace T, Janse CJ, Birago C, Ramesar J, Picci L, Ponzi M, Waters AP. Interspecies conservation of gene order and intron-exon structure in a genomic locus of high gene density and complexity in Plasmodium. Nucleic Acids Res. 2001;29:2059–2068. doi: 10.1093/nar/29.10.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dooren GG, Su V, DiOmbrain MC, McFadden GI. Processing of an apicoplast leader sequence in Plasmodium falciparum, and the identification of a putative leader cleavage enzyme. J Biol Chem. 2002;277:23612–23619. doi: 10.1074/jbc.M201748200. [DOI] [PubMed] [Google Scholar]
- Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Horrocks P, Dechering K, Lanzer M. Control of gene expression in Plasmodium falciparum. Mol Biochem Parasitol. 1998;95:171–181. doi: 10.1016/S0166-6851(98)00110-8. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Brown PO. DNA arrays for analysis of gene expression. Methods Enzymol. 1999;303:179–205. doi: 10.1016/s0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Microarrays: source for microarray protocols and software http://derisilab.ucsf.edu/microarray/index.html
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 5: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Figure 5: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Figure 5: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Figure 5: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Figure 5: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Figure 5: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy3 and schizont RNA labeled with Cy5
Additional hybridization: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Additional hybridization: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Additional hybridization: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3
Additional hybridization: hybridization with trophozoite RNA labeled with Cy5 and schizont RNA labeled with Cy3