

Abstract
Identification of targets and delivery platforms for gene therapy of neurodegenerative disorders is a clinical challenge. We describe a novel paradigm in which the neuroprotective gene is the herpes simplex virus type 2 (HSV-2) anti-apoptotic gene ICP10PK and the vector is the growth compromised HSV-2 mutant ΔRR. ΔRR is delivered intranasally. It is not toxic in rats and mice. ICP10PK is expressed in the hippocampus of the ΔRR treated animals for at least 42 days in the absence of virus replication and late virus gene expression. Its expression is regulated by an AP-1 amplification loop. Intranasally delivered ΔRR prevents kainic acid (KA) induced seizures, neuronal loss and inflammation, in both rats and mice. The data suggest that ΔRR is a promising therapeutic platform for neurodegenerative diseases.
Keywords: HSV, ICP10PK, neuroprotection, gene therapy, kainic acid, seizures, glia
INTRODUCTION
Recent progress in understanding molecular mechanisms of cell fate determination has imparted increased validity to gene-based therapies for CNS disorders. However, the paucity of genes with a therapeutically effective neuroprotective profile, and the difficulties associated with the construction of appropriate vectors for gene delivery to the CNS, pose a major clinical challenge. Members of anti-apoptotic gene families, heat shock proteins, and neurotrophins and various delivery platforms were studied, with relatively limited success [1, 2]. The advantage of the vectors based on herpes simplex virus is that they are neurotropic. However, safety concerns linger, as presently available vectors are based on the type 1 virus (HSV-1) that induces apoptosis in CNS neurons, causes severe and often fatal encephalitis in immunocompetent humans and has been associated with epileptiform seizures [3-6]. Replication incompetent and genome-free vectors were developed, but high doses that retain residual dose-related toxicity are required for efficient transgene delivery, and vector administration is invasive [7, 8]. Although viruses have evolved potent anti-apoptotic genes for their own survival, their neuroprotective profile, is still poorly understood. We have recently shown that the HSV type 2 (HSV-2) gene, known as ICP10PK, has anti-apoptotic activity in virus infected primary hippocampal cultures [5]. Here we report that ΔRR delivered by the non-invasive intranasal route, protects rats and mice from KA induced seizures and neuronal loss in the absence of any evident toxicity.
RESULTS
ΔRR is replication incompetent in neurons
ΔRR is deleted in the ICP10 RR domain, which is required for virus replication in neurons [9]. We used organotypic hippocampal cultures (OHC), which retain much of the in vivo hippocampal anatomy and physiology [10], to examine whether ΔRR replicates and causes cytopathic effects (CPE) in neurons. HSV-2, HSV-1 and the ICP10PK deleted HSV-2 mutant ΔPK, which is replication compromised in neurons [5,11] were studied in parallel and served as controls. Single step growth curves indicated that ΔRR and ΔPK are replication incompetent in OHC (Fig. 1A). They do not cause cell death, as determined by staining with ethidium homodimer (Fig. 1B). Virus growth and cell death were seen for HSV-2 and HSV-1 (Fig. 1A,B). The % dead cells was 87 ± 11 % and 4 ± 1% for HSV-2 and ΔRR, respectively (p<0.001; Fig 1C). The data indicate that ΔRR does not replicate and is not toxic in OHC, at least at the infectious dose used in these experiments However, staining with the LacZ substrate C12FDG indicated that ICP10PK-LacZ was expressed in the dentate gyrus and hippocampus, with primary localization in the CA3 and CA1 fields (Fig. 1D).
Fig 1.
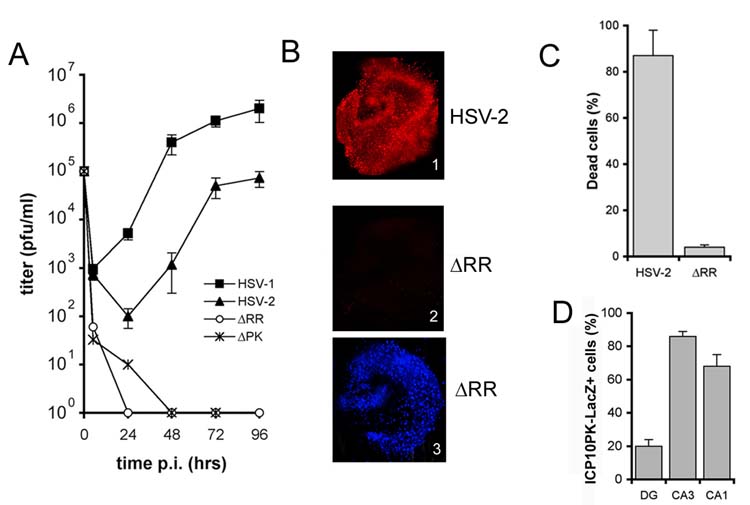
ΔRR does not replicate nor cause cell death in neurons. (A) Single step growth curves of HSV-1, HSV-2, ΔPK and ΔRR in OHC were done as described in materials and methods. (B) OHC infected with HSV-2 (panel 1) or ΔRR (panels 2 and 3) as in (A) were stained with ethidium homodimer (cell death) at 96 hr p.i. (panels 1 and 2) and counterstained with DAPI (panel 3). (C) Ethidium staining cells in (B) were counted and the results are expressed as % dead cells ± SEM. (D) ΔRR infected OHC were stained with C12FDG and the % staining cells in the dentate gyrus (DG) and the CA1 and CA3 hippocampus fields calculated relative to DAPI staining as described in materials and methods.
ICP10PK delivered by intranasal instillation of ΔRR reaches the hippocampus
Previous studies have shown that HSV gains access to the temporal lobes by the olfactory route, presumably by axonal transport [12]. Because this property holds the therapeutic promise of non-invasive vector delivery, we wanted to know whether a similar distribution is evidenced by ΔRR, which is growth compromised in neurons. Mice and rats were given ΔRR by intranasal instillation and tissues along the lateral olfactory tract [piriform cortex, amygdala and entorhinal cortex (which projects to the hippocampus) (13) (Fig 2A)] were examined for infectious virus (by plaque assay) and for ICP10-LacZ expression by immunohistochemistry and/or immunoblotting with ICP10 antibody on days 1-8 post infection (p.i.).
Fig. 2.
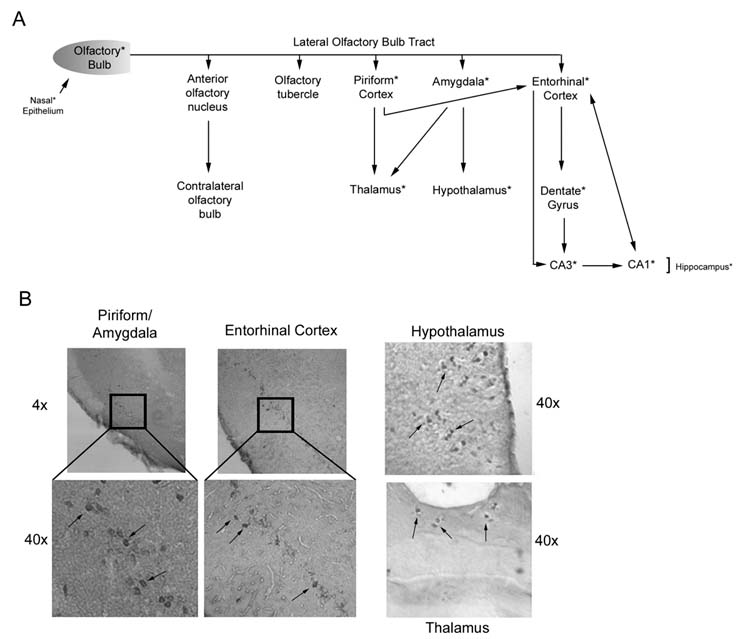
ICP10PK-LacZ reaches the hippocampus through the lateral olfactory bulb tract. (A) Schematic representation of the tract with relevant connections. Asterisk indicates tissues examined for ICP10PK and found to be positive. (B) Immunohistochemical staining of brain tissue along the lateral olfactory bulb tract with ICP10 antibody.
Infectious virus was not found in any tissues throughout the study interval. However, ICP10PK-LacZ was seen in the nasal epithelium at very low levels (confirmed by densitometric scanning) and only on day 1 p.i. (Fig.3A,B). While the levels of protein in these tissues is relatively low, this is unlikely to explain the failure to detect ICP10PK-LacZ at later times p.i., since the same protein levels were loaded in all lanes and the levels of actin were virtually identical for all samples (Fig. 3A, lanes 1-3). Staining with ICP10 antibody was also seen in the olfactory bulb, piriform cortex, thalamus, hypothalamus and enthorinal cortex [lateral olfactory tract (Fig. 2A)] on day 2 p.i. (Fig. 2B). In the hippocampus, ICP10PK-LacZ expression was first seen on day 2 p.i.. (Fig. 3D, panels 1, 2) and it primarily localized in the CA3 and CA1 fields (Fig. 3D, panel 1). Staining was not seen with preimmune IgG (Fig. 3D, panel 4) nor in mock-infected animals (data not shown). In the hippocampus and olfactory bulb, expression was still high on day 8 p.i. (Fig. 3A, lanes 4-7). Densitometric scanning and data analysis relative to actin, indicate that the levels of ICP10PK in the hippocampus are significantly higher than in the nasal epithelium (Fig. 3B). Supporting the conclusion that ΔRR does not replicate, duplicate samples of the olfactory bulb and hippocampal tissues were negative for the major capsid protein VP5 (Fig. 3A,C), the expression of which requires viral DNA replication [14]. Interestingly, VP5 was also not seen in the nasal epithelium tissues (Fig. 3A). This likely reflects the composition of the nasal epithelium, which consists of over 80% neurons [15], with virus replication potentially occurring only in the remaining 20% epithelial cells.
Fig. 3.
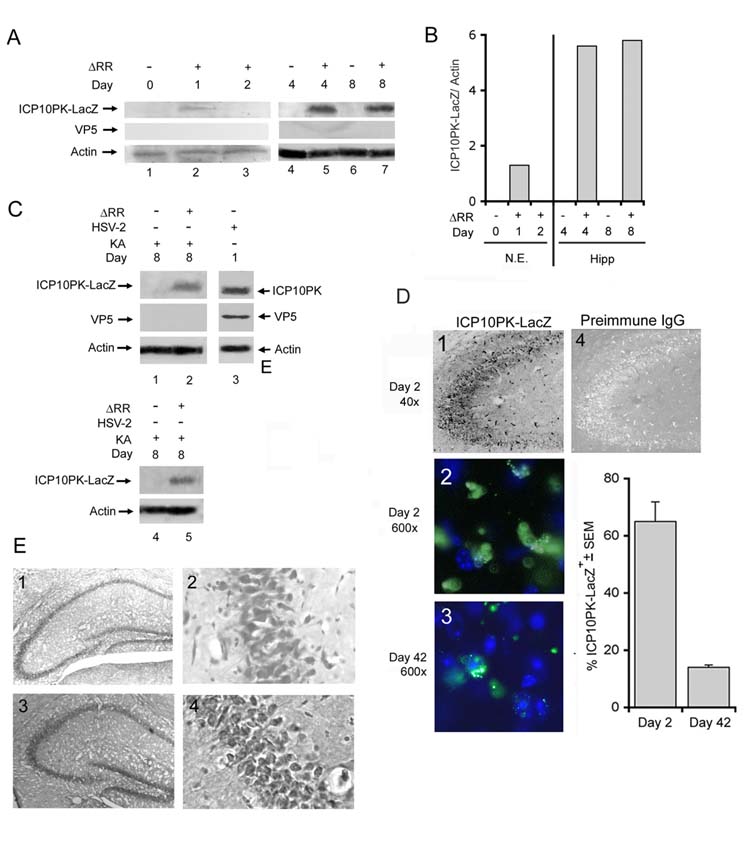
ICP10PK is expressed in the hippocampus after intranasal ΔRR instillation. (A) C57BL/6 mice were given ΔRR (3 doses; 3×106 pfu each) and extracts of nasal epithelia (lanes 1-3), and hippocampi (lanes 4-7) were immunoblotted with antibody to ICP10, VP5 and actin. Blots were stripped and reprobed between each antibody. (B) Fold increase in ICP10PK-LacZ expression was determined by densitometric scanning. Results are ICP10PK-LacZ/actin ratios (N.E.= nasal epithelia; Hipp = hippocampi). (C) Mice treated with ΔRR or PBS as in (A) were given KA (30mg/kg, i.p.) 24 hours after the last treatment (day 0). Extracts of olfactory bulbs (lanes 1, 2) and hippocampi (lanes 4, 5) collected on day 8 were immunoblotted with ICP10 antibody and the blots were sequentially stripped and re-probed with antibodies to VP5 and actin. Extracts of HSV-2 infected Vero cells (MOI=2; 24 hrs p.i.) (lane 3) served as control. (D) Brains from animals treated with ΔRR as in (A) were harvested on days 2 and 42 after the final treatment, coronally sectioned and stained by immunohistochemistry with ICP10 antibody (panel 1) or preimmune IgG (panel 4) or with FITC-conjugated ICP10 antibody (panels 2, 3). The % staining cells in the latter sections was calculated relative to the total number of cells determined by DAPI in 3 randomly selected fields of 29μm2 (at least 250 cells) from 5 serial sections for all animals. Results are % ICP10PK-LacZ + cells ± SEM. Similar results were obtained in Sprague Dawley rats. (E) Brains from animals treated with PBS (panels 1,2) or ΔRR (panels 3,4) as in (A) were harvested on day 42, coronally sectioned and thionin stained.
ICP10PK was still expressed in the hippocampus on day 42 p.i., albeit in a smaller number of cells (14 ± 1% and 65 ± 7% positive cells on days 42 and 2, respectively) (Fig. 3D, panels 2,3). We conclude that this decrease is not due to toxicity resulting from residual expression of viral genes, because the 42-days old sections were indistinguishable from those obtained from mock-infected animals by thionin staining (Fig. 3E), late viral genes (viz. VP5) were not expressed, and clinically evident ill effects were not seen at any time after ΔRR infection. Collectively, the data indicate that ICP10PK delivered by intranasal instillation of ΔRR gains access to the hippocampus, where it is expressed for a relatively long time (at least 42 days) in the absence of virus replication or any evidence of neuronal toxicity.
Intranasally delivered ΔRR prevents KA induced seizures
Systemic KA injection causes epileptiform seizures which propagate from the CA3 to the CA1 field and other limbic structures, and are followed by a pattern of neuronal cell loss which is similar to that seen in patients with temporal lobe epilepsy [16]. We used both rats (Sprague Dawley) and mice (C57BL/6) in order to examine whether ΔRR prevents KA-induced seizure development, because the disease may be somewhat different in the two animal models [17, 18]. Three inoculations of ΔRR (3 and 5×106 pfu each for rats and mice, respectively) were given intranasally, as described in materials and methods. KA was administered 24 hrs after the last virus inoculation. Animals, mock-treated with PBS or given the replication incompetent HSV-2 mutant ΔPK, which is deleted in ICP10PK [5, 11], served as controls. Clinical symptoms were seen with mock or ΔPK treatment within 30 minutes of KA administration and there was no difference between rats and mice. The animals were catatonic and immobilized. At the end of one hour, they progressed to more severe clinical symptoms. While the groups averaged a score of 2 on the clinical scale, clinical response was variable, with individual animals showing severe seizures. Tonic-clonic activity was seen in 20% of the ΔPK treated rats, 40% of the PBS treated rats, 33% of the ΔPK treated mice, and 40% of the PBS treated mice. Sustained tonic-clonic seizure activity and an increase in the associated behavioral symptoms were seen with time post KA administration, with 100% of the rats and 80% of the mice exhibiting tonic-clonic seizure activity (behavioral scale = 4) at 3 hrs after KA. Symptoms began to abate after 5 hours and all animals were symptom-free by 12 hrs after treatment. By contrast, ΔRR treated animals never progressed beyond a score of 1 on the clinical scale, and the symptoms completely resolved between 2 and 3 hours after KA administration. Not one of the ΔRR-treated animals displayed tonic-clonic seizure activity (Fig. 4). The data indicate that ΔRR has neuroprotective activity in vivo, and it is mediated by ICP10PK.
Fig. 4.
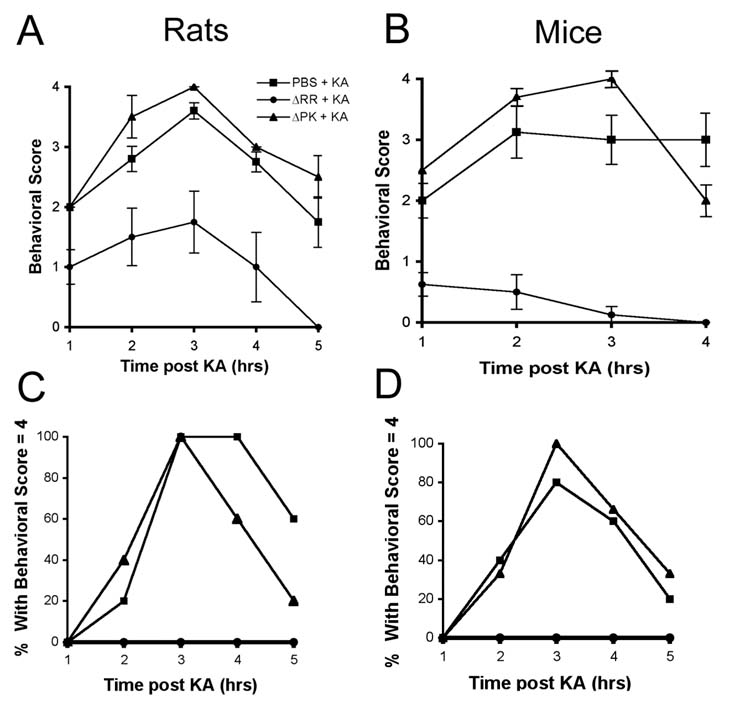
Intranasally delivered ΔRR protects from KA induced seizures. Sprague Dawley rats and C57BL/6 mice were given 3 intranasal doses of ΔRR or ΔPK (5×106 pfu and 3×106 pfu each for rats and mice, respectively) or PBS, and given of KA (15mg/kg and 30mg/kg for rats and mice, respectively) 24 hrs later by i.p. injection. They were examined for behavioral changes for 5 hours and rated on a scale of: 0, normal; 1, catatonic staring and immobilization; 2, ‘wet-dog shakes’, abnormal ambulation, stretching of limbs; 3, rearing and falling behavior; 4, tonic-clonic seizure activity; 5, death. Average behavioral score ± SEM is presented for each hour of observation, for rats (A) and mice (B). The % animals in each treatment group experiencing a behavioral score = 4 at any time during the observation period is shown for rats (C) and mice (D).
ΔRR prevents KA-induced neuronal loss, formation of reactive oxygen species (ROS), astrogliosis and microglia activation
Previous studies had shown that KA induces neuronal loss primarily in the CA1 field in rats [17], and in the CA3 field in mice [18]. Having seen that ΔRR has clinical activity, we wanted to know whether it is associated with the prevention of neuronal loss in these fields. Brains from PBS- or ΔPK-treated animals that had experienced seizures with clinical scores of at least 3, and their ΔRR-treated matched pairs (clinical scores = 1 or less) were collected at 24 hrs after KA administration and stained with thionin, that recognizes the Nissl substance in neurons [19]. Cell counts were done in the CA1 (rats) or CA3 (mice) fields, consistent with the respective lesion sites [17, 18]. As shown in Fig. 5A,B for the rat model, significant (p<0.001) neuronal loss was seen in the mock- (58 ± 2.5 %) and ΔPK- (62 ± 3.4 %), but not ΔRR-treated brains at 2 days (10 ± 4.6 %) or 42 days (14 ± 1.2 %) after treatment. The absence of neuroprotective activity in ΔPK is not due to its failure to reach the hippocampus, because staining with ICP10 antibody indicated that the PK deleted ICP10 protein was expressed as well as ICP10PK-LacZ (Fig. 5C).
Fig. 5.
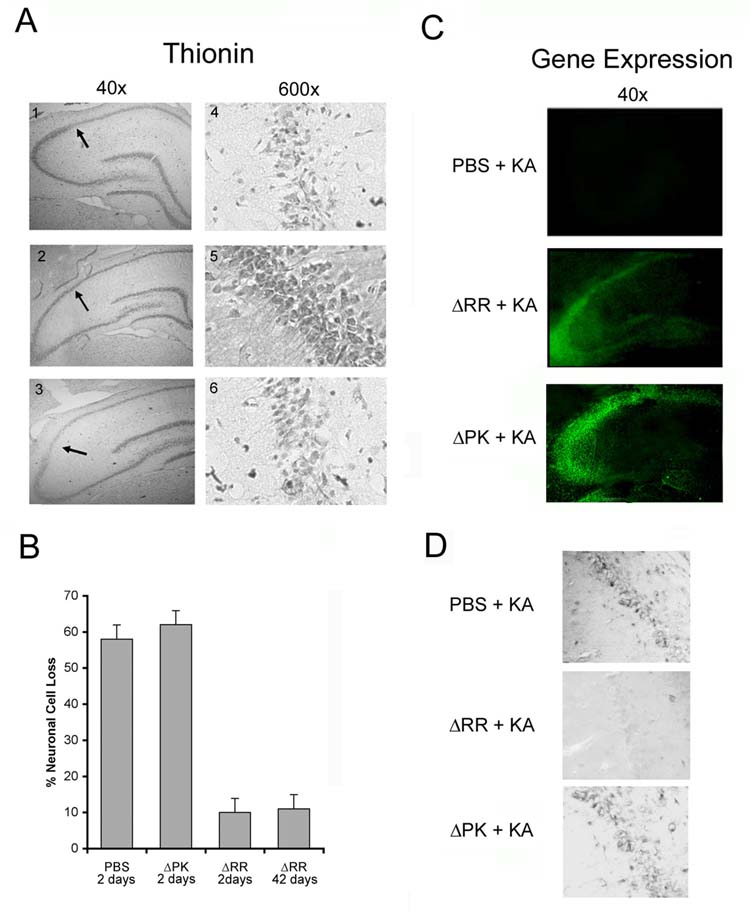
ΔRR prevents KA-induced neuronal loss and oxidative stress. Sprague Dawley rats were treated with PBS (panels 1,4) ΔRR (panels 2,5), or ΔPK (panels 3,6) and given KA as in Fig. 2. Coronal sections of brains collected 2 days later were stained with thionin (A). The numbers of neurons were counted in 3 randomly selected fields of 29μm2 (at least 250 cells) from 5 serial sections for all animals and the data are expressed as % neuronal loss ± SEM relative to untreated brains (B). Duplicate sections were stained with FITC-conjugated ICP10 antibody (C). To visualize oxidative stress, coronal sections were stained with NITT (D). Similar results were obtained in mice for the CA3 region.
Previous studies had shown that KA-induced neurodegeneration is associated with the formation of ROS and the generation of inflammatory responses related to reactive astrocytes (astrogliosis) and activated microglia [20]. We conclude that, at least in part, protection is associated with the inhibition of reactive oxygen species (ROS), because brain sections from ΔRR treated animals given KA did not stain with antibody to nitrotyrosine (NITT), a marker of oxidative stress [21], while staining was seen in the PBS or ΔPK-treated animals (Fig. 5D). Immunoblotting with antibody to glial fibrillary acid protein (GFAP), a marker of astrocyte activation [22] indicated that KA induced a transient increase in GFAP levels in hippocampi from mock- or ΔPK-, but not ΔRR-treated animals (Fig. 6A). Moreover, hippocampal sections from ΔRR-treated mice did not stain with antibody to the inflammatory molecule TNFα nor the complement receptor CD11b, a marker of activated microglia [18]. This is in contrast to the KA-exposed mock-treated animals, which evidenced increased staining for both TNFα and CD11b, peaking on day 4 post-treatment (Fig. 6B). Similar results were obtained in both animal models. The data indicate that the neuroprotective activity of ΔRR is associated with inhibition of both KA-induced ROS and astrogliosis/microglia activation. However, we still do not know whether this inhibition is the result of seizure-blocking by ΔRR, a direct effect of ΔRR on astrocytes and/or microglia, or an indirect effect of ΔRR-modulated neuronal factors that trigger astrogliosis and/or microglia activation.
Fig. 6.

ΔRR inhibits KA-induced astrogliosis and microglia activation. (A) C57BL/6 mice were treated with ΔRR, ΔPK, or PBS and given KA as in Fig. 2. Hippocampi were collected on days 0, 4, and 8 and extracts were immunoblotted with GFAP antibody, stripped and re-probed with antibody to actin. Fold increase was determined by densitometric scanning and results are presented as GFAP/actin ratio normalized to untreated. (B) Brains from mice treated with ΔRR, ΔPK, or PBS and given KA as in Fig. 2, were harvested on days 4 and 6, sectioned coronally, and serial sections were stained with antibodies to TNFα or CD11b. Day 8 sections were negative.
ICP10PK expression is regulated by AP-1 and is required for ΔRR neuroprotection
Two series of experiments were done in order to evaluate the relationship between ICP10PK expression and neuroprotection. In the first series we asked whether ICP10PK expression is regulated by activated AP-1. This question follows on previous findings that AP-1 is induced/activated by ICP10PK [23], and it, in turn, activates the ICP10 promoter [24, 25]. Extracts from olfactory bulbs and hippocampi collected at 8 days after administration of PBS or ΔRR [respectively negative and positive for ICP10PK (Fig. 3A)] were immunoblotted with antibody to phosphorylated (activated) c-Jun (P-c-Jun). One band was seen in the mock-treated tissues (Fig 7A, lane 1), but an additional, slower migrating band (hyperphosphorylated) was seen only in the ΔRR treated tissues and also in the presence of KA (Fig. 7A, lanes 2, 3). We conclude that hyperphosphorylation (activation) is due to ΔRR infection, because c-Jun was also hyperphosphorylated in neuronally differentiated PC12 cells infected with ΔRR (Fig. 7B). In these cells, ΔRR activates the ICP10 promoter (pJW24), but not a promoter mutated in the AP-1 cis-response elements (pJZ34), as determined by CAT assays (Fig. 7C). Hyperphosphorylation of c-Jun and activation of the ICP10 promoter were not seen with ΔPK (data not shown).
Fig. 7.
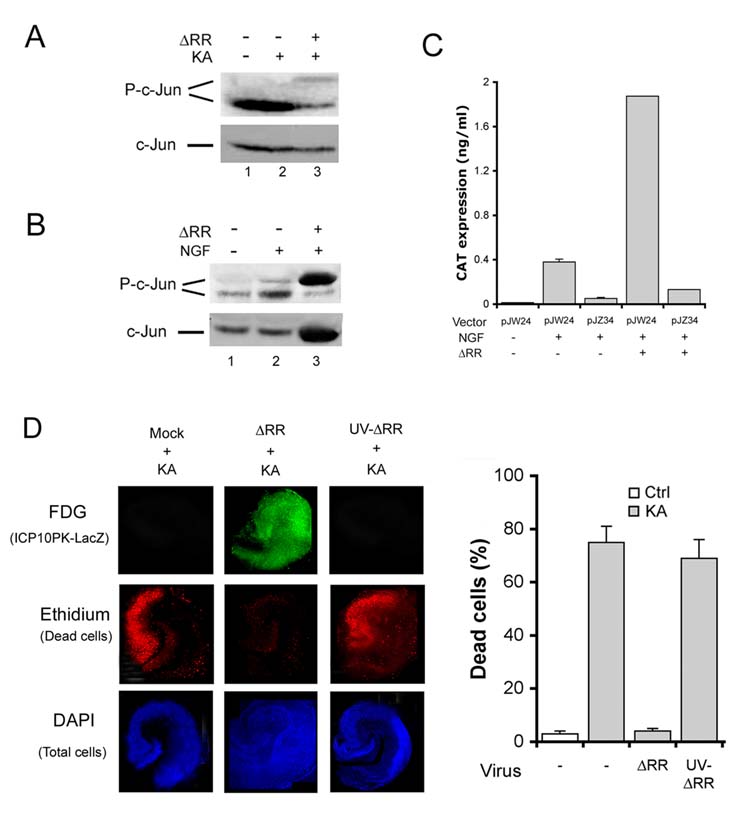
ICP10PK expression is regulated by an AP-1 feedback amplification loop. (A) Sprague Dawley rats were treated with ΔRR or PBS and given KA (15mg/kg; i.p.) at 24 hrs after the final treatment (day 0). Extracts of olfactory bulbs (day 8) were immunoblotted with P-c-Jun antibody. Blots were stripped and re-probed with antibody to total c-Jun. (B) Neuronally differentiated PC12 cells were infected with ΔRR (MOI=2) and extracts obtained at 8 hrs p.i. were immunoblotted with P-c-Jun followed by c-Jun antibodies. (C) Neuronally differentiated PC12 cells were transfected with pJW24 or pJZ34 (2 μg) using Fugene 6 transfection reagent and infected (or not) with ΔRR (MOI=2) at 24 hrs post transfection. CAT expression was measured by ELISA 24 hrs later and results are expressed as ng/ml (D). OHC treated (2hrs, 37°C) with ΔRR, UV-inactivated ΔRR (105 pfu), or mock treated with PBS and exposed to KA (5μM, 24 hrs) on day 2 were stained C12FDG (ICP10PK-LacZ expression) and ethidium homodimer (dead cells) and the number of ethidium homodimer staining cells counted as described in Materials and Methods. The % positive cells ± SEM was calculated relative to DAPI staining.
In the second series of experiments, we asked whether protection requires de novo ICP10PK expression. This question is relevant, because ICP10PK is located within the virion tegument, and is brought into the cells with the infecting virus particles [26]. OHC were infected with 105 pfu of ΔRR or UV-inactivated ΔRR (UV-ΔRR), the transcription of which is blocked due to UV-induced DNA strand breaks. They were simultaneously treated with KA (3μM; 24 hrs) and examined for cell death by staining with ethidium homodimer on day 3 p.i. Because ICP10PK is fused in frame with LacZ, its expression was determined by staining with the fluorescent LacZ substrate C12FDG. Protection was seen in ΔRR-treated OHC, with only 3 ± 1 % detectable dead cells, but a similar % dead cells was seen in the CA3 fields from UV-ΔRR (70 ± 8%) and PBS (75 ± 5%) treated OHC. The absence of neuroprotection for UV-ΔRR correlated with the absence of ICP10PK-LacZ expression (Fig. 7D). The data indicate that protection requires de novo ICP10PK expression, involving a feedback amplification loop in which ΔRR-delivered ICP10PK activates AP-1 (notably c-Jun) and it, in turn, activates the ICP10 promoter.
DISCUSSION
The salient feature of our data is the finding that intranasal delivery of ΔRR prevents KA-induced seizures and neuronal loss in rats and mice, apparently related to ICP10PK-mediated inhibition of ROS, astrogliosis and microglia activation.
ΔRR is based on HSV-2, which is less virulent in the CNS than HSV-1, does not trigger apoptosis in CNS neurons, nor cause epileptiform seizures in animals and encephalitis in immunocompetent adult humans [3-6]. ΔRR is replication incompetent in neurons and does not cause cell death in OHC. Intranasal delivery takes advantage of previous findings that HSV is efficiently transported from the site of inoculation to connected sites within the nervous system [12]. This is a desirable property for gene therapy applications, particularly where target neurons are in regions of the nervous system that are inaccessible to surgical techniques. After intranasal delivery of ΔRR, ICP10PK gained rapid (2 days) access to the hippocampus, apparently through the lateral olfactory bulb tract, with expression detected in the olfactory bulb, piriform cortex, amygdala, thalamus, hypothalamus and entorhinal cortex, a major input and output site for the hippocampus (13, 27). In the hippocampus, ICP10PK-LacZ was expressed in the CA1 and CA3 fields in the absence of late virus proteins (viz. VP5), the expression of which depends on viral DNA replication [14]. Notable was the absence of toxicity (as late as day 42 p.i.), as determined by the failure to detect neuronal loss or clinically evident ill effects. This likely reflects the failure of ΔRR to replicate in neurons, both in OHC and in vivo. Ongoing studies are designed to examine whether traffic also extends to other secondary sites along the lateral olfactory bulb tract and central pathways.
The relatively high number of ICP10PK+ hippocampal neurons in animals given a total of 9×106 to 1.5×107 pfu of ΔRR (or ΔPK) is intriguing. The precise mechanisms of nose to brain transport remain incompletely understood [28]. If access to the hippocampus is by axonal transport, the virus must cross synaptic junctions, which may require limited virus replication. We find no evidence of virus replication in any of the studied tissues, as determined both by plaque assay and VP5 expression. Virus replication in interneurons is also ruled out, because GABA+/ICP10PK+ neurons were VP5- as determined by immunofluorescent staining (data not shown). Moreover, astrocytes (GFAP+) are negative for the HSV receptor Nectin-1 and unable to support virus replication, and ΔRR does not replicate in microglia, which are resident macrophages that can migrate in the brain [29]. However, virus is retained by the microglia for up to 2 weeks, suggesting that they may act as a reservoir for ΔRR shuttling in the hippocampus (data not shown). An alternative pathway for virus traffic after intranasal delivery is the extracellular diffusion along the open intercellular clefts in the olfactory epithelium with subsequent diffusion to the olfactory bulb and CSF circulation, bypassing the blood-brain barrier [28]. While we still do not know whether ΔRR uses this pathway, nor whether pathway selection depends on the virus dose, it is important to point out that a genetically engineered and non-replicating filamentous bacteriophage has proved to be an efficient and non-toxic viral delivery vector to the brain using the intranasal administration route [30]. However, we cannot rule out non-toxic ΔRR amplification in other non-neuronal support cells. Virus titers could also be minimally amplified (still undetectable) through one round of replication in the nasal epithelial cells (20%). Indeed, ICP10PK was seen in the nasal epithelium only on day 1 p.i., at very low levels and in the absence of detectable VP5. Alternatively, since ICP10PK is a tegument protein [26], its expression in the olfactory bulb and hippocampus tissues may reflect intercellular trafficking, on its own or attached to the tegument protein VP22, which is known to traverse cell membranes independent of classical endocytosis [31, 32].
ΔRR prevented KA-induced seizures and neuronal loss in both the rat and mouse models that involved KA-induced neuronal loss primarily in the CA3 (mice) and CA1 (rats) fields, as described [17, 18]. The dose of ΔRR used in these studies is within the range previously described for transgene delivery with HSV-1 vectors [8]. At present, we do not know whether lower doses would be equally effective. However, because ICP10PK functions upstream of caspase activation [5], we are also developing second generation vectors in which LacZ is replaced by XIAP or another anti-apoptotic gene that functions downstream of caspase activation. This should achieve synergistic neuroprotective activity, for example by reducing the dose required for protection. Although ΔRR is not toxic at the doses used in these studies, such reduction is always desirable from a therapeutic standpoint. Protection was mediated by ICP10PK, because animals given ΔPK were not protected. ΔPK is a particularly stringent control for ΔRR, because: (i) it was constructed from the same HSV-2 strain, (ii) the PK-deleted ICP10 protein is driven by the same promoter and is expressed as well as ICP10PK-LacZ, and (iii) the virus is also replication compromised [11].
ICP10PK-mediated protection appears to involve inhibition of KA-induced ROS formation [33], a finding consistent with our previous reports that it inhibits oxidative stress [34]. It is also associated with inhibition of KA-induced astrogliosis and microglial activation (notably production of the inflammatory cytokine TNF-α [35]), both of which are involved in neuronal damage [18]. At present we do not know whether protection is due to the ability of ICP10PK to inhibit KA-induced seizure activity, or a direct effect on either one of these components. However, protection was lost upon UV inactivation of the ΔRR vector, suggesting that it requires sustained ICP10PK synthesis. In this context, it is important to point out that ICP10PK was expressed in the brain of the ΔRR infected animals for a relatively long time (at least 42 days) in the absence of virus replication and the expression of late virus proteins (viz. VP5). We conclude that sustained expression is likely related to the generation of an AP-1 amplification loop, because: (i) olfactory bulbs and hippocampi from ΔRR infected animals were positive for hyperphosphorylated (activated) c-Jun, and (ii) hyperphosphorylation was also seen in neuronally differentiated PC12 cells infected with ΔRR, and it was associated with activation of the ICP10 promoter. These findings are consistent with our previous reports about the relationship between AP-1 and ICP10PK expression [24, 25]. They are particularly relevant to the use of ΔRR as a neuroprotective agent because neurotoxic stimuli activate/upregulate AP-1 [25, 36]. Still, ICP10PK expression decreased with time p.i., suggesting that repeat vector administration may be necessary for long term clinical success. In this context, it should be mentioned that HSV-specific immunity should not be a major impediment towards repeated administration, because ΔRR retains the viral genes responsible for immune evasion [37, 38].
In summary, our data suggest that ΔRR is a promising platform for the treatment of chronic neurodegenerative diseases and the prevention of neuronal loss secondary to an acute insult. Ongoing studies are designed to further evaluate the validity of this interpretation, with particular emphasis on the contribution of the glial cells, the state of the viral genome in the tissues and the trafficking of ICP10PK-LacZ delivered by intranasal administration of ΔRR. Future studies will also examine the therapeutic activity of ΔRR in the kindling model of epilepsy and determine whether ΔRR treatment prevents seizure recurrences and blocks behavioral deficits due to loss of neuronal function.
MATERIALS AND METHODS
Cells and organotypic hippocampal cultures (OHC)
Vero cells were grown in minimal essential medium (MEM) with 1 mM sodium pyruvate, 2 mM L-glutamine, 100 μM non-essential amino acids and 10% fetal bovine serum (FBS). PC12 (rat pheochromocytoma) cells were grown in DMEM/F12 [Dulbecco's modified Eagle medium (Invitrogen, Carlsbad, CA) and F12 nutrient mixture (Invitrogen) at 1:1 ratio, 7.75 g/L of glucose, and 7.5 mg/L sodium bicarbonate] with 10% FBS. They were differentiated into neurons by culture (24 hrs, 37 °C) in NB/B27 medium [neurobasal medium with 1X concentration of B27 serum-free supplement (Invitrogen)] with 100 ng/ml nerve growth factor (NGF) (Invitrogen) [25]. OHC were prepared as described [10]. Briefly, hippocampi were dissected from 5-7-day old Sprague Dawley rat pups, and slices (400 μm) attached to glass coverslips in a chicken plasma clot were cultured (14 days; 36°C) on a roller-drum in medium consisting of 50% Basal Eagle Medium (Invitrogen, Carlsbad, CA), 25% Hank's balanced salt solution and 25% heat inactivated horse serum with 55.6 mM glucose and 1mM L-glutamine. Cytosine-β-D-arabino-furanoside (Sigma), uridine (Sigma), and 5-fluoro-2'-deoxyuridine (Sigma) (0.8 μg/ml) were added after 5 days in culture to inhibit glial cell growth.
Viruses
HSV-1 (F), HSV-2 (G) and the HSV-2 mutant ΔPK that is deleted in the protein kinase (PK) domain of the large subunit of ribonucleotide reductase (R1, also known as ICP10) (Fig 8A), were described [11]. To construct ΔRR, the Stu I/Bgl II fragment encompassing the ICP10PK domain was replaced with the BsA I/BamHI fragment from pSVβgal (Promega, Madison, WI), which includes the LacZ coding region and stop codon. The final construct contains ICP10PK-LacZ driven by the authentic ICP10 promoter, which is regulated with IE kinetics (independent of virus replication) and responds to AP-1 [24,25]. It was co-transfected into Vero cells with 1 μg of infectious HSV-2 DNA at 20-fold molar excess using lipofectamine (Invitrogen) and recombinant virus (ΔRR) was isolated as blue plaques after staining with X-gal (Fig. 8B). UV inactivation was as previously described [39].
Fig. 8.
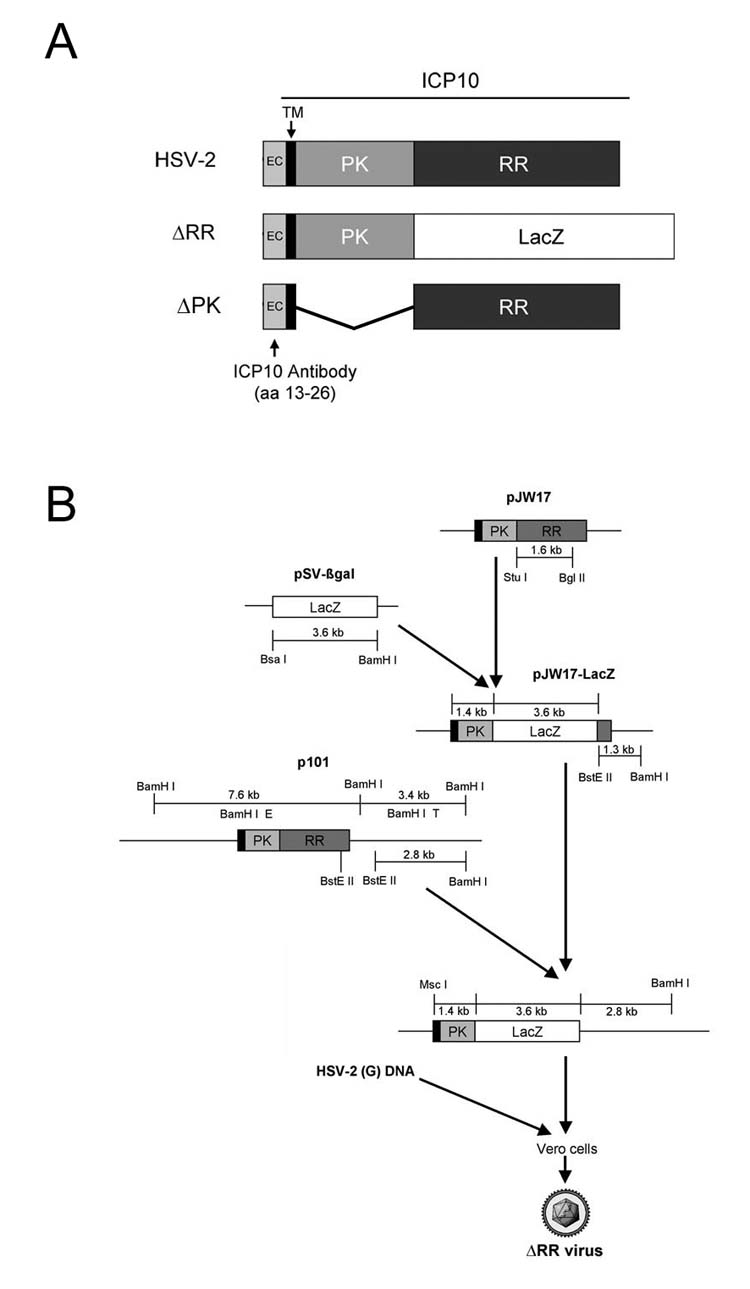
Schematic representation of the ICP10 gene and ΔRR construction. (A) In ΔRR, the ribonucleotide reductase (RR) domain was replaced with the ß-galactosidase gene (LacZ); in ΔPK, the protein kinase (PK) domain was deleted. Both mutants retain the transmembrane (TM) and extracellular (EC) domains and amino acids 13-26, which are recognized by the ICP10 antibody. (B) The RR sequences in the ICP10 expression vector (pJW17) were replaced with LacZ and flanking HSV-2 DNA sequences from plasmid p101 were added. The ICP10PK-LacZ chimera was introduced into HSV-2 by recombination and ΔRR virus was isolated as blue plaques after staining with X-gal.
Single step growth curves
Single step growth curves were done as previously described [11]. Briefly, OHC were exposed (2hrs, 36 °C) to 105 pfu of virus, washed and re-fed with fresh medium (0 hrs). Cultures collected at various times thereafter (in triplicate) were subjected to 7 freeze-thaw cycles and sonicated for 60 seconds at 25% output power using a Sonicator/Ultrasonic processor (Misonix, Inc., Farmingdale, NY) and virus titers determined by plaque assay [11]. Virus titers are expressed as the mean plaque forming units (pfu)/ml ± SEM.
Antibodies and reagents
The preparation and specificity of the rabbit ICP10 antibody were described [5, 11, 23, 26, 39]. It recognizes an epitope located within amino acids 13-26, which are retained by both ΔPK and ΔRR. The following antibodies were purchased and used according to the manufacturer's instructions: Monoclonal antibodies to GFAP (Dako Corporation, Carpinteria, CA), HSV VP5 (Virusys Corporation, Sykesville, MD), TNFα (R&D Systems, Minneapolis, MN), CD11b (Leinco Technologies, St. Louis, MO), and NITT (specific for nitrotyrosine residues) (Alpha Diagnostics, San Antonio, TX). Polyclonal antibodies to activated (phosphorylated on Ser63) c-Jun (P-c-Jun) (Cell Signaling Technology, Beverly, MA), and c-Jun (Cell Signaling Technology). FITC conjugated goat anti rabbit IgG (BD Biosciences, San Jose, CA), and Texas Red conjugated goat anti rat IgG (Jackson ImmunoResearch, West Grove, PA).
ΔRR administration and KA induced seizures
Sprague Dawley male rats and C57BL/6 male mice were obtained from Charles River Laboratories (Wilmington, MA). Animals were housed on a 12 hour light/ dark cycle with water and food supplied ad libitum. All procedures were performed in accordance with the University of Maryland, Baltimore Institutional Animal Care and Use Committee. Animals were partially anesthetized with Isoflo (Abbott Laboratories, Abbott Park, IL) and positioned on their backs. ΔRR, ΔPK or PBS were slowly and gently dropped in alternating naris with a micropipette tip in 5 μl aliquots until the total volume was reached [50μl (5×106 pfu) and 20μl (3×106 pfu) for rats and mice, respectively]. Delivery was over 15 minutes with 1 min breaks between instillation into each naris. Three inoculations were given at 24 hour intervals, with the last instillation considered day 0 p.i. KA (A.G. Scientific, San Diego, CA) was administered 24 hrs later (day 1) by i.p. injection. The route and doses of KA administration (15 mg/kg and 30 mg/kg for rats and mice respectively) were previously shown to elicit a well-characterized seizure activity followed by cell loss in the hippocampus [2, 17, 18]. Clinical response was scored as an average behavioral score for each animal every hour using the previously defined scale: 0, normal; 1, catatonic staring and immobilization; 2, ‘wet-dog shakes’, abnormal ambulation, stretching of limbs; 3, rearing and falling behavior; 4, tonic-clonic seizure activity; 5, death [17]. Results are expressed as the mean behavioral score/hour for each treatment group ± SEM. In addition, the % animals in each treatment group that experienced tonic-clonic seizure activity (score = 4) was recorded for each hour.
Cell death and LacZ expression
Cell death was determined by staining with ethidium homodimer (Molecular Probes), a fluorescent nuclear stain in the red spectrum that penetrates dead cells and increases intensity after binding to DNA. Staining was done according to manufacturer's instructions. Staining cells were counted in 5 randomly selected microscopic fields (at least 250 cells) and the % positive cells was calculated relative to the total number of cell visualized by permeabilizing the cultures with 5 % triton X-100 for 30 seconds followed by incubation with the fluorescent nuclear stain, DAPI (Sigma). Data are expressed as the mean % positive cells ± SEM. ICP10PK-LacZ expression was examined by staining with the fluorescent β-galactosidase substrate, C12-fluorescein di-β-D-galactopyranoside (FDG; Molecular Probes, Eugene, OR) according to the manufacturer's instructions, or with the FITC-conjugated ICP10 antibody, as described [5]. For assessment of neuronal cell loss in the hippocampus, brain sections were fixed with 4% PAF in PBS (30 minutes at RT) and stained with thionin (J.T. Baker, Phillipsburg, NJ) for 30 minutes. Sections were dehydrated and mounted in Permount (Fisher Scientific, Fair Lawn, NJ) and visualized by bright field microscopy.
Plasmids, transfection and CAT ELISA
Activation of the ICP10 promoter was quantified using the previously described pJW24 and pJZ34 constructs and the CAT (chloramphenicol acetyltransferase) reporter gene assay as previously described [23-25]. pJW24 contains the wild type 649 bp ICP10 promoter (−544 to +105 relative to the mRNA cap site) inserted 5' to the CAT structural gene. In pJZ34 the two AP-1 cis-response elements (TGACTCA) in the ICP10 promoter were mutated (TGAAGCA) by oligonucleotide-directed mutagenesis [25]. Constructs were transfected into neuronally differentiated PC12 cells using Fugene 6 transfection reagent (Roche) and CAT expression was assayed with the CAT ELISA (Roche), as described [25].
Immunofluorescent and immunohistochemistry staining
OHC were blocked (1 hr, 25 °C) with 10% goat serum in PBS, exposed overnight at 4 °C to ICP10-specific antibody followed by incubation (1 hr, 25 °C) with FITC-labeled anti-rabbit antibody. For staining of brain sections, animals were fully anesthetized with Isoflo and cardiac perfusion was done with PBS and 4% paraformaldehyde (PAF). The brains were removed, allowed to sit in PAF for 24 hours at 4°C, moved to 30% sucrose (in PBS) for 24 hours and embedded in Tissue-Tek OCT (Sakura Finetechnical, Tokyo, Japan). They were sectioned coronally at a thickness of 20 μm and sections were stored at −80 °C until used. For immunofluorescent staining, frozen sections were permeabilized with 0.1% triton X-100 in 0.1% sodium citrate buffer for 15 min at room temperature (RT) and blocked with 5% normal goat serum and 5% BSA (for 30 min at RT). They were incubated with primary antibody overnight at 4°C, washed in PBS with 0.1% Tween 20 and exposed to fluorochrome labeled secondary antibodies (37°C, 1 hour). After washing and mounting in Vectashield with DAPI (Vector, Burlingame, CA) the slides were visualized with a Nikon E4100 fluorescent microscope utilizing FITC (330-380nM), UV (for DAPI) (465-495nM), and Texas Red (540-580nM) cubes. For immohistochemistry, the secondary antibody was biotinylated and the immunolabeled sections were subsequently exposed to avidin conjugated alkaline phosphatase and alkaline phosphatase substrate [Dako LSAB-2 AP kit (Dako Corp)] according to manufacturer's instructions. Staining cells were counted in 3 randomly selected 29 μm2 fields (at least 250 cells) from 5 serial sections for all animals and the % positive cells was calculated relative to the total number of cells visualized by DAPI. Data are expressed as the mean % positive cells ± SEM.
Immunoblotting
Nasal epithelia, olfactory bulbs, and hippocampi dissected from animals brains were flash frozen in liquid nitrogen prior to protein extraction and lysed. Olfactory bulbs and hippocampi samples were lysed using Laemmli buffer (Sigma) supplemented with protease and phosphatase inhibitior cocktails (Sigma) while nasal epithelia were extracted in RIPA buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 % Nonidet P-40, 1 % sodium deoxycholate, 0.1 % SDS) with protease inhibitor cocktail. PC12 cells were extracted in RIPA buffer supplemented with protease and phosphatase inhibitior cocktails and sonicated for 60 seconds at 25 % output power using the Sonicator/Ultrasonic Processor (Misonix, Inc., Farmingdale, NY). Protein denaturation was at 95°C for 5 min , the entire tissue samples were loaded onto SDS-PAGE gels and the resolved proteins were transferred onto nitrocellulose membranes. When using P-c-Jun antibody, blots were blocked (1hr at RT) in 1% bovine serum albumin (fraction V) (w/v) in TN-T buffer (0.01M Tric-HCl pH7.4, 0.15 M NaCl, 0.05% Tween-20) and incubated (overnight at 4°C) in primary antibody diluted in blocking buffer. With all other antibodies 5% nonfat milk (w/v) in TN-T buffer was used as blocking buffer. After washing with TN-T buffer, the blots were incubated (1hr, RT) with secondary antibodies. Detection was with ECL Reagents (Amersham Life Science, Arlington Heights, IL) followed by exposure to Hyperfilm ECL (Amersham Life Sciences) developed in a Kodak OXmat processor. Quantification was by densitometry using the Bio Rad GS-700 Imaging Densitometer.
Statistical analyses
Analysis of variance (ANOVA) was performed with GraphPad Instat version 3.0 for Mac OS 8.6 (or higher) (GraphPad Software, San Diego, CA)
ACKNOWLEDGEMENTS
We thank Walter Benton (University of Maryland Medical Center) for help with the histology sections. These studies were supported by Public Health Service grants NS45169 (LA) and NS40338 (SMT) and by a grant from the Maryland Technology Development Corporation (LA). JML and MDG were supported by grant ES07263 from NIEHS, NIH.
REFERENCES
- 1.Natsume A, et al. Bcl-2 and GDNF delivered by HSV-mediated gene transfer act additively to protect dopaminergic neurons from 6-OHDA-induced degeneration. Exp. Neurol. 2001;169:231–238. doi: 10.1006/exnr.2001.7671. [DOI] [PubMed] [Google Scholar]
- 2.Kalwy SA, Akbar MT, Coffin RS, de Belleroche J, Latchman DS. Heat shock protein 27 delivered via a herpes simplex virus vector can protect neurons of the hippocampus against kainic-acid-induced cell loss. Brain Res. Mol. Brain Res. 2003;111:91–103. doi: 10.1016/s0169-328x(02)00692-7. [DOI] [PubMed] [Google Scholar]
- 3.Najioullah F, et al. Diagnosis and surveillance of herpes simplex virus infection of the central nervous system. J. Med. Virol. 2000;61:468–73. doi: 10.1002/1096-9071(200008)61:4<468::aid-jmv9>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 4.Perkins D, Gyure KA, Pereira EF, Aurelian L. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J. Neurovirol. 2003;9:101–111. doi: 10.1080/13550280390173427. [DOI] [PubMed] [Google Scholar]
- 5.Perkins D, Pereira EF, Aurelian L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) functions as a dominant regulator of apoptosis in hippocampal neurons involving activation of the ERK survival pathway and upregulation of the antiapoptotic protein Bag-1. J. Virol. 2003;77:1292–1305. doi: 10.1128/JVI.77.2.1292-1305.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu HM, Huang CC, Chen SH, Liang YC, Tsai JJ, Hsieh CL, Hsu KS. Herpes simplex virus type 1 inoculation enhances hippocampal excitability and seizure susceptibility in mice. Eur. J. Neurosci. 2003;18:3294–3304. doi: 10.1111/j.1460-9568.2003.03075.x. [DOI] [PubMed] [Google Scholar]
- 7.Olschowka JA, Bowers WJ, Hurley SD, Mastrangelo MA, Federoff HJ. Helper-free HSV-1 amplicons elicit a markedly less robust innate immune response in the CNS. Mol. Ther. 2003;7:218–227. doi: 10.1016/s1525-0016(02)00036-9. [DOI] [PubMed] [Google Scholar]
- 8.Monville C, et al. HSV vector-delivery of GDNF in a rat model of PD: partial efficacy obscured by vector toxicity. Brain Res. 2004;1024:1–15. doi: 10.1016/j.brainres.2004.06.082. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein DJ, Weller SK. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology. 1988;166:41–51. doi: 10.1016/0042-6822(88)90144-4. [DOI] [PubMed] [Google Scholar]
- 10.Gähwiler BH, Capogna M, Debanne D, McKinney RA, Thompson SM. Organotypic slice cultures: a technique has come of age. Trends Neurosci. 1997;20:471–477. doi: 10.1016/s0166-2236(97)01122-3. [DOI] [PubMed] [Google Scholar]
- 11.Smith CC, Peng T, Kulka M, Aurelian L. The PK domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) is required for immediate-early gene expression and virus growth. J. Virol. 1998;72:9131–9141. doi: 10.1128/jvi.72.11.9131-9141.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomlinson AH, Esiri MM. Herpes simplex encephalitis. Immunohistological demonstration of spread of virus via olfactory pathways in mice. J. Neurol. Sci. 1983;60:473–484. doi: 10.1016/0022-510x(83)90158-2. [DOI] [PubMed] [Google Scholar]
- 13.Kandel ER. Learning and Memory. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principals of Neural Science. 4th ed. Vol. 13. McGraw Hill; NY: 2000. pp. 1227–1246. [Google Scholar]
- 14.Roizman B, Sears AE. Herpes simplex viruses and their replication. In: Fields BN, Knipe DM, Howley PM, editors. Virology. 3rd ed. Lippincott-Raven Publishers; Philadelphia: 1996. pp. 2231–2295. [Google Scholar]
- 15.Sourisseau T, et al. Alteration of the stability of Bag-1 protein in the control of olfactory neuronal apoptosis. J. Cell Sci. 2001;114:1409–1416. doi: 10.1242/jcs.114.7.1409. [DOI] [PubMed] [Google Scholar]
- 16.Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;22:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- 17.Sperk G, Lassmann H, Baran H, Kish SJ, Seitelberger F, Hornykiewicz O. Kainic acid induced seizures: neurochemical and histopathological changes. Neuroscience. 1983;10:1301–1315. doi: 10.1016/0306-4522(83)90113-6. [DOI] [PubMed] [Google Scholar]
- 18.Hu RQ, Koh S, Torgerson T, Cole AJ. Neuronal stress and injury in C57/BL mice after systemic kainic acid administration. Brain Res. 1998;810:229–240. doi: 10.1016/s0006-8993(98)00863-4. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, et al. Increased microglial activation and astrogliosis after intranasal administration of kanic acid in C57BL/6 mice. J. Neurobiol. 2005;62:207–218. doi: 10.1002/neu.20099. [DOI] [PubMed] [Google Scholar]
- 20.Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol. Neurobiol. 2005;31:3–16. doi: 10.1385/MN:31:1-3:003. [DOI] [PubMed] [Google Scholar]
- 21.Penkowa M, et al. Metallothionein reduces central nerous system inflammation, neurodegeneration, and cell death following kainic acid-induced epileptic seizures. J. Neurosci. Res. 2005;79:522–534. doi: 10.1002/jnr.20387. [DOI] [PubMed] [Google Scholar]
- 22.Little AR, O'Callagha JP. Astrogliosis in the adult and developing CNS: is there a role for proinflammatory cytokines? Neurotoxicology. 2001;22:607–618. doi: 10.1016/s0161-813x(01)00032-8. [DOI] [PubMed] [Google Scholar]
- 23.Smith CC, Nelson J, Aurelian L, Gober M, Goswami BB. Ras-GAP binding and phosphorylation by herpes simplex virus type 2 RR1 PK (ICP10) and activation of the Ras/MEK/MAPK mitogenic pathway are required for timely onset of virus growth. J. Virol. 2000;74:10417–10429. doi: 10.1128/jvi.74.22.10417-10429.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu J, Aurelian L. AP-1 cis-response elements are involved in basal expression and Vmw110 transactivation of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) Virology. 1997;231:301–312. doi: 10.1006/viro.1997.8522. [DOI] [PubMed] [Google Scholar]
- 25.Gober MD, Wales SQ, Hunter JC, Sharma BK, Aurelian L. Stress upregulates neuronal expression of the HSV-2 large subunit of ribonucleotide reductase (R1; ICP10) by activating AP-1. J. Neurovirol. 2005;11:329–336. doi: 10.1080/13550280591002423. [DOI] [PubMed] [Google Scholar]
- 26.Smith CC, Aurelian L. The large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) is associated with the virion tegument and has PK activity. Virology. 1997;234:235–242. doi: 10.1006/viro.1997.8645. [DOI] [PubMed] [Google Scholar]
- 27.Sewards TV, Sewards MA. Input and output stations of the entorhinal cortex: superficial vs. deep layers or lateral vs. medial divisions? Brain Res. Brain Res. Rev. 2003;42:243–251. doi: 10.1016/s0165-0173(03)00175-9. [DOI] [PubMed] [Google Scholar]
- 28.Thorne RG, Pronk GJ, Padmanabhan V, Frey WH., 2nd Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127:481–496. doi: 10.1016/j.neuroscience.2004.05.029. [DOI] [PubMed] [Google Scholar]
- 29.Carbonell WS, Murase S, Horwitz AF, Mandell JW. Migration of perilesional microglia after focal brain injury and modulation by CC chemokine receptor 5: an in situ time-lapse confocal imaging study. J. Neurosci. 2005;25:7040–7047. doi: 10.1523/JNEUROSCI.5171-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frenkel D, Solomon B. Filamentous phage as vector-mediated antibody delivery to the brain. Proc. Natl. Acad. Sci. U. S. A. 2002;99:5675–5679. doi: 10.1073/pnas.072027199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elliott G, O'Hare P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell. 1997;88:223–233. doi: 10.1016/s0092-8674(00)81843-7. [DOI] [PubMed] [Google Scholar]
- 32.Cashman SM, Sadowski SL, Morris DJ, Frederick J, Kumar-Singh R. Intercellular trafficking of adenovirus-delivered HSV VP22 from the retinal pigment epithelium to the photoreceptors--implications for gene therapy. Mol Ther. 2002;6:813–823. doi: 10.1006/mthe.2002.0806. [DOI] [PubMed] [Google Scholar]
- 33.Milatovic D, Gupta RC, Dettbarn WD. Involvement of nitric oxide in kainic acid-induced excitotoxicity in rat brain. Brain Res. 2002;957:330–337. doi: 10.1016/s0006-8993(02)03669-7. [DOI] [PubMed] [Google Scholar]
- 34.Aurelian L. HSV-induced apoptosis in herpes encephalitis. Curr. Top. Microbiol. Immunol. 2005;289:79–111. doi: 10.1007/3-540-27320-4_4. [DOI] [PubMed] [Google Scholar]
- 35.Shinoda S, et al. Formation of a tumour necrosis factor receptor 1 molecular scaffolding complex and activation of apoptosis signal-regulating kinase 1 during seizure-induced neuronal death. Eur. J. Neurosci. 2003;17:2065–2076. doi: 10.1046/j.1460-9568.2003.02655.x. [DOI] [PubMed] [Google Scholar]
- 36.Pennypacker KR, Thai L, Hong JS, McMillian MK. Prolonged expression of AP-1 transcription factors in the rat hippocampus after systemic kainate treatment. J. Neurosci. 1994;14:3998–4006. doi: 10.1523/JNEUROSCI.14-07-03998.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahn K, et al. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 1996;15:3247–3255. [PMC free article] [PubMed] [Google Scholar]
- 38.Salio M, Cella M, Suter M, Lanzavecchia A. Inhibition of dendritic cell maturation by herpes simplex virus. Eur. J. Immunol. 1999;29:3245–3253. doi: 10.1002/(SICI)1521-4141(199910)29:10<3245::AID-IMMU3245>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 39.Perkins D, Pereira EF, Gober M, Yarowsky PJ, Aurelian L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J. Virol. 2002;76:1435–1449. doi: 10.1128/JVI.76.3.1435-1449.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]