

Abstract
Proline utilization A (PutA) from Escherichia coli is a multifunctional flavoprotein that is both a transcriptional repressor of the proline utilization (put) genes and a membrane-associated enzyme which catalyzes the 4e- oxidation of proline to glutamate. Previously, proline was shown to induce PutA-membrane binding and alter the intracellular location and function of PutA. To distinguish the roles of substrate binding and FAD reduction in the mechanism of how PutA changes from a DNA-binding protein to a membrane-bound enzyme, the kinetic parameters of PutA-membrane binding were measured under different conditions using model lipid bilayers and surface plasmon resonance (SPR). The effects of proline, FAD reduction, and proline analogues on PutA-membrane associations were determined. Oxidized PutA shows no binding to E. coli polar lipid vesicles. In contrast, proline and sodium dithionite induce tight binding of PutA to the lipid bilayer with indistinguishable kinetic parameters and an estimated dissociation constant (KD) of < 0.01 nM (pH 7.4) for the reduced PutA-lipid complex. Proline analogues such as L-THFA and DL-P5C also stimulate PutA binding to E. coli polar lipid vesicles with KD values ranging from about 3.6 – 34 nM (pH 7.4) for the PutA-lipid complex. The greater PutA-membrane binding affinity (> 300-fold) generated by FAD reduction relative to the nonreducing ligands demonstrates that FAD reduction controls PutA-membrane associations. On the basis of SPR kinetic analysis with differently charged lipid bilayers, the driving force for PutA-membrane binding is primarily hydrophobic. In the SPR experiments membrane-bound PutA did not bind put control DNA confirming that the membrane-binding and DNA-binding activities of PutA are mutually exclusive. A model for the regulation of PutA is described in which the overall translocation of PutA from the cytoplasm to the membrane is driven by FAD reduction and the subsequent energy difference (∼ 24 kJ/mol) between PutA-membrane and PutA-DNA binding.
The multifunctional proline utilization A (PutA) flavoprotein from Escherichia coli and Salmonella typhimurium is both a transcriptional repressor and a membrane associated enzyme (1-5). PutA regulates the put regulon, which contains the genes putA and putP (Na+/proline transporter) that are transcribed in opposite directions from the put control intergenic DNA (1, 5-7). PutA represses transcription of the put genes by binding to specific sequences in the put control DNA region (2, 8). The enzyme action of PutA coordinates step-wise proline dehydrogenase (PRODH) and Δ1-pyrroline-5-carboxylate dehydrogenase (P5CDH) activities, allowing bacteria to utilize proline as a sole nitrogen and energy source. The PRODH active site couples the oxidation of proline to the reduction of noncovalently bound flavin adenine dinucleotide (FAD) to form Δ1-pyrroline-5-carboxylate (P5C) (9). Reduced FAD is subsequently oxidized by an electron acceptor in the membrane to regenerate oxidized FAD and complete the PRODH catalytic cycle (3, 10). Next, P5C is hydrolyzed to γ-glutamic acid semialdehyde followed by a NAD-dependent oxidation to glutamate catalyzed by the P5CDH domain (3, 11).
PutA has been most extensively studied from E. coli and S. typhimurium. From these bacteria, PutA is a polypeptide of 1320 amino acids with 91.3% sequence identity (5). PutA from E. coli, the subject of this study, purifies predominately as a dimer with a molecular mass of 293 kDa (2). The PRODH and DNA binding domains of PutA have been identified by structural and biochemical characterization of truncated PutA proteins (12-14). The X-ray crystal structure of a truncated form of E. coli PutA containing residues 1-669 (PutA669) complexed to the competitive inhibitor L-lactate was solved to 2.0 Å resolution (13). The crystal structure included residues 87-612 of PutA669 and revealed that the PRODH domain is a β8α8 barrel comprised of residues 261-612, with the FAD bound at the C-terminal ends of the β-strands of the barrel. The DNA-binding activity of PutA involves N-terminal residues 1-47 which also appear to be important for PutA dimerization as truncation mutants (e.g., PutAΔ85) that lack these residues purify as a monomer (14). Primary structure analysis predicts the P5CDH domain includes residues 650-1130 while the location of the membrane binding domain is still unknown (5).
The availability of proline governs the intracellular location of PutA and whether PutA functions as a transcriptional repressor or a membrane-bound enzyme (10, 15-18). The general model is that in the absence of proline, PutA accumulates in the cytoplasm and represses transcription of the put regulon while in the presence of proline, PutA associates with the membrane. Because proline reduces FAD, the FAD redox state is thought to have a key role in regulating PutA. A redox dependent mechanism for PutA-membrane binding was first proposed by Wood (1987) after observing enhanced steady-state kinetics of membrane association with proline and with NADH and D-lactate under anaerobic conditions (10). A redox mechanism was also implied from sedimentation experiments in which PutA binding to membrane vesicles and reduction of PutA-bound FAD were shown to occur at similar concentrations of proline (16). Surber and Maloy (1999) have also reported that PutA-membrane binding is dependent on proline reduction of FAD from studies with PutA from S. typhimurium using synthetic lipid vesicles and sucrose step-gradient centrifugation (15). Corresponding with the aforementioned studies, Wood and Brown (1993) showed that PutA undergoes a conformational change in the presence of proline that is coincident with PutA-membrane binding (16). Thus, proline induces a conformational change in PutA that is critical for enhancing PutA-membrane associations.
We seek to dissect the roles that proline binding and FAD reduction have in switching the intracellular location and function of PutA. Previously, controlled potentiometric proteolysis of PutA showed that FAD reduction is the chief determinant of conformational changes in PutA (19). However, substrate analogues that do not reduce FAD, such as L-tetrahydro-2-furoic acid (THFA), also induce a conformational change but the resulting structure is distinct from reduced PutA (19). Thus, three different conformers of PutA have been identified which correspond to oxidized PutA (1), substrate analogue bound PutA (2), and reduced FAD (3). In addition to conformational studies, we have shown that substrate binding and the FAD redox state have little influence on PutA-DNA interactions. The dissociation constant of the PutA-DNA complex increases by only 2-fold in the presence of proline or upon reduction of the PutA-bound FAD (20). The nominal influence of FAD reduction on PutA-DNA affinity is consistent with previous studies that have shown proline dehydrogenase activity and membrane binding are necessary for the induction of put gene expression (15, 17, 21, 22).
In this report, we distinguish the impact that substrate binding and FAD reduction have on PutA-membrane associations. The effects of proline, FAD reduction, and ligand binding to the PRODH active site on the kinetics of PutA membrane-binding are explored using surface plasmon resonance (SPR) technology. SPR technology is well suited to measure the interactions of macromolecules, because association and dissociation of protein to the immobilized lipid on the surface of the sensor chip can be monitored in real time (23-26). Kinetic measurements of PutA binding to the model lipid bilayers clearly show that FAD reduction alone regulates PutA membrane-binding, proving Wood's (1987) original redox dependent hypothesis (10). Our work strengthens previous studies which proposed that the disruption of the PutA-DNA complex in the cytoplasm and therefore activation of the put genes is due to favorable binding of reduced PutA to the membrane (15, 16, 21). Surprisingly, LTHFA and other ligands also induce membrane binding but the membrane associations appear to be insufficient to meaningfully perturb the PutA-DNA complex. Altering the overall charge of the lipid surface has no affect on proline reduced PutA-membrane binding suggesting PutA-membrane interactions are mainly hydrophobic. We believe the described experimental system closely mimics cellular PutA-membrane interactions and provides unique insights into the regulation of PutA function.
Materials and Methods
Chemicals. E. coli polar lipid extracts, phosphatidyl glycerol (PG), and 1, 2-dioleoyl-sn-glycerol-3-ethylphosphocholine (EDOPC) were purchased from Avanti Polar Lipids Inc. and used without purification. 3-[(3-cholamidopropyl)dimethylammonio]-1-propane sulfonate (CHAPS) was purchased from Novachem Biochem Corp. The L1 sensor chip and buffers for preparation of the surface were from Biacore AB (Piscataway, NJ). DL-P5C was synthesized as previously described and quantitated using o-aminobenzaldehyde (27, 28). The 21-bp synthetic oligonucleotide 5'-TTTGCGGTTGCACCTTTCAAA-3' and its complement were purchased from Integrated DNA Technologies. The fluorescently labeled 21-bp oligonucleotide was purchased from LI-COR, Inc. The 21-bp duplex DNA was prepared by annealing the complementary oligonucleotides in buffer (10 mM Tris, pH 8.0, 50 mM NaCl, 1 mM EDTA) by first heating at 95 °C for 5 min and then gradually cooling the sample to room temperature. The put intergenic DNA (419 bp) was prepared as described previously using genomic DNA from E. coli strain JT31 (20). All other buffers and chemicals were purchased from Fisher Scientific or Sigma-Aldrich Inc. All experiments used NanoPure water.
Preparation of PutA and Lipid Vesicles. PutA was overexpressed from the pET14b-PutA construct in E. coli strain BL21 DE3 pLysS and purified as previously described for wild-type PutA (19). The N-terminal hexahistidine tag was retained after purification. Size exclusion chromatography (Superdex-200 column) was then used to separate the dimeric and monomeric forms of PutA. The dimeric form was collected and used for all of the experiments. Subsequent analysis of the PutA dimer by size exclusion chromatography showed that the PutA preparation remained in the dimeric form. The concentration of PutA was determined using the BCA method (Pierce) with bovine serum albumin as the standard and spectrophotometrically using a molar extinction coefficient at 451 nm of 12,700 M−1 cm−1 (20).
In this work small unilamellar vesicles (SUV) of various phospholipids were used to prepare model bilayer membrane surfaces on the L1 sensor chip. The phospholipids were received as chloroform solutions. To prepare lipid vesicles, the lipid solutions were dried by carefully flushing with N2 gas and resuspended in 10 mM HEPES containing 150 mM NaCl at pH 7.4 (HEPES-N buffer) to a concentration of 20 mM lipid. The lipid suspensions were then subjected to ten freeze-thaw cycles and stored frozen in liquid nitrogen. Upon thawing, the lipid suspension was diluted 100-fold with HEPES-N buffer to a final concentration of 0.2 mg/ml, except EDOPC lipid, which was prepared at a final concentration of 0.1 mg/ml. SUV of all phospholipids were prepared by passing the lipid suspension through a 100 nm polycarbonate filter thirty times using a LiposoFast microextruder. This extrusion technique has been shown to generate unilamellar vesicles from a variety of phospholipids (29). EDOPC, a cationic analogue of phosphatidylcholine, also forms stable unilamellar vesicles and has been used previously to study peptide-membrane interactions (30, 31). All SUVs were prepared fresh on the day of the experiment. The lipid vesicles were visualized by electron microscopy (HITACHI, Japan) and shown to have an average diameter 100-200 nm.
Immobilization of Lipid Vesicles on L1 Chip. PutA lipid vesicle binding experiments were performed on a Biacore 2000 instrument at 25 °C in the Genomics Core Facility in the Center for Biotechnology at the University of Nebraska. HEPES-N buffer was used as running buffer in all SPR experiments. The L1 sensor chip was washed with 20 mM CHAPS at 30 μl/min for 2 min prior to loading any lipid vesicles. Previously, Erb et al. (2000) demonstrated that lipid vesicles loaded onto a L1 sensor surface fuse together to form a lipid bilayer (32). For the lipid vesicles made from E. coli polar extracts, the sensor surface was coated with 40 μl of lipid vesicles at 5 μl/min. For PG and EDOPC lipid vesicles, the sensor surface was coated with 15 and 10 μl lipid vesicles at 30 μl/min, respectively. After coating with lipid vesicles, the surface was washed with 10 mM NaOH which was injected at a flow rate of 60 μl/min for 1 min to wash away multilayer and loosely immobilized vesicles on the sensor surface. The total response (RU) for the lipid vesicle loading after NaOH washing was about 4500 RU, 6000 RU and 1000 RU for the E. coli polar lipid extract, EDOPC, and PG, respectively. Finally, 40 μl of fatty acid free BSA (0.1 mg/ml) was injected at a flow rate 5 μl/min to block possible nonspecific protein-lipid interactions. The response observed from the BSA injection was about 1200 RU and 100 RU for reference and lipid vesicle coated sample flow cells, respectively. In addition to the aformentioned surface preparation, the surfaces were tested prior to each PutA-binding experiment by injecting 60 μl of BSA (0.1 mg/ml) at 30 μl/min followed by an identical injection of HEPES-N buffer. For a well-prepared reference cell, BSA and buffer injections cause response signals < 5 RU.
Surface Plasmon Resonance Analysis. PutA (13.7 μM dimer) in 10 mM HEPES buffer with 50 mM NaCl (pH 7.4) was diluted to appropriate concentrations using HEPES-N buffer. All buffers were degassed and filtered by passing through a 0.22 μm filter prior to binding experiments. The effects of L-proline, L-THFA, L-lactate, and DL-P5C on PutA-lipid binding were tested by adding the ligands to the PutA sample at a final concentration of 5 mM for 15 min prior to injection. To form the PutA-DNA complexes, put control DNA and 21-bp duplex DNA were added to the PutA samples at final concentrations of 300 nM and 1.8 μM, respectively, prior to injection. To test the effect of FAD reduction alone, 10 mM sodium dithionite was added to the PutA sample under anearobic conditions. Unlike proline reduced PutA, PutA reduced with dithionite is readily reoxidized in the presence of air (t½ < 0.5 min), which necessitated careful anaerobic techniques (20). The PutA sample was first made anaerobic by applying several cycles of argon and vacuum. Next, sodium dithionite was added to the PutA sample under a nitrogen atmosphere in a glove box (Belle Technology). Reduced PutA samples were then put in a vial and sealed by a rubber cap to keep PutA in the reduced state. The running buffer was also made anaerobic by applying several cycles of argon and vacuum. During the Biacore experiment, the HEPES-N running buffer was continuously flushed with nitrogen to prevent oxygenation of the buffer.
In all kinetic experiments, 120 μl of the PutA samples were injected at a flow rate 60 μl/min. The association and dissociation phases were monitored for 120 and 300 s, respectively. Different flow rates from 20 – 80 μl/min were also used to confirm there were no mass transfer effects during the kinetic experiments. After each protein injection, PutA and the lipid vesicles were removed from the L1 surface by applying 60 μl of CHAPS at 30 μl/min. For the simultaneous PutA-DNA and PutA-membrane binding experiments, slower flow rates from 5 to 20 μl/min were used to allow enough time for PutA to bind to the DNA or to the membrane.
The sensorgrams of PutA-lipid associations were analyzed by Biaevaluation 4.1 software. Changes in refractive index due to buffer changes were subtracted prior to kinetic analysis. Global fitting to a 1:1 Langmuir PutA-membrane binding model
was used to calculate all the kinetic and equilibrium binding constants. The association and dissociation phases of the binding reaction are described by the following equations:
where R is the time dependent response unit, ka is the association rate constant, kd is the dissociation rate constant, C is the concentration of analyte, Rmax is the theoretical binding capacity, t0 is the time at the start of the association or dissociation phase, and RI is the refractive index change of the bulk samples. The dissociation constant KD was calculated from the following equation:
DNA Binding Assays. The binding of PutA to the 21-bp oligonucleotide duplex DNA was analyzed by gel mobility shift assays using a synthetic oligonucleotide that was 5' end-labeled with IRdye-700 (LI-COR, Inc.). The gel-shift assays were performed as previously described except that HEPES-N buffer was used to mimic the conditions of the SPR experiments (14). Calf thymus competitor DNA (100 μg/ml) was added to the binding mixtures to prevent nonspecfic PutA-DNA interactions. The PutA-DNA mixtures were seperated using a polyacrylamide (8%) native gel at 4 °C and visualized with a LI-COR Odyssey Imager as described (14).
Results
Proline-Dependent Membrane Binding. The phospholipid bilayer model system was first tested by assessing PutA-lipid interactions in the absence and presence of proline. Previously, proline was observed to cause about a 2-fold increase in PutA-membrane binding in sedimentation and sucrose step-gradient centrifugation experiments using inverted membrane vesicles (15, 16). Sucrose-step gradient assays also showed proline induces PutA binding to synthetic lipid vesicles (15). Thus, it is apparent that proline induces PutA-membrane binding and that association is generally comprised of protein-phospholipid interactions. Our initial SPR experiments with E. coli polar lipid vesicles confirmed these earlier results but showed a more striking proline induction of PutA-membrane binding than was previously described. Figure 1A shows that in the absence of proline, PutA generates no repsonse indicating that oxidized PutA does not bind to the lipid bilayer surface. After the injection of PutA is complete, the sensorgram response returns to the intial value. In stark contrast, a large response was observed for PutA in the presence of 5 mM proline (Figure 1A). Clearly, proline promotes PutA binding to the lipid vesicles. Furthermore, after injection of PutA with proline is complete and the dissociation phase begins, no significant decrease in the sensorgram signal is observed. This unique sensorgram suggests that the proline-dependent binding of PutA on the lipid bilayer is nearly irreversible or the dissociation rate is too slow to be detected by SPR.
Figure 1.

SPR sensorgrams showing proline and ligand induced PutA-lipid binding. Panel A: Sensorgrams of PutA (20 nM) in the oxidized state (a) and in the presence of 5 mM proline (b) binding to a lipid bilayer of E. coli lipid polar extracts. The arrows indicate the beginning and end of the protein sample injection. In sensorgram a, the rapid change in response units (RU) at the injection start point is due to a refractive index change of the sample, not by protein binding to the lipid bilayer. The RU returned to the initial value after the injection of the sample was complete. Panel B: Oxidized PutA (80 nM) in the presence of 5 mM L-THFA ( ○ ), 5 mM L-lactate ( □ ) and 5 mM DL-P5C ( ◇ ) was injected at 60 μl/min for 120 s onto a L1 chip coated with E. coli polar lipid vesicles. The dissociation phase was observed by the flow of HEPES-N buffer at 60 μl/min for 300 s.
The kinetics of PutA-lipid binding in the presence of proline were determined by measuring the senorgram response with a series of PutA concentrations ranging from 2.5 to 40 nM (Figure 2A). Because proline was kept in excess to PutA and proline reduced PutA reacts slowly with air (t1/2 ∼ 125 min), the FAD cofactor remained in the reduced state during the entire kinetic analysis (20). A global fit of the binding data with a 1:1 Langmuir isotherm by the Biacore evaluation software was used to estimate the kinetic constants. The association (ka) and dissociation (kd) rate constants were determined to be 1.4 (± 0.2)×105 M−1s−1 and 10−6 − 10−10 s−1, respectively. An upper limit for the apparent equilibrium dissociation constant (KD) for the PutA-lipid complex is thus < 0.01 nM (Table 1). Since the limit of kd values measured by Biacore 2000 is 10−6, the proline-dependent binding of PutA to the lipid bilayer is considered to be irreversible, thus, the apparent KD has a only a relative meaning for this specific study (33). Attempts to release PutA from the lipid surface by injecting an artifical electron acceptor such as phenazine methosulfate (5 mM) onto the surface of the PutA-lipid complex were unsuccessful. Therefore, PutA appears to be trapped on the lipid bilayer even under oxidizing conditions.
Figure 2.

SPR sensorgrams of the association and dissociation kinetics of proline and sodium dithionite reduced PutA-lipid binding. Panel A: From bottom to top, increasing concentrations of PutA (2.5, 5, 10, 20, and 40 nM) in the presence of 5 mM proline (HEPES-N buffer, pH 7.4) were injected onto a L1 chip coated with E. coli polar lipid vesicles. Panel B: From bottom to top, increasing concentrations of PutA (2.5, 5, 10, 20, and 40 nM) in the presence of 10 mM sodium dithionite (HEPES-N buffer, pH 7.4) were injected onto a L1 chip coated with E. coli polar lipid vesicles. Association phase (a) corresponds to the injection of PutA at 60 μl/min for 120 s and the dissociation phase (b) corresponds to the flow of HEPES-N buffer at 60 μl/min for 300 s. The data were fit by global analysis to a 1:1 Langmuir binding isotherm. Signals from the control surface have been subtracted.
Table 1.
Kinetic and Equilibrium Binding Constants of PutA Binding to E. coli Polar Lipid Vesicles Determined on Biacore 2000.
| Supplement | ka (M−1s−1) | kd (s−1) | KD (nM) | χ2 |
|---|---|---|---|---|
| None | No binding | |||
| L-proline | 1.4 (± 0.2) ×105 | < 10−6 | < 0.01 | 0.8 |
| L-THFA | 2.3 (± 0.32) ×104 | 7.8 (± 0.93) ×10−4 | 34 ± 0.5 | 1.4 |
| L-lactate | 2.8 (± 0.13) ×104 | 3.1 (± 0.22) ×10−4 | 11 ± 0.5 | 0.6 |
| DL-P5C | 7.0 (± 0.4) ×104 | 2.5 (± 0.6) ×10−4 | 3.6 ± 0.7 | 3.3 |
| sodium dithionite | 1.7 (± 0.14) ×105 | < 10−6 | < 0.01 | 1.1 |
Kinetic constants were determined in HEPES-N buffer (pH 7.4) at 25 °C.
Effects of Ligand Binding and FAD Reduction on PutA-membrane Interactions. We next sought to distinguish the effects of ligand binding (PutA conformation 2) and FAD reduction (PutA conformation 3) on PutA-membrane binding. To mimic substrate binding, we complexed PutA with L-THFA, a nonreducing substrate analogue and a competitive inhibitor of PRODH activity in PutA (Ki = 0.2 mM) (34). We also tested other nonreducing compounds such as the competitive inhibitor sodium L-lactate (Ki = 1.4 mM) and the product of proline oxidation, P5C (34). In these experiments, PutA was incubated with 5 mM of L-THFA, L-lactate, and DL-P5C for 15 min prior to injection onto the lipid bilayer surface. Figure 1B shows representative sensorgrams of PutA-lipid binding in the presence of each molecule. Each of the ligands induced PutA-membrane binding. The kinetic parameters of PutA-lipid binding in the presence of each ligand were measured as described above and are summarized in Table 1. The estimated KD values for the PutA-lipid complex in the presence of L-THFA, L-lactate, and DL-P5C varied by 10-fold within the range of 3.6 -34 nM. Thus, complexation of substrate/product analogues and inihibitors promotes analogous PutA binding to the lipid bilayer. However, marked differences in the lipid binding properties of PutA in the presence of ligands and proline are evident. First, ligand induced PutA-lipid binding displays reversible behavior with a noticeable dissociation phase once the injection of the PutA-ligand complex is complete. Second, the KD values (3.6-34 nM) of the PutA-lipid complex in the presence of ligands is > 300-fold higher than the upper limit of the KD value (< 0.01 nM) estimated for the PutA-lipid complex in the presence of proline (Table 1).
To study how reduction of FAD affects PutA-membrane binding, we performed SPR experiments under anearobic conditions in the presence of 10 mM sodium dithionite. Figure 2B shows that reduction of the PutA-bound FAD by sodium dithionite promotes PutA binding to the lipid bilayer surface. Interestingly, the lipid-binding behavior of PutA in the presence of sodium dithionite appears similar to that observed in the presence of proline (Figure 2). The kinetic parameters for PutA-lipid binding in the presence of dithionite and proline are indistinguishable (see Table 1). In addition, after injection of dithionite reduced PutA is complete, no dissociation of PutA is observed from the lipid surface. The reoxidation of dithionite reduced PutA with air-saturated buffer has a t1/2 ∼ 5 min, thus, dithionite reduced PutA is anticipated to readily oxidize upon injecting air-saturated HEPES-N buffer. However, no significant dissociation of PutA was observed under conditions predicted to reoxidize lipid-bound PutA. The close similarities of the PutA-membrane binding properties in the presence of proline and dithionite demonstrate that FAD reduction alone can induce PutA-lipid binding.
PutA Binding to Differently Charged Lipids. SUVs prepared from E. coli polar lipid extracts are negatively charged overall with about a 6:2:1 ratio (67%:23%:10%) of zwitterionic PE lipid, negatively charged PG lipid, and negatively charged cardiolipin, respectively. To study the role of phospholipid charge in PutA-lipid binding, we prepared bilayer surfaces that were either negatively charged (PG) or postively charged (EDOPC). Lipid-binding of proline reduced PutA was observed with both of the differently charged lipid bilayers. Figure 3A shows the sensorgram of proline reduced PutA binding to positively charged EDOPC lipids. The lipid binding of proline reduced PutA to the negatively and positively charged lipids is characterized by similar KD values (see Table 2) and no significant dissociation of PutA from the lipid surface. Thus, it appears proline-reduced PutA-membrane binding is primarily hydrophobic in nature and is not significantly influenced by electrostatic interactions. Hydrophobic interactions with the membrane are also suggested by previous detergent partitioning studies that demonstrated the relative hydrophobicity of PutA increases upon FAD reduction (15, 21).
Figure 3.

Sensorgrams of PutA binding to positively charged lipid vesicles. Panel A: Sensorgrams of proline (5 mM) reduced PutA (10, 20, 40, 80, 160 nM, bottom to top) injected onto a L1 chip coated with EDOPC lipid vesicles. Panel B: Sensorgrams of oxidized PutA (20, 40, 80, 160, 320 nM, bottom to top) injected onto a L1 chip coated with EDOPC lipid vesicles. For all sensorgrams, the association phase (a) corresponds to the injection of the PutA sample at 60 μl/min for 120 s and the dissociation phase (b) corresponds to the flow of HEPES-N buffer at 60 μl/min for 300 s. The data were fit by global analysis to a 1:1 Langmuir binding isotherm. Signals from the control surface have been subtracted.
Table 2.
Kinetic and Equilibrium Binding Constants of PutA Binding to Different Lipid Vesicles.
| PutA and lipid surface | ka (M−1s−1) | kd (s−1) | KD (nM) | χ2 |
|---|---|---|---|---|
| Proline reduced PutA |
||||
| E. coli lipid | 1.4 (± 0.14)×105 | < 10−6 | < 0.01 | 1.6 |
| PG lipid | 4.2 (± 0.43)×104 | < 10−6 | < 0.01 | 0.3 |
| EDOPC lipid | 1.3 (± 0.04)×105 | < 10−6 | < 0.01 | 0.8 |
| Oxidized PutA |
||||
| E. coli lipid | No Binding | |||
| PG lipid | No Binding | |||
| EDOPC lipid | 4.9 (± 0.29)×104 | 1.5 (± 0.48)×10−4 | 3.1 ± 0.9 | 1.6 |
Kinetic constants were determined in HEPES-N buffer (pH 7.4) at 25 °C.
In contrast, oxidized PutA-membrane interactions are highly dependent on the overall charge of the lipids. As previously observed with lipid vesicles from E. coli polar extracts, oxidized PutA did not bind to a negatively charged lipid bilayer. However, oxidized PutA was observed to bind to the positively charged lipids. Figure 3B shows the sensorgram of oxidized PutA binding to the positvely charged lipid surface. The association of oxidized PutA with the lipids displays reversible binding behavior with a KD value of 3 nM estimated for the oxidized PutA-lipid (EDOPC) complex. The kinetic parameters of oxidized PutA binding to the different lipid vesicles are summarized in Table 2. PutA is an acidic protein with an isoelectric point below pH 7.0, thus, the binding of oxidized PutA to the positively charged lipid surface appears to be influenced by electrostatic interactions, which were further explored by evaluating the influence of divalent cations.
Divalent cations such as Ca2+ and Mg2+ usually play a key role in the physiological function of protein-membrane associations and can alter the surface charge of the membrane (35, 36). Because oxidized PutA interacts with positively charged lipids, we studied how Mg2+ may affect PutA-membrane interactions by measuring kinetic parameters of PutA-lipid binding at different Mg2+ concentrations. At Mg2+ concentrations of < 0.1 mM, both oxidized PutA and proline-reduced PutA were observed to associate with lipid vesicles from E. coli polar extracts (data not shown). At concentrations of Mg2+ > 1 mM, however, only proline reduced PutA bound to the lipids. The binding of oxidized PutA to the lipids is highly influenced by Mg2+ indicating a strong dependence on electrostatic interactions. At low concentrations, Mg2+ ions probably alter the negatively charged surface of the membrane creating favorable electrostatic interactions between oxidized PutA and the membrane. At higher and perhaps more physiologically relevant Mg2+ concentrations (i.e. ∼ 1-2 mM), the Mg2+ ions interfere with oxidized PutA-membrane ionic interactions. Thus, under favorable ionic conditions oxidized PutA associates with the lipids.
PutA-Membrane vs. PutA-DNA Binding. PutA-DNA and PutA-membrane binding were reported to be mutually exclusive by Muro-Pastor et al. (1997) on the basis that a PutA-DNA complex was not observed in gel mobility shift assays when proline reduced PutA was incubated simultaneously with put control DNA and membrane vesicles (17). Sedimentation experiments also showed that in the presence of proline the put control DNA remained in the soluble fraction and did not precipitate with the PutA-membrane complex (17). We sought to explore this further by testing whether PutA complexed with the lipid bilayer could bind put control DNA. Figure 4A shows the immoblization of PutA onto the lipid vesicles in the presence L-lactate. Different concentrations of the put control DNA region (419 base pairs) from 10 to 40 nM were then injected at 5 μl/min for 15 min. No binding reponse was observed with the put control DNA since after the DNA injection the sensorgram returned to the intial value of lactate-bound PutA immobilized on the lipid surface (Figure 4A). The same result was also obtained with PutA bound to lipids in the presence of proline (data not shown). The order of binding was then reversed by first complexing PutA with DNA followed by injection of the PutA-DNA complex onto the lipid bilayer surface in the presence of proline. Figure 4B (lower trace) again shows no response in the sensorgram upon applying the proline-reduced PutA-DNA complex onto the lipid surface. Thus, the order of binding events does not influence the inability of PutA to associate concurrently with the put control DNA and the membrane. These results agree with the previous conclusion from gel-mobility shift assays and further demonstrate that PutA-DNA and PutA-membrane binding are mutually exclusive (17).
Figure 4.
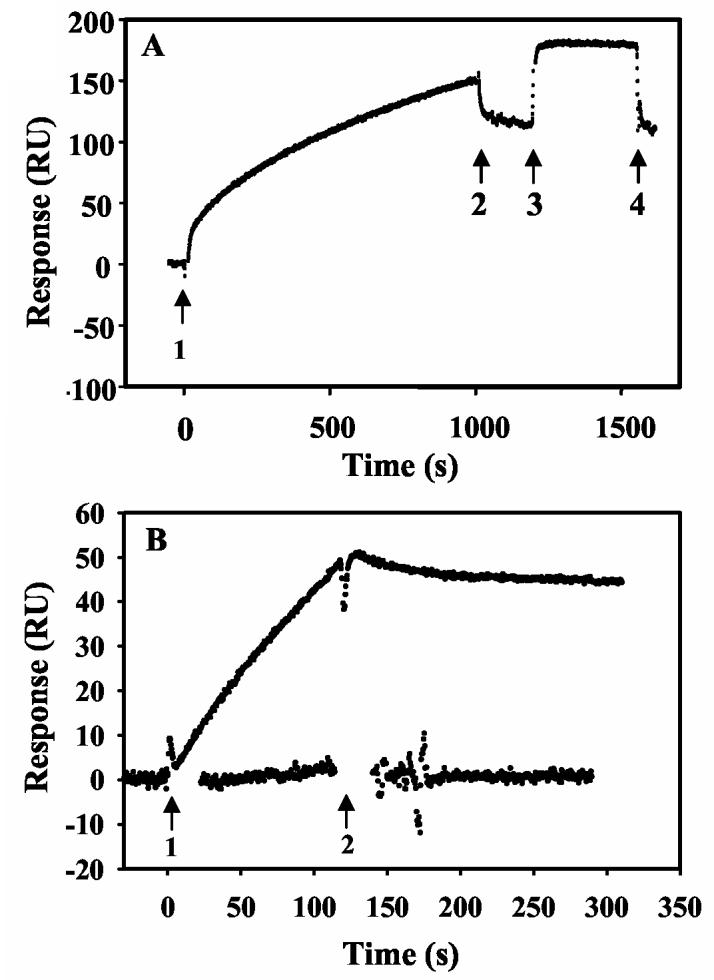
Effects of DNA binding on PutA membrane associations. Panel A: Oxidized PutA (100 nM) in the presence of sodium lactate (5 mM) was injected onto a L1 chip coated with E. coli polar lipid vesicles (arrows 1 and 2). The lipid-bound PutA was then washed with HEPES-N buffer at 60 μl/min for 180 s (arrows 2-3) followed by injection of put control DNA (419 bp, 20 nM) (arrows 3-4). The rapid change in RU at the beginning of the injection of DNA is due to the refractive index change of DNA sample, not to PutA-DNA binding. The RU returned to the initial value after the injection of the DNA sample was complete. Panel B: Sensorgrams of the PutA (10 nM)-oligonucleotide (1.8 μM) complex (upper trace) and the PutA (10 nM)-put control DNA (300 nM) complex (lower trace) injected onto a L1 chip coated with E. coli polar lipid vesicles in the presence of 5 mM proline (arrows 1 and 2).
The inability of PutA to bind the DNA and membrane simulatenously may indicate that recognition of a second macromolecule is not physically possible once PutA is bound to the membrane or to the put control DNA. PutA has been shown to have muliple DNA binding sites and bend the put control DNA suggesting the formation of a mulitmeric PutA-DNA complex (18). Thus, the membrane binding domain(s) of PutA could be constrained in the PutA-DNA complex and not accessible to the membrane. Likewise, membrane-bound PutA would constrain the DNA binding domain of PutA and prohibit DNA recognition. On the other hand, the DNA binding and membrane binding domains of PutA may overlap. The DNA-binding site of PutA is located in the N-terminal residues 1-47 while the location of the membrane binding region is still unknown (14).
To explore possible mechanisms of exclusive PutA-DNA and PutA-membrane binding, we tested whether PutA complexed with a 21-bp duplex oligonucleotide binding site (5'-TTTGCGGTTGCACCTTTCAAA-3') could associate with the membrane. Gel mobility shift analysis in Figure 5 shows that PutA binds specifically to the 21-bp duplex DNA forming a low mobility complex upon increasing the concentration of PutA in the binding reactions. The PutA-DNA complex is estimated to have a KD value of < 200 nM in HEPES-N buffer. To form the PutA-DNA complex 10 nM of PutA was incubated with 1.8 μM of the oliognucleotide DNA for 15 min. On the basis of the upper limit for the KD of the PutA-DNA oligonucleotide complex, > 90% of the PutA is bound to the DNA. Figure 4B (upper trace) shows the sensorgram of the PutA-DNA oligonucleotide complex injected at 5 μl/min onto the lipid surface for 2 min. In contrast to the PutA-DNA complex with put control DNA, the PutA-DNA oligonucleotide complex associates with the lipid bilayer. However, if the order of binding is reversed, that is, when PutA is immobilized on the lipid surface prior to DNA complexation, lipid-bound PutA is not able to bind the 21-bp oligonucleotide (data not shown). Thus, the DNA and membrane binding sites do not overlap but instead are physically contrained when complexed to the put control DNA or the membrane.
Figure 5.
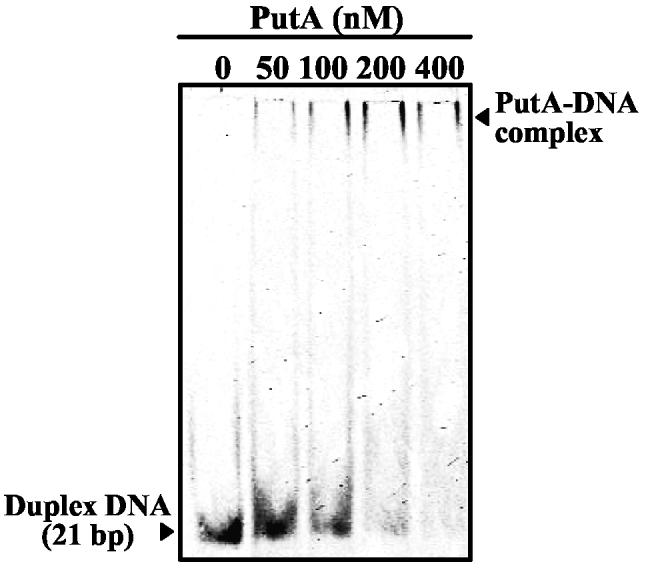
Gel mobility shift assay of the oligonucleotide binding site complexed with PutA. IRdye-700 labeled 21-bp duplex DNA (5 nM) and varying concentrations of PutA (0-400 nM dimer) were incubated in binding mixtures (HEPES-N buffer, pH 7.4) containing 100 μg/ml of nonspecific calf thymus DNA at 20 °C for 20 min. The complexes were separated using a nondenaturing polyacrylamide gel (8%).
Dissusion
FAD Reduction Activates PutA-membrane Binding. The effects of substrate binding and FAD reduction on PutA-membrane interactions have been evaluated separately for the first time. By SPR analysis we have defined quantitatively the role of substrate binding and FAD reduction in modulating PutA-membrane interactions. The membrane binding induced by proline and dithionite reduction of PutA have the same kinetic parameters (see Table 1) demonstrating that PutA-membrane interactions are controlled by FAD redox signaling. These results confirm previous studies which proposed FAD reduction is required for PutA-membrane associations (10, 15, 16, 21). Nevertheless, membrane binding is also induced by complexation of PutA with nonreducing ligands such as L-THFA, L-lactate, and DL-P5C. The membrane binding behavior of PutA in the presence of these ligands, however, is reversible and considerably weaker with KD values > 300-fold higher than that estimated for the reduced PutA-lipid complex. In previous studies by Wood (1987), D-lactate and NADH were observed to promote PutA membrane association under anaerobic conditions (10). The induction of membrane-binding was explained by reduction of PutA either directly or via the respiratory chain. Our results provide an alternative explanation and show that the binding of lactate to the PRODH domain induces PutA-membrane associations. Similar to L-lactate (Ki = 1.4 mM), D-lactate (Ki = 2.1 mM) is a competitive inhibitor of PRODH activity in PutA and would also be anticipated to bind to the PRODH active site and induce PutA-membrane binding (9).
A distinguishing characterteristic of reduced PutA-membrane binding is that the dissociation rate is beyond the lower limits of the SPR analysis. Consequently, PutA-membrane binding in the presence of proline appears to be irreversible. Even under conditions anticipated to oxidize membrane-bound PutA, dissociation of PutA from the membrane was not observed. The data supports Wood's previous suggestion that PutA-membrane associations are irreversible because under oxidizing conditions they did not detect soluble PutA protein in equilibrium with membrane vesicles harboring endogenous PutA (10, 16). Irreversible PutA-membrane binding would explain how PutA remains membrane-bound during catalysis, when it is presumably cycling between redox states with high (reduced) and low (oxidized) affinity for the membrane. However, since we have no direct evidence of the FAD redox state during the SPR experiments, it is possible that when PutA is immobilized on the membrane surface FAD is inaccessible and remains in the reduced state regardless of the oxidizing environment.
The nature of PutA-membrane interactions appears to be largely hydrophobic based on the SPR experiments with differently charged lipids. Upon FAD reduction, protein-membrane binding is likely promoted by interactions between hydrophobic region(s) of PutA and the lipid bilayer. Indeed, a conformational change that increases the overall hydrophobic nature of PutA has been proposed to be a mechanism by which the affinity of PutA for the membrane is enhanced upon reduction of FAD (15, 16, 21). Surber and Maloy (1999) have also shown that proline dehydrogenase activity can be reconstituted with lipid vesicles containing ubiquinone and cytochrome bo indicating that PutA-membrane association involves primarily protein-lipid interactions and is not dependent on other membrane proteins (15). A potential membrane partner for PutA is the Na+/proline transporter PutP. PutP has been extensively characterized and a method for reconstituting PutP into proteoliposomes has been developed by Jung et al. (1998) (37-39). We have begun exploring the binding of proline reduced PutA to E. coli polar lipid vesicles reconstituted with PutP. So far using SPR analysis, however, we have not detected any relative increase in the binding of PutA to PutP proteoliposomes which is consistent with protein-lipid contacts being largely responsible for PutA-membrane associations (data not shown).
A useful parallel for our studies on PutA is pyruvate oxidase from E. coli in which reduction of the FAD cofactor also enhances membrane associations. Pyruvate oxidase catalyzes the thiamin pyrophosphate (TPP) dependent oxidation of pyruvate to acetic acid and carbon dioxide and the subsequent reduction of ubiquinone in the cytoplasmic membrane (40). Pyruvate or chemical reduction of the FAD cofactor in the presence of TPP exposes a lipid-binding site in the C-terminal domain of the enzyme that enables pyruvate oxidase to associate with the membrane (41-45) . Because lipid binding is dependent on TPP, it is thought that changes in the FAD binding domain upon reduction are signaled to the C-terminal region of pyruvate oxidase via the TPP binding site (45). Communication between the FAD domain and the C-terminus is evident from spectroscopic studies, which show that the removal of the C-terminus decreases the hydrophobic character of the FAD environment (46). Thus, similar to that proposed for PutA, changes in the FAD redox site results in an overall conformational change that is conducive for membrane binding and converts a soluble flavoenzyme into a hydrophobic membrane-bound enzyme (42, 43, 45, 47).
Membrane and DNA Binding are Exclusive. PutA must switch between a transcriptional repressor and a membrane bound enzyme to upregulate proline utilization in bacteria, thus, it seems unlikely that properly functioning PutA would bind simultaneously to the DNA and the membrane. Even so, we explored this possibility. SPR analysis with the entire put control DNA illustrated that PutA exclusively interacts with the DNA and the membrane. Changing the order of the binding events had no effect as PutA complexed to either the put control DNA or the membrane was unable to recognize an additional macromolecule in the SPR experiments (Figure 4). The exclusive macromolecular binding seems to be caused by inaccessibility of the DNA or membrane binding domains in the corresponding PutA-DNA and PutA-membrane complexes. Alternatively, since solution continuously flows over the lipid surface in the SPR experiments, the PutA-membrane binding reaction is not fast enough to distrupt the PutA-DNA complex equilibrium in bulk solution.
To test whether the DNA and membrane binding domains of PutA overlap, we repeated the experiments using a single oligonucleotide binding site from the put control DNA region. Again, PutA immobilized on the lipid surface did not bind the DNA signifying membrane binding prevents access of the oligonucleotide to the DNA binding domain. However, reversing the order of binding events elicited the binding of the PutA-oligonucleotide complex to the lipid surface (Figure 4B). These results suggest that the DNA and membrane binding regions are sufficiently separated so that the binding of a 21-bp oligonucleotide to PutA does not interfere with membrane binding. We cannot exclude the possibility, however, that secondary PutA-DNA contacts occur immediately outside the 21-bp region which could interrupt PutA-membrane associations.
Regulation of PutA Intracellular Location. Until now, quantitative data describing PutA-membrane binding has not been available to provide details concerning the distribution of PutA between the DNA and the membrane. On the basis of the SPR kinetic analysis of PutA-membrane binding and previous PutA-DNA binding studies, a model for how the intracellular location and function of PutA is regulated by proline reduction of FAD is described in Figure 6. The mechanism of PutA translocation from the cytoplasm to the membrane in Figure 6 assumes that the PutA-DNA complex does not bind to the membrane which is supported by our work and previous observations (17). When PutA is in the oxidized state (conformer 1), an equilibrium exists in the cytoplasm between PutA, DNA and the PutA-DNA complex (KDox ∼ 45 nM) (20). Upon reduction of PutA-bound FAD (conformer 3), the equilibrium of PutA, DNA, and the PutA-DNA complex (KDred ∼ 100 nM) still exists but now uncomplexed reduced PutA will strongly associate with the membrane (20). The large energy difference (∼ 24 kJ/mol) between PutA-membrane and PutA-DNA binding subsequently distrupts the PutA-DNA equilibrum in the cytoplasm. As a result, the PutA-DNA complex dissociates generating more reduced uncomplexed PutA that can bind to the membrane. Since the PutA-DNA complex cannot bind the membrane directly, PutA must first dissociate from the PutA-DNA complex before binding to the membrane. Therefore, the translocation of PutA to the membrane will depend on the dissociation rate of the PutA-DNA complex.
Figure 6.
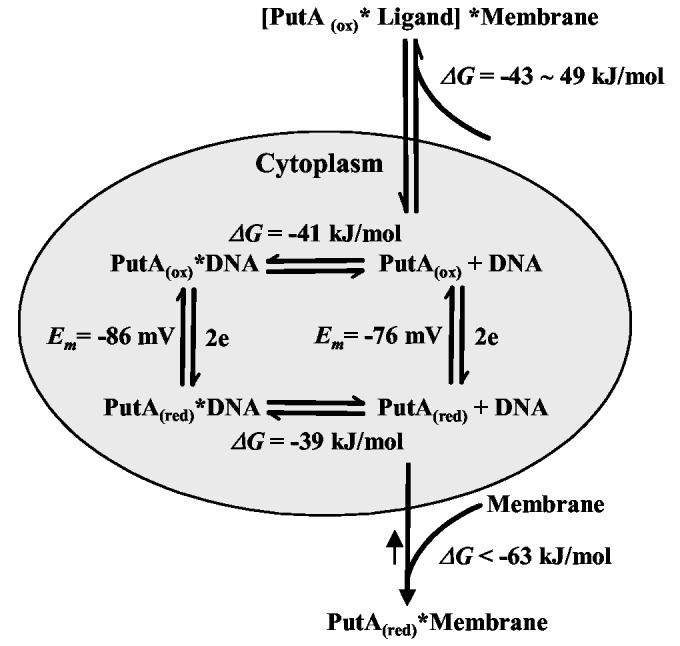
Thermodynamic model for how FAD reduction and ligand binding regulate PutA intracellular location and function (transcriptional repressor vs. membrane associated enzyme). Free energy of PutA-DNA binding and midpoint potential (Em) values are from reference (20) which were determined in 70 mM Tris buffer (pH 7.5) at 20 °C.
The surprising results with nonreducing ligands such as L-THFA and L-lactate evokes the question of what physiological impact does ligand binding have on PutA macromolecular associations? The estimated energy difference (≤ 8 kJ/mol) between PutA-membrane and PutA-DNA binding in the presence of ligands seems minor compared to that generated by proline. The lack of a significant driving force for PutA binding to the membrane relative to PutA-DNA binding predicts that nonreducing ligands are much less influential in regulating PutA than proline. Furthermore, PutA-membrane binding is reversible in the presence of ligands, implying that the PutA-membrane associations are in equilibrium with the PutA-DNA complex. Thus, nonreducing ligands have only a limited effect on the intracellular location and function of PutA.
From this study and previous work, it is clear that reduction of FAD bound to the PRODH active site of PutA elicits global conformational changes that direct PutA-membrane associations. Oxidized PutA is the non-membrane binding form (conformer 1) while reduction of FAD generates the membrane binding form (conformer 3). Ligand bound PutA which has a distinct conformation (conformer 2) is also capable of binding the membrane but the association is much weaker relative to PutA conformer 3. X-ray crystallography of PutA669 and more recently PutA86-669, has revealed detailed structural information of PutA conformer 2 and the PRODH active site complexed with L-THFA, L-lactate, and acetate (Tanner, personal communication). Important ionic bond interactions are formed between the carboxylate group of each ligand and Arg555, Arg556 and Lys329 (13). Since these ligands all generate the same conformation of PutA, the carboxylate group is the minimal element required to induce PutA conformation 2. Therefore, other molecules with carboxylate groups that can bind to the PRODH active site such as pyruvic acid (Ki = 3.3 mM) are predicted to generate PutA conformer 2 (9). The mechanism for how carboxylate binding to the PRODH active site affects PutA conformation is not known but since residues Arg555 and Arg556 are part of helix α8 of the β8α8-barrel in the PRODH domain helix α8 most likely is involved in transmitting signals out of the FAD active site. Work is ongoing to discover the molecular interactions that are critical for generating the different conformers of PutA and how PutA-membrane associations are controlled.
ACKNOWLEDGMENTS
We thank Dr. John J. Tanner, Department of Chemistry, University of Missouri-Columbia (Columbia, MO) for sharing information concerning the crystal structure of PutA86-669 prior to publication. We thank Dr. Yuannan Xia for assistance with the SPR experiments and the Genomics Core Facility in the Center for Biotechnology for maintenance of the Biacore 2000. We also thank Dr. Jeng-Jong Shieh and Dr. James Bashkin for help in the intial stages of this SPR study and Dr. Heinrich Jung (Ludwig-Maximilians-University, Munich, Germany) for the generous gift of PutP proteoliposomes.
ABBREVIATIONS
- FAD
flavin adenine dinucleotide
- put
proline utilization
- NAD
nicotinamide adenine dinucleotide
- PRODH
proline dehydrogenase
- P5CDH
Δ1-pyrroline-5-carboxylate dehydrogenase
- P5C
Δ1-pyrroline-5-carboxylate
- THFA
tetrahydro-2-furoic acid
- DCPIP
dichlorophenolindophenol
- HEPES
N-2-hydroxyethylpiperazine-N'-2-ethane sulfonic acid
- Em
midpoint potential
- SPR
surface plasmon resonance
- SUV
small unilamellar vesicle
- EDOPC
1, 2-dioleoyl-sn-glycerol-3-ethylphosphocholine
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propane sulfonate.
Footnotes
This research was supported in part by NSF MCB0091664 and NIH GM61068, University of Nebraska Biochemistry Department and Redox Biology Center, and the Nebraska Agricultural Research Division, Journal Series No. 14680. This publication was also made possible by NIH Grant Number P20 RR-017675-02 from the National Center for Research Resources. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
References
- 1.Wood JM. Genetics of L-proline utilization in Eschericia coli. J. Bacteriol. 1981;146:895–901. doi: 10.1128/jb.146.3.895-901.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brown E, Wood JM. Redesigned purification yields a fully functional PutA protein dimer from Escherichia coli. J. Biol. Chem. 1992;267:13086–13092. [PubMed] [Google Scholar]
- 3.Abrahamson JLA, Baker LG, Stephenson JT, Wood JM. Proline dehydrogenase from Escherichia K12, properties of the membrane-associated enzyme. Eur. J. Biochem. 1983;134:77–82. doi: 10.1111/j.1432-1033.1983.tb07533.x. [DOI] [PubMed] [Google Scholar]
- 4.Graham S, Stephenson JT, Wood JM. Proline dehydrogenase from Escherichia coli K12, reconstitution of functional membrane association. J. Biol. Chem. 1984;259:2656–2661. [PubMed] [Google Scholar]
- 5.Ling M, Allen SW, Wood JM. Sequence analysis identifies the proline dehydrogenase and pyrroline-5-carboxylate dehydrogenase domains of the multifunctional Escherichia coli PutA protein. J. Mol. Biol. 1994;245:950–956. doi: 10.1006/jmbi.1994.1696. [DOI] [PubMed] [Google Scholar]
- 6.Chen CC, Tsuchiya T, Yamane Y, Wood JM, Wilson TH. Na+ (Li+)-proline cotransport in Escherichia coli. J. Membr. Biol. 1985;84:157–164. doi: 10.1007/BF01872213. [DOI] [PubMed] [Google Scholar]
- 7.Chen CC, Wilson TH. Solubilization and functional reconstitution of the proline transport system of Escherichia coli. J. Biol. Chem. 1986;261:2599–2604. [PubMed] [Google Scholar]
- 8.Nakao T, Yamato I, Anraku Y. Nucleotide sequence of putC, the regulatory region for the put regulon of Escherichia coli K12. Mol. Gen. Genet. 1987;210:364–368. doi: 10.1007/BF00325707. [DOI] [PubMed] [Google Scholar]
- 9.Scarpulla RC, Soffer RL. Membrane-bound proline dehydrogenase from Escherichia coli. J. Biol. Chem. 1978;253:5997–6001. [PubMed] [Google Scholar]
- 10.Wood J. Membrane association of proline dehydrogenase in Escherichia coli is redox dependent. Proc. Natl. Acad. Sci. USA. 1987;84:373–377. doi: 10.1073/pnas.84.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menzel R, Roth J. Enzymatic properties of the purified putA protein from Salmonella typhimurium. J. Biol. Chem. 1981;256:9762–9766. [PubMed] [Google Scholar]
- 12.Vinod MP, Bellur P, Becker DF. Electrochemical and functional characterization of the proline dehydrogenase domain of the PutA flavoprotein from Escherichia coli. Biochemistry. 2002;41:6525–6532. doi: 10.1021/bi025706f. [DOI] [PubMed] [Google Scholar]
- 13.Lee YH, Nadaraia S, Gu D, Becker DF, Tanner JJ. Structure of the proline dehydrogenase domain of the multifunctional PutA flavoprotein. Nat. Struct. Biol. 2003;10:109–114. doi: 10.1038/nsb885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu D, Zhou Y, Kallhoff V, Baban B, Tanner JJ, Becker DF. Identification and characterization of the DNA-binding domain of the multifunctional PutA flavoenzyme. J. Biol. Chem. 2004;279:31171–31176. doi: 10.1074/jbc.M403701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Surber MW, Maloy S. Regulation of flavin dehydrogenase compartmentalization: Requirements for PutA-membrane association in Salmonella typhimurium. Biochim. Biophys. Acta. 1999;1421:5–18. doi: 10.1016/s0005-2736(99)00104-2. [DOI] [PubMed] [Google Scholar]
- 16.Brown ED, Wood JM. Conformational change and membrane association of the PutA protein are coincident with reduction of its FAD cofactor by proline. J. Biol. Chem. 1993;268:8972–8979. [PubMed] [Google Scholar]
- 17.Muro-Pastor AM, Ostrovsky P, Maloy S. Regulation of gene expression by repressor localization: Biochemical evidence that membrane and DNA binding by the PutA protein are mutually exclusive. J. Bacteriol. 1997;179:2788–2791. doi: 10.1128/jb.179.8.2788-2791.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ostrovsky De Spicer P, O'Brian K, Maloy S. Regulation of proline utilization in Salmonella typhimurium: a membrane-associated dehydrogenase binds DNA in vitro. J. Bacteriol. 1991;173:211–219. doi: 10.1128/jb.173.1.211-219.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu W, Becker DF. Flavin redox state triggers conformational changes in the PutA protein from Escherichia coli. Biochemistry. 2003;42:5469–5477. doi: 10.1021/bi0272196. [DOI] [PubMed] [Google Scholar]
- 20.Becker DF, Thomas EA. Redox properties of the PutA protein from Escherichia coli and the influence of the flavin redox state on PutA-DNA interactions. Biochemistry. 2001;40:4714–4722. doi: 10.1021/bi0019491. [DOI] [PubMed] [Google Scholar]
- 21.Ostrovsky De Spicer P, Maloy S. PutA protein, a membrane-associated flavin dehydrogenase, acts as a redox-dependent transcriptional regulator. Proc. Natl. Acad. Sci. USA. 1993;90:4295–4298. doi: 10.1073/pnas.90.9.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muro-Pastor AM, Maloy S. Proline dehydrogenase activity of the transcriptional repressor PutA is required for induction of the put operon by proline. J. Biol. Chem. 1995;270:9819–9827. doi: 10.1074/jbc.270.17.9819. [DOI] [PubMed] [Google Scholar]
- 23.Stahelin RV, Cho W. Differential roles of ionic, aliphatic, and aromatic residues in membrane-protein interactions: A surface plasmon resonance study on phospholipase A2. Biochemistry. 2001;40:4672–4678. doi: 10.1021/bi0020325. [DOI] [PubMed] [Google Scholar]
- 24.Bitto E, Li M, Tikhonov AM, Schlossman ML, Cho W. Mechanism of annexin I-mediated membrane aggregation. Biochemistry. 2000;39:13469–13477. doi: 10.1021/bi001275u. [DOI] [PubMed] [Google Scholar]
- 25.Papo N, Shai Y. Exploring peptide membrane interaction using surface plasmon resonance: Differentiation between pore formation versus membrane disruption by lytic peptides. Biochemistry. 2003;42:458–466. doi: 10.1021/bi0267846. [DOI] [PubMed] [Google Scholar]
- 26.Papo N, Shai Y. Effect of drastic sequence alteration and D-amino acid incorporation on the membrane binding behavior of lytic peptides. Biochemistry. 2004;43:6393–6403. doi: 10.1021/bi049944h. [DOI] [PubMed] [Google Scholar]
- 27.Mezel VA, Knox WE. Properties and analysis of a stable derivative of pyrroline-5-carboxylic acid for use in metabolic studies. Anal. Biochem. 1976;74:430–440. doi: 10.1016/0003-2697(76)90223-2. [DOI] [PubMed] [Google Scholar]
- 28.Strecker HJ. The interconversion of glutamic acid and proline. J. Biol. Chem. 1957;225:825–834. [PubMed] [Google Scholar]
- 29.Hope MJ, Bally MB, Webb G, Cullis PR. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain membrane potential. Biochim. Biophys. Acta. 1985;812:55–65. doi: 10.1016/0005-2736(85)90521-8. [DOI] [PubMed] [Google Scholar]
- 30.MacDonald RC, Ashley GW, Shida MM, Rakhmanova VA, Tarahovsky YS, Pantazatos DP, Kennedy MT, Pozharski EV, Baker KA, Jones RD, Rosenzweig HS, Choi KL, Qiu R, McIntosh TJ. Physical and biological properties of cationic triesters of phosphotidylcholine. Biophys. J. 1999;77:2612–2629. doi: 10.1016/S0006-3495(99)77095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis RNA, Prenner EJ, Kondejewski LH, Flach CR, Mednelsohn R, Hodges RS, McElhaney RN. Fourier transform infrared spectroscopic studies of the interaction of the antimicrobial peptide gramicidin S with lipid micelles and with lipid monolayer and bilayer membranes. Biochemistry. 1999;38:15193–15203. doi: 10.1021/bi9912342. [DOI] [PubMed] [Google Scholar]
- 32.Erb EM, Chen X, Allen S, Roberts CJ, Tendler SJB, Davies MC, Forsen S. Characterization of the surfaces generated by liposome binding to the modified dextran matrix of a surface plasmon resonance sensor chip. Anal. Biochem. 2000;280:29–35. doi: 10.1006/abio.1999.4469. [DOI] [PubMed] [Google Scholar]
- 33.Myszka DG. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Cur. Opin. Biotech. 1997;8:50–57. doi: 10.1016/s0958-1669(97)80157-7. [DOI] [PubMed] [Google Scholar]
- 34.Zhu W, Gincherman Y, Docherty P, Spilling CD, Becker DF. Effects of proline analog binding on the spectroscopic and redox properties of PutA. Arch. Biochem. Biophys. 2002;408:131–136. doi: 10.1016/s0003-9861(02)00535-0. [DOI] [PubMed] [Google Scholar]
- 35.Bittova L, Sumandea M, Cho W. A structure-function study of the C2 domain of cytosolic phospholipase A2. J. Biol. Chem. 1999;274:9665–9672. doi: 10.1074/jbc.274.14.9665. [DOI] [PubMed] [Google Scholar]
- 36.Ermakov YA, Averbakh AZ, Yusipovich AI, Sukharev S. Dipole potentials indicate restructuring of the membrane interface induced by gadolinium and beryllium ions. Biophys. J. 2001;80:1851–1862. doi: 10.1016/S0006-3495(01)76155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung H, Tebbe S, Schmid R, Jung K. Unidirectional reconstitution and characterization of purified Na+/proline transporter of Escherichia coli. Biochemistry. 1998;37:11083–11088. doi: 10.1021/bi980684b. [DOI] [PubMed] [Google Scholar]
- 38.Jung H, Rubenhagen R, Tebbe S, Leifker K, Tholema N. Topology of the Na+/proline transporter of Escherichia coli. J. Biol. Chem. 1998;273:26400–26407. doi: 10.1074/jbc.273.41.26400. [DOI] [PubMed] [Google Scholar]
- 39.Pirch T, Landmeier S, Jung H. Transmembrane domain II of the Na+/proline transporter PutP of Escherichia coli forms part of a conformationally flexible, cytoplasmic exposed aqueous cavity within the membrane. J. Biol. Chem. 2003;278:42942–42949. doi: 10.1074/jbc.M308253200. [DOI] [PubMed] [Google Scholar]
- 40.Koland JG, Miller MJ, Gennis RB. Reconstitution of the membrane-bound, ubiquinone-dependent pyruvate oxidase respiratory chain of Escherichia coli with the cytochrome d terminal oxidase. Biochemistry. 1984;445 doi: 10.1021/bi00298a008. [DOI] [PubMed] [Google Scholar]
- 41.Russell P, Hager LP, Gennis RB. Characterization of the proteolytic activation of pyruvate oxidase. J. Biol. Chem. 1977;252:7877–7882. [PubMed] [Google Scholar]
- 42.Schrock HL, Gennis RB. High affinity lipid binding sites on the peripheral membrane enzyme pyruvate oxidase. J. Biol. Chem. 1977;252:5990–5995. [PubMed] [Google Scholar]
- 43.Schrock HL, Gennis RB. The binding of a fluorescent activator 2-(N-decyl) aminonaphthalene-6-sulfonic acid to pyruvate oxidase. Biochim. Biophys. Acta. 1980;615:10–18. doi: 10.1016/0005-2744(80)90003-0. [DOI] [PubMed] [Google Scholar]
- 44.Chang Y-Y, Cronan JEJ. Detection by site-specific disulfide cross-linking of a conformational change in binding of Escherichia coli pyruvate oxidase to lipid bilayers. J. Biol. Chem. 1995;270:7896–7901. doi: 10.1074/jbc.270.14.7896. [DOI] [PubMed] [Google Scholar]
- 45.Chang Y-Y, Cronan JEJ. Sulfhydryl chemistry detects three conformations of the lipid binding region of Escherichia coli pyruvate oxidase. Biochemistry. 1997;36:11564–11573. doi: 10.1021/bi9709102. [DOI] [PubMed] [Google Scholar]
- 46.Recny MA, Hager LP. Isolation and characterization of the protease-activated form of pyruvate oxidase. Evidence for a conformational change in the environment of the flavin prosthetic group. J. Biol. Chem. 1983;258:5189–519. [PubMed] [Google Scholar]
- 47.Russell P, Schrock HL, Gennis RB. Lipid activation and protease activation of pyruvate oxidase. J. Biol. Chem. 1977;252:7883–7887. [PubMed] [Google Scholar]