

Abstract
Motivation: The emerging glycomics and glycoproteomics projects aim to characterize all forms of glycoproteins in different tissues and organisms. Tandem mass spectrometry (MS/MS) is the key experimental methodology for high-throughput glycan identification and characterization. Fragmentation of glycans from high energy collision-induced dissociation generates ions from glycosidic as well as internal cleavages. The cross-ring ions resulting from internal cleavages provide additional information that is important to reveal the type of linkage between monosaccharides. This information, however, is not incorporated into the current programs for analyzing glycan mass spectra. As a result, they can rarely distinguish from the mass spectra isomeric oligosaccharides, which have the same saccharide composition but different types of sequences, branches or linkages.
Results: In this paper, we describe a novel algorithm for glycan characterization using MS/MS. This algorithm consists of three steps. First, we develop a scoring scheme to identify potential bond linkages between monosaccharides, based on the appearance pattern of cross-ring ions. Next, we use a dynamic programming algorithm to determine the most probable oligosaccharide structures from the mass spectrum. Finally, we re-evaluate these oligosaccharide structures, taking into account the double fragmentation ions. We also show the preliminary results of testing our algorithm on several MS/MS spectra of oligosaccharides.
Availability: The program GLYCH is available upon request from the authors.
Contact: hatang@indiana.edu
1 INTRODUCTION
Glycosylation is a common post-translational modification that covalently attaches sugar chains (called oligosaccharides or glycans) to the cell surface or secreted proteins. Glycans (either attached to proteins or sometimes in free form) can mediate cell-cell, cell-matrix and cell-environment interactions, which are crucial to many biological functions in a complex multicellular organism (Varki et al., 1999). The emerging glycomics and glycoproteomics projects aim to identify all the carbohydrate molecules, the `glycome', in an organism (e.g. human) and to profile their changes under different conditions, e.g. healthy versus disease.
Owing to the structural complexity of glycans, the technology for determining glycan structure has lagged far behind those for the other classes of biological macromolecules, such as nucleic acids and proteins. In recent years, mass spectrometry (MS) has become the key tool for glycan structural analysis because of its high sensitivity. A number of glycan analytical methods using MS have been published which differ in various characteristics ranging from the ionization method (e.g. MALDI/TOF-MS or ESI/MS) to the choice of derivatives (or ions) (e.g. paralkyl or native structures, protonated or metal-cationized ions) (Mechref and Novotny, 2002; Zaia, 2004). In this paper, we will focus on the experimental results from MALDI/TOF/TOF-MS, but the same algorithm can also be applied to the other MS platforms by adjusting the fragmentation model appropriately. Fragmentation of glycans in MALDI/MS can result from the post-source decay (PSD) or collision-induced dissociation (CID). Low energy PSD spectra from neutral glycans are dominated by the glycosidic cleavages with very weak cross-ring ions. On the other hand, in high energy CID spectra, in addition to glycosidic cleavages, cross-ring ions are often observed, which provide valuable information for oligosaccharide characterization (Mechref et al., 2003), as we show in the following sections.
Despite the rapid progress in techniques in the field of glycomics, the tools for automatic glycan mass spectrum interpretation are still some way off (von der Lieth et al., 2004). There are several programs that are used to deduce glycan structures from their tandem mass spectrometry (MS/MS) spectra. Glycomod is one of the most commonly used tools, which attempts to find all possible compositions of a glycan molecule from its determined total mass (Cooper et al., 2001). However, this program was not designed for interpreting MS/MS spectra and hence cannot distinguish between isomeric oligosaccharides, i.e. oligosaccharides with the same monosaccharide composition but different structures. GlycosidIQ (Joshi et al., 2004) and GlycoSearchMS (Lohnmann et al., 2004) each implemented a SEQUEST-like (Eng et al., 1994) database searching algorithm to compare an experimental MS/MS spectrum against the theoretical spectra of a collection of oligosaccharides from a curated database, SweetDB (Loss et al., 2002) and GlycoSuiteDB (Cooper et al., 2003), respectively. Despite being successful in some cases, these approaches are not suitable for a large-scale glycomics project, because unlike the protein database (which can be derived from completely sequenced genomes), the glycan database is still largely incomplete. One of the specific aims of the glycomics project is to collect oligosaccharides from model organisms. STAT (Gaucher et al., 2000), StrOligo (Eithier et al., 2002) and a recently developed algorithm (Shan et al., 2004) have attempted to reconstruct the glycan structure from MS/MS spectra de novo by exhaustively searching all possible glycan structures that match the experimental total mass. Owing to the limitation of computational complexity, these algorithms can analyze only glycans consisting of no more than 10 monosaccharides. Sophisticated algorithms are required to analyze relatively large oligosaccharides efficiently. Finally, none of the above programs utilizes the information provided by the cross-ring ions to identify the correct oligosaccharides among many isomers.
In this paper, we propose a dynamic programming algorithm in conjuction with a novel scoring scheme for de novo oligosaccharide characterization from MS/MS spectra. This algorithm is a variant of the de novo peptide sequencing algorithm (Dancik et al., 1999; Chen et al., 2001; Bafna and Edwards, 2003; Ma et al., 2003), except that it allows branching in the polymer structure that the glycans may have.
2 OLIGOSACCHARIDES AND THEIR FRAGMENTATION PATTERNS
There are two main types of glycosylation for glycoproteins in vivo: N-linked glycosylation, in which N-linked oligosaccharides are linked to the amide N atom in the side-chain of amino acid residue Asn, and O-linked glycosylation, in which O-linked oligosaccharides are linked to the hydroxyl O atom in the side-chain of amino acid residues Ser or Thr. The glycosylation consists of a series of reactions catalyzed by many different glycan transferases. For each reaction, one monosaccharide is linked to the end of the present oligosaccharide by forming a glycosidic bond. The common monosaccharides found in higher animal oligosaccharides, which are considered in this study, are listed in Table 1. Several others can be found in lower animals, bacteria or plants. Even in higher animals, the monosaccharides found in different species may be slightly different. For example, two predominant forms of sialic acid in animals are N-acetyl-neuraminic acid (NeuAc) and N-glycolyl-neuraminic acid (NeuGc). Because of a missing enzyme, humans express only NeuAc, which is different from NeuGc only by one less oxygen atom (Chou et al., 1998). Therefore, we should design a specific set of monosaccharides in the analysis of the organism from which the oligosaccharides are sampled. In this paper, we use the alphabet, consisting of six monosaccharides, in which either NeuAcA or NeuGc is considered depending on the organism of interest (human or the other higher animals).
Table 1.
The common monosaccharides in animal oligosaccharides
| Name (abbreviation) | Epimers a | Mass | Symbol |
|---|---|---|---|
| Hexose (Hex) | Glucose (Glc) | 162.05 | ○ |
| Mannose (Man) | |||
| Galactose (Gal) | |||
| HexNAc | N-acetylglucosamine (GlcNAc) | 203.08 | □ |
| N-acetylgalactosamine (GalNAc) | |||
| HexA | Glucuronic acid (GlcA) | 176.03 | ◇ |
| Iduronic acid (IdcA) | |||
| Xylose (Xyl) | — | 132.04 | ▽ |
| Fucose (Fuc) | — | 146.06 | △ |
| N-acetyl neuraminic acid (NeuAc) | — | 291.10 | ◇ |
| N-glycolyl neuraminic acid (NeuGc) | — | 307.09 | ◇ |
Epimers, two monosaccharides that differ only in their configuration, cannot be distinguished by mass spectrometry.
Free monosaccharides can exist in an open or cyclic form (Fig. 1a, b). The numbering of the carbon atoms follows the rules of organic chemistry nomenclature, such that the aldehyde carbon is referred to as C1 (Fig. 1a). The cyclic form of monosaccharides has a ring structure that consists of six covalent bonds, conventionally denoted from `0' to `5' in the clockwise direction (Fig. 1b). Two monosaccharides can react with each other, forming a glycosidic bond between the hemiacetal group (i.e. the C1 group) from one monosaccharide and the alcohol group from the other while releasing a water molecule. Depending on which alcohol group participates in the reaction, the set of possible linkage types, denoted as, has four different types of glycosidic bonds, i.e. 1-2, 1-3, B 1-4 and 1-6 bonds. The first number in the notation (e.g. 1 in 1-2 bond) represents the numbering of the carbon in the hemiacetal group, and hence is always 1. The second number in the notation (e.g. 2 in 1-2 bond) represents the numbering of the carbon that the alcohol group is linked to, and hence could be 2, 3, 4 or 6. Figure 1c shows a disaccharide consisting of two glucoses forming a 1-4 glycosidic bond. Since a monosaccharide has more than one alcohol group, it may react with more than one other monosaccharide. As a result, an oligosaccharide can form branching structures. Figure 1d shows a tetrasaccharide consisting of four glucoses, in which one forms 1-4 and 1-6 glycosidic bonds with two others. This branching structure can be drawn as a tree (Fig. 1e), in which each monosaccharide is represented by a symbol (Table 1) and each glycosidic bond is represented by an edge. In theory, there could be at most four branches in the tree; in reality, in higher animals, the branchings are always binary, except in one special case, called `bisecting GlcNAc' (Stanley, 2002).
Fig. 1.

The structure of oligosaccharides. (a) The open (acyclic) form structure of glucose, an epimer of hexose, one of the monosaccharides, i.e. the basic unit of an oligosaccharide. (b) The cyclic form structure of glucose. The carbon atoms in the ring are not shown. (c) A diglucose, consisting of two glucoses forming a 1-4 glycosidic bond. (d) A tetraglucose, consisting of four glucoses with a branching. (e) The symbolic representation of the tetraglucose shown in (d). The numbers show the linkage types.
Above all, in addition to sequence variation, oligosaccharides can also exhibit variations of branching and bond linkages. As a result, monosaccharides generate much higher linkage complexity than amino acids and nucleotides. For example, three amino acids can generate six different peptide sequences; in comparison, three monosaccharides can generate >100 trisaccharide configurations. Therefore, oligosaccharide characterization is more complex than protein identification.
The fragmentation patterns for oligosaccharides have been studied in detail using different ionization techniques. According to the definitions of Domon and Costello (1988), the corresponding ions are denoted Yn/Bl-n and Zn/Cl n, where l is the length of the oligosaccharides and n denotes the cleavage site (Fig. 2a). In high energy CID spectra, in addition to the glycosidic cleavages, cross-ring cleavages, in which two covalent bonds in the ring structure are broken, can often be observed (Fig. 2b). The corresponding ions are denoted p,qXn and p,q An, where p and q are sites for cross-ring bond (designated from 0 to 5, Fig.1b) cleavages. Similar to peptide MS/MS spectra, the ions corresponding to more than one glycosidic bond cleavage are often weak. The X/A ions in the CID spectra provide valuable information to distinguish between isomeric oligosaccharides and to determine the branching and bond linkages. Table 2 shows mass differences of all nine types of potential X/A ions compared with the Y/B ions cleaved at the same residue. Clearly, different monosaccharides tend to have different X/A ion patterns. If we assume one peak in the MS/MS spectrum is from a Y/B ion, based on its surrounding peaks, we may be able to distinguish at which type of monosaccharide this cleavage is. On the other hand, given an oligosaccharide, no matter how complicated its structure is, we can construct its theoretical MS/MS spectrum by (1) computing the mass values of the Y/B and Z/C ions and (2) computing the corresponding mass values of X/A ions corresponding to each Y/B ion. Moreover, in a monosaccharide in the middle of an oligosaccharide (i.e. when it forms glycosidic bonds with two other monosaccharides), some X/A ions will not appear in the spectra, even though the corresponding fragmentation could happen. Figure 2c illustrates this using the fragmentation of triglucose as an example. We call the occurrences of X/A ions in a particular type of linkage the occurrence pattern. Among four types of linkages in B, any one has a unique occurrence pattern different from the others. Table 3 summarizes the X/A ion occurrence patterns of all linkages. Finally, some monosaccharides cannot form certain linkages, since they have no alcohol group at that position. Table 4 summarizes the linkage types that each monosaccharide can form.
Fig. 2.
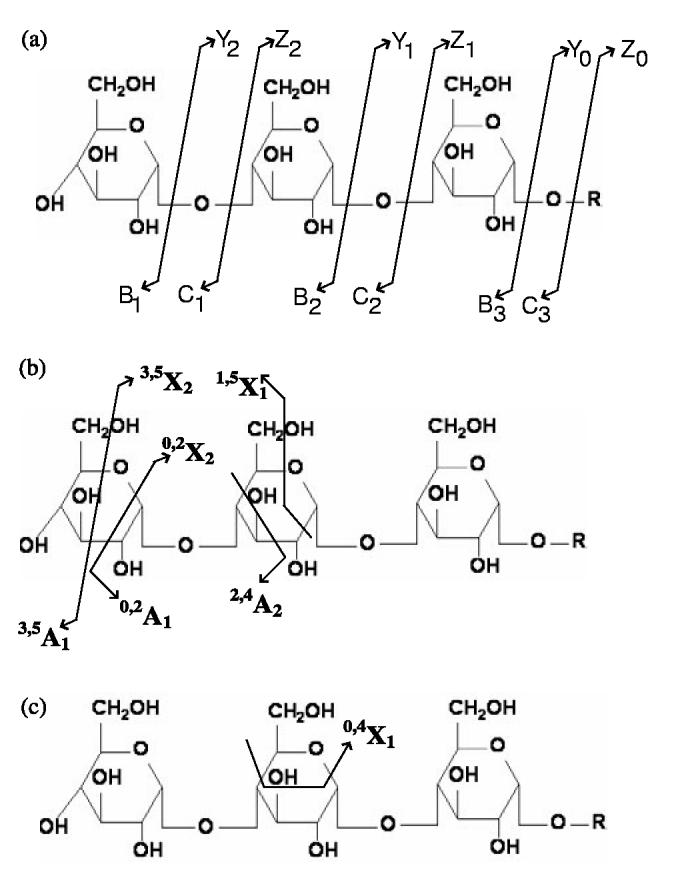
The fragmentation patterns of oligosaccharides. (a) The Y/b and Z/C ions are from the glycosidic cleavages. (b) The cross-ring cleavages create X/A ions. (c) Since the middle unit of the triglucose is linked to two other glucoses, some cleavages create fragment ions with small masses that are not close to the corresponding Y/b ions, e.g. 0,4X1. These ions will be ignored in our program.
Table 2.
The mass difference of X / A ions and the corresponding Y / B ion
| Monosaccharide | 0,2A | 0,3A | 0,4A | 1,3A | 1,4A | 1,5A | 2,4A | 2,5A | 3,5A |
|---|---|---|---|---|---|---|---|---|---|
| Hex | 43.02 | 73.03 | 103.04 | 103.04 | 73.03 | 29.00 | 103.04 | 59.01 | 89.03 |
| HexA | 43.02 | 73.03 | 103.04 | 116.01 | 86.00 | 29.00 | 116.01 | 59.01 | 89.03 |
| HexNAc | 84.04 | 114.05 | 144.06 | 103.04 | 73.02 | 29.00 | 144.06 | 100.04 | 130.04 |
| Fuc | 43.02 | 73.03 | 103.04 | 87.04 | 57.03 | 29.00 | 87.04 | 59.01 | 89.03 |
| Xyl | 43.02 | 73.03 | 103.04 | 103.04 | 73.03 | 29.00 | 103.04 | 59.01 | 89.03 |
| NeuAc | 71.01 | 101.03 | 172.06 | 232.08 | 161.04 | 72.99 | 175.06 | 87.01 | 117.03 |
| NeuGc | 87.01 | 101.03 | 172.06 | 232.08 | 161.04 | 72.99 | 190.05 | 103.00 | 133.03 |
Table 3.
The list of occurring X / A ions for monosaccharide residues that are linked to two or more other monosaccharidesa
| X / A ions | (2, n)b | (3, n) | (4, n) | (6, n) | (2, 3)3 | (2, 4) | (2, 6) | (3, 4) | (3, 6) | (4, 6) |
|---|---|---|---|---|---|---|---|---|---|---|
| 0,2 X / A | − | + | + | + | + | + | + | − | − | − |
| 0,3 X / A | − | − | + | + | − | + | + | + | − | − |
| 0,4 X / A | − | − | − | + | − | − | + | − | + | + |
| 1,3 X / A | + | + | − | − | − | + | + | + | − | − |
| 1,4 X / A | + | + | − | − | − | − | + | − | + | + |
| 1,5 X / A | + | + | − | − | − | − | − | − | + | − |
| 2,4 X / A | + | + | − | − | + | + | + | − | − | + |
| 2,5 X / A | + | + | − | − | + | + | + | − | + | − |
| 3,5 X / A | + | + | − | − | − | + | + | + | + | − |
`+' represents presence; `−' represents absence. For those monosaccharides that are linked to more than two other ones, only the A / X ions that occurs in all the bi-linkage cases should occur.
(2,n) represents that one of two linkages is 1-2; the other is any of four linked to the monosaccharide's first carbon; (2,3) shows that the two linkages are to the monosaccharide's second and third carbons, i.e. a branching with 1-2 and 1-3 linkages.
Table 4.
The list of occurring X/A ions for monosaccharide residues in difference linkagesa
| X / A ions | 1-2 | 1-3 | 1-4 | 1-6 |
|---|---|---|---|---|
| Hex | + | + | + | + |
| HexA | + | + | + | − |
| HexNAc | − | + | + | + |
| Fuc | + | + | + | − |
| Xyl | + | + | + | − |
| NeuAc | − | + | − | − |
| NeuGc | − | + | − | − |
`+' represents presence; `−' represents absence.
Utilizing the rules given in Tables 2, 3, 4 for a given oligosaccharide, we can compute its theoretical MS/MS spectrum, i.e. the mass values of all its potential ions under high energy CID fragmentation, including Y/B, Z/C and X/A ions. This procedure can be integrated into a glycan database searching program such as GlycosidIQ (Joshi et al., 2004) or GlycoSearchMS (Lohnmann et al., 2004), comparing an experimental spectrum with the theoretical spectrum of each known oligosaccharide in the database and determining an optimal match among them. However, given the incompleteness of the current glycan database, we are more interested in solving this problem assuming no preliminary knowledge. Therefore, we need to solve the following problem.
De novo oligosaccharide characterization problem. Given an experimental MS/MS spectrum and the total mass of the oligosaccharide, reconstruct the structure of the oligosaccharide such that its theoretical spectrum optimally matches the given experimental spectrum.
Note that there are different levels of structures to be characterized for oligosaccharides: monosaccharide composition, sequence, branching structure and linkage. In the context of this paper, we aim to characterize the complete structure of the oligosaccharides, i.e. all the above four should be determined by the computational procedure.
3 ALGORITHM
An oligosaccharide can be represented by a tree structure of n monosaccharide residues (Fig. 3a), r1, r2, ..., rn, in which the root residue rn has an open hemiacetal group and the leaf residues have open alcohol groups. Similar to the approach employed for peptide sequencing (Dancik et al., 1999; Bandeira et al., 2004), we define the prefix residue mass (PRM) mi as the total mass of residues in the subtree rooted by residue ri (Fig. 3b); in the special case, mn is referred to as the parent mass Mparent, i.e. the total mass of the oligosaccharide. Generally, the series of PRMs corresponds to the series of B ions in the MS/MS spectrum, with a constant offset. However, unlike the peptide sequences, in addition to the sequence variants, oligosaccharides have linkage variants. To reflect this, we define the prefix residue feature (PRF), a combination of three properties: (1) the PRM, (2) the monosaccaride type and (3) the linkage type of the residue. For instance, (mi, ri, bi) is the PRF at position i, where mi is the PRM, ri is the monosaccharide type of residue i and bi is the linkage type between residue i and the residue at the root side to which it links.1 Therefore, any olicosaccharide with n residues can be represented by a series of PRFs, (m1, r1, b1), (m2, r2, b2), ..., (mn, rn, bn).
Fig. 3.
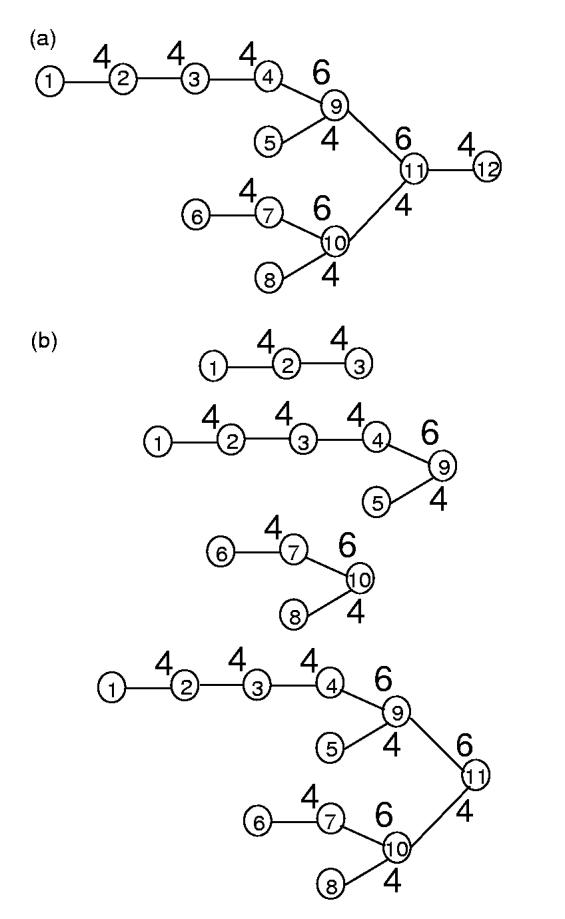
The PRMs of an oligosacchrides. (a) An oligosaccharide consisting of 12 glucoses. The glucose residues are indexed according to the partial order of nodes of the tree, from leafs to the root. For any two residues ri and rj, if ri is within the subtree rooted by rj, i must be indexed smaller than j.(b) The subtrees rooted by residues r3, r9, r10, r11.
Our strategy to solve the oligosaccharide characterization problem is first to find and score potential PRFs by evaluating the local fragmentation patterns and then to deduce the oligosaccharide structure from these PRFs. Every peak in an experimental MS/MS spectrum should correspond either to one of the fragmentation ions (B/Y, C/Z, A/X) or to a noisy ion.2 Assume that one peak p is the fragmentation ion type i of residue r ∈ A with linkage b ∈, we can compute the corresponding PRM Mir as
| (1) |
| (2) |
where Mp is the mass of p and δMir is the mass difference between ion type i and the B ion for the residue r (Table 2). We call p an i-support peak of PRF (Mir, r, b) for any b . B Therefore, every peak in the experimental spectrum can∈ be In practice, since MS has mass accuracy of measurement, we allow a certain tolerance for this support peak computation. For MALDI/TOF/TOF-MS, 0.2 Da.
We score PRFs depending on=how their fragmentation patterns are supported by the experimental ion peaks. Currently, we use a simple scoring scheme. We define the supporting score of a PRF as the number of its support peaks Nf. After deducing a list of PRFs, each associated with a supporting score, we attempt to find a series of PRFs (M1, r1, b1), (M2, r2, b2), ..., (Mn = Mparent, rn, bn)(M1 M2 M) that correspond to one oligosaccharide's tree structure, n and have the maximal total score
| (3) |
where S(Mi, ri, bi) is the score of PRF (Mi, ri, bi).
In order to solve this problem, we first sort all PRFs according to their PRMs. For each PRF (m, r, b), we define the score V(m, r, b) as the maximal total score of a series of (M1, r1, b1), (M2, r2, b2),... , (Ml =, m, rj, bj) (M1 ≤ M2 ≤ ... ≤ Mj) that corresponds to one oligosaccharide's tree structure with a root residue rj and a parent mass M. Clearly, the global optimal solution for the problem described above is maxr∈A V(Mparent, r,1).
The optimal score V(m, r, b) can be computed by dynamic programming over all PRFs with the ascending order of their PRMs:
| (4) |
where mass(r) is the mass of the monosaccharide residue r (Table 1). The computational complexity of this algorithm is O(N3), where N is the number of PRFs, which is proportional to the number of peaks in the MS/MS spectrum. Note that the above iteration formula takes into consideration only binary branching in the tree structure of the oligosaccharide. This is a realistic assumption since there are very few exceptions in a real situation. On the other hand, it is easy to modify this algorithm to consider more complex branching. At each step in the process of the forwarding iteration, we need to record the optimal trace. After we get the optimal score, we can retrieve the entire series of PRFs using a traceback procedure.
The algorithm described above maximizes a simple scoring function: the number of support peaks, i.e. the number of theoretical ions of an oligosaccharide that match the mass of peaks in the experimental spectrum. It does not consider the relative intensities of the matched peaks. Nor does it consider the potential redundantly matched peaks (i.e. one peak can support several different theoretical ions) if their masses are the same. We note that this is different from what was considered in peptide de novo sequencing algorithms (Dancik et al., 1999; Chen et al., 2001; Bafna and Edwards, 2003), in which it is forbidden to use one peak to explain more than one fragment ion. To consider this constraint, de novo peptide sequencing methods employed a dynamic programming algorithm to solve the anti-symmetric longest path problem. However, a similar problem formulation and solution allowing for branching stuctures, e.g. oligosaccharides, remains open. What is more, in oligosaccharide characterization, there are often actual redundant ions (i.e. ions with the same theoretical mass values) because oligosaccharides have a smaller alphabet than proteins and hexose is the most frequent monosaccharide. Therefore, in our scoring framework we allow the redundant usage of the peaks. A more sophisticated scoring scheme, e.g. intensity-based scores, may alleviate this problem and will be considered in the future.
Because of the complexity of oligosaccharide structures, there are often multiple solutions which match the experimental spectrum equally well in terms of the matching score, especially those oligosaccharides that differ only in linkages. Our approach can report all the optimal solutions from the dynamic programming algorithm by recording the k best solutions at each iteration step (by default k 200). We adopt a post-processing step to re-evaluate each =of them and finally rank these oligosaccharides based on their re-evaluation score. In the re-evaluation scheme, we generate a theoretical spectrum from the oligosaccharides, taking into consideration both the single cleavage (the glycosidic and the cross-ring) and the double cleavage ions. Then we compare this spectrum with the experimental spectrum, count the number of common peaks between them and use this score to rank the candidate oligosaccharides.
4 RESULTS
The algorithm described above is implemented in a program entitled GLYCH (GLYcan CHaracterization), written in the C language on a UNIX/LINUX system. We have tested this program for several experimental MS/MS spectra of oligosaccharides (shown in Fig. 4). In these experiments, N-glycans were released enzymatically by ribonuclease B (Mechref and Novotny, 1998). All oligosaccharides were permethylated before the MS/MS analysis. MS/MS spectra were obtained by MALDI/TOF/TOF-MS, using the procedure described by Mechref et al. (2003). All MS/MS spectra were pre-processed before being analyzed using GLYCH. A sliding window including 20 peaks was moved along the experimental spectrum. Inside every window, all peaks with an intensity below the average intensity minus three times the standard deviation of all peak intensities were ignored.
Fig. 4.

The structures of the oligosaccharides used for testing in this paper.
The results are summarized in Table 5. In all cases, the real structure is among the best solutions GLYCH identified. The dynamic programming approach can identify the real oligosaccharide among many other which have very similar structures, often differing by only one linkage. The re-evaluation procedure can dramatically reduce the number of possible optimal solutions, while keeping the real structure among the optimal solutions. For more complicated structures, e.g. the oligomannose with branching structure, the real oligosaccharide is not ranked best by the dynamic programming approach; after the re-evaluation, however, it is among the optimal solutions.
Table 5.
Performance of GLYCH on oligosaccharide characterization
| Category | Oligosaccharides | DP solutions Rank | OS | Score | Re-evaluation Rank | OS | Score |
|---|---|---|---|---|---|---|---|
| Linear | Hexaose | 1 | 369 | 19 | 1 | 26 | 26 |
| 3-Sialyllactose | 1 | 31 | 11 | 1 | 2 | 19 | |
| 6-Sialyllactose | 1 | 35 | 12 | 1 | 2 | 21 | |
| Tetraose-a | 1 | 61 | 13 | 1 | 3 | 20 | |
| Tetraose-c | 1 | 58 | 14 | 1 | 2 | 20 | |
| Branching | Oligomannose | 13 | 177 | 20 | 1 | 17 | 25 |
DP solutions: solutions identified by the dynamic programming approach; re-evaluation: solutions ranked by the re-evalution score; OS: number of optimal solutions; score: score of the optimal solution. The structures ranked at the first two positions are selected for the re-evaluation.
4.1 Linear oligosaccharides
We first tested GLYCH on the MS/MS spectrum of oligomannose, consisting of six mannoses linked by 1-4 glycasilic bonds. Our dynamic programming algorithm successfully determined the linear sequence of this oligosaccharide. Among the 107 optimally ranked solutions, which all have the same score (19), 37 of them give the correct linear sequence of the oligosaccharide and the real structure is among them. The other 70 solutions have the correct monosaccharide composition but wrong (branching) structures. Our re-evaluation procedure significantly reduced the redundancy of the optimal solutions. After re-evalution, only six were ranked with the highest score (26), including the real structure. All of these six optimal solutions have a linear structure and they differ only by the linkage types (1-4 or 1-6) in the middle of the chain.
Next we tested GLYCH on the MS/MS spectra of several oligosaccharides with hybrid linkage type. 3-Sialyllactose and 6-sialyllactose are two trisaccharides, both consisting of two hexoses and one sialic acid (NeuAc) at the end. The difference between them is that the linkages between the sialic acid and the hexose are 1-3 and 1-6, respectively. GLYCH correctly identified the real structure and ranked it in first place even before re-evalution. For two oligosaccharides that have more complicated structures, tetraose-a and tetraose-c, both consisting of one sialic acid, three hexoses and one acetylglucosamine, GLYCH again performed well. It identified the real structure of the oligosaccharides and ranked it in first place (among several others) after the re-evaluation step.
4.2 Oligosaccharides with branching structure
Finally, we tested GLYCH on the MS/MS spectra of a branching hexaose, an N-glycan. This oligosaccharide consists of seven monosaccharides, four mannoses, two acetylglucosamines and one sialic acid (Fig. 4), of which three mannoses and the two acetylglucosamines form the core `pentamer' structure (see Discussion section). The dynamic programming approach identified the real structure, but did not rank it in first place; its score is smaller than those of 12 other oligosaccharides. However, after the re-evalution, the real structure was re-ranked to first place among 16 other oligosaccharides. It is worth pointing out that among the 16 oligosaccharides, 13 have the linear structure with no branching, which implies that the current GLYCH algorithm prefers linear structure to branching structure. This issue has to be addressed by the future improvement of the scoring function used in GLYCH.
5 DISCUSSION
We proposed a novel algorithm to interpret the MS/MS spectra of oligosaccharides de novo. The preliminary performance tests show that our scoring function, which simply counts the number of support peaks, gives encouraging results. However, in most cases, the real structure is scored similarly to several other oligosaccharides. The more complicated the oligosaccharide structure was, the more optimal solutions we got. Furthermore, this simple scoring function prefers linear structure to branching structure, maybe because linear structures often have more hypothetical cross-ring cleavages, and hence have more (random) chances of matching experimental peaks. In addition, we found that our post-processing procedure often improved the rank of the real structure. This is because the real structure can match more peaks from double cleavages. However, it is not easy to incorporate double cleavage ions into the scoring function used in the dynamic programming algorithm. In order to improve scoring function, a probabilistic model is required to describe how likely it is that one fragmentation could happen and one ion could be captured by the MS. In this way, we will be able to model not only the appearance but also the intensity of the peaks. Such a model has recently been described for peptide fragmentation in low energy MS (Zhang, 2004). But similar models have not been well studied for glycans, except that the case study showed that the relative intensities of different cross-ring peaks were useful to distinguish 1-4 and 1-6 linkages of isomeric oligosaccharides (Mechref et al., 2003). We are going to generate plenty of experimental MS/MS spectra of glycans in order to perform a systematic study of this topic. The long-term objective will be to develop an intensity-based scoring function that helps distinguish different linkages and branchings in high resolution. We expect that this new scoring scheme will significantly reduce the redundancy in the current solutions and therefore can be used in a large-scale benchmarking test.
The examples used in this paper are for testing purposes and not from a real glycomics study. We must address some practical issues when applying this algorithm to high-throughput glycomics studies, in which we may encounter large glycans with complex branching structures. One of them is how to incorporate the knowledge from other glycobiology experiments. For instance, in higher animals, N-linked glycosylation starts from the additions of five oligosaccharides (two acetylglucosamines and three mannoses) forming a unique branching core structure, often called a `pentamer', which is covalently linked to the amino acid residue. Additional variant oligosaccharides can be added to this core structure and form different branching configurations (e.g. the hexaose in Fig. 4). In other words, the structures of oligosaccharides from in vivo studies are often variants of some regular structures, which are largely determined by the glycan transferases present in the organism. By incorporating this knowledge, on the one hand, we may reduce the search space of the optimal solutions; on the other hand, the scoring function needs to be modified according to those glycan structures that are favored in cells.
The ultimate goal of glycomics and glycoproteomics is to annotate the structure of glycoproteins, including
identification of the glycoproteins;
characterization of the modification of oligosaccharides;
determination of the modification site, i.e. the residues to which the oligosaccharides are attached.
In this paper, we partially addressed the second problem. In fact, the other two problems are equally important, but more difficult computationally. For example, so far no method has been proposed to interpret the MS/MS spectra of glycopeptides. We are going to study these computational problems in the future.
ACKNOWLEDGEMENTS
This work was partially funded by Grant No. (GM24349) from the National Institute of General Medical Sciences, US Department of Health and Human Services, and the Indiana Genomics Initiative, which is funded in part by the Lilly Endowment, Inc.
Footnotes
We note by this definition that the root residue N does not have the feature of linkage type; thus we always (arbitarily) assign bn to 1 (1-2 linkage).
Some noisy ions may be explained by the products of double cleavages, i.e. two cleavages at different sites. At this step, we ignore the effect of these ions and treat them as noisy peaks.
REFERENCES
- Bafna V, Edwards N. On de novo interpretation of peptide mass spectra. Proceedings of Research in Computational Biology (RECOMB).2003. pp. 9–18. [Google Scholar]
- Bandeira N, Tang H, Bafna V, Pevzner PA. Shotgun protein sequencing by tandem mass spectra assembly. Anal. Chem. 2004;76:7221–7233. doi: 10.1021/ac0489162. [DOI] [PubMed] [Google Scholar]
- Chen T, Kao MY, Tepel M, Rush J, Church GM. A dynamic programming approach to de novo peptide sequencing via tandem mass spectrometry. J. Comput. Biol. 2001;8:325–337. doi: 10.1089/10665270152530872. [DOI] [PubMed] [Google Scholar]
- Chou H, Takematsu H, Diaz S, Iber J, Nickerson E, Wright KL, Muchmore EA, Nelson DL, Warren ST, Varki A. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc. Natl Acad. Sci. USA. 1998;95:11751–11756. doi: 10.1073/pnas.95.20.11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CA, Gasteiger E, Packer NH. GlycoMod—a software tool for determining glycosylation compositions from mass spectrometric data. Proteomics. 2001;1:340–349. doi: 10.1002/1615-9861(200102)1:2<340::AID-PROT340>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Cooper CA, Joshi HJ, Harrison MJ, Wilkins MR, Packer NH. GlycoSuiteDB: a curated relational database of glycoprotein glycan structures and their biological sources. Nucleic Acids Res. 2003;31:511–513. doi: 10.1093/nar/gkg099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancik V, Vddona TA, Clauser KR, Vath JE, Pevzner PA. de novo peptide sequencing via tandem mass spectrometry. J. Comput. Biol. 1999;6:327–342. doi: 10.1089/106652799318300. [DOI] [PubMed] [Google Scholar]
- Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconjugate. 1988;5:397–409. [Google Scholar]
- Eithier M, Saba JA, Spearman M, Krokhin O, Butler M, Ens W, Standing KG, Perreault H. Application of the StrOligo algorithm for the automated structure assignment of complex N-linked glycans from glycoproteins using tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2002;17:2713–2720. doi: 10.1002/rcm.1252. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequencesin a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Gaucher SP, Morrow J, Leary JA. STAT: a saccharide topology analysis tool used in combination with tandem mass spectrometry. Anal. Chem. 2000;72:2332–2336. doi: 10.1021/ac000096f. [DOI] [PubMed] [Google Scholar]
- Joshi HJ, Harrison MJ, Schultz BL, Cooper CA, Packer NH, Karlsson NG. Development of a mass fingerprinting tool for automated interpretation of oligosaccharide fragmentation data. Proteomics. 2004;4:1650–1664. doi: 10.1002/pmic.200300784. [DOI] [PubMed] [Google Scholar]
- von der Lieth CW, Bohne-Lang A, Lohmann KK, Frank M. Bioinformatics for glycomics: status, methods, requirements and prespective. Brief Bioinform. 2004;5:164–178. doi: 10.1093/bib/5.2.164. [DOI] [PubMed] [Google Scholar]
- Lohnmann KK, von der Lieth CW. GlycoFragment and GlycoSearchMS: web tools to support the interpretation of mass spectra of complex carbohydrates. Nucleic Acids Res. 2004;32:W261–W266. doi: 10.1093/nar/gkh392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loss A, Bunsmann PB, Ohne A, Loss ,A, Schwarzer E, von der Lieth CW. Sweet-DB: an attempt to create annotated data collections for carbohydrates. Nucleic Acids Res. 2002;30:405–408. doi: 10.1093/nar/30.1.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, Lajoie G. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003;17:2337–2342. doi: 10.1002/rcm.1196. [DOI] [PubMed] [Google Scholar]
- Mechref Y, Novotny MV. Mass spectrometric mapping and sequencing of N-linked oligosaccharides derived from submicrogram amounts of glycoproteins. Anal. Chem. 1998;1998:455–463. doi: 10.1021/ac970947s. [DOI] [PubMed] [Google Scholar]
- Mechref Y, Novotny MV. Structural investigations of glycoconjudates at high sensitivity. Chem. Rev. 2002;2002:321–369. doi: 10.1021/cr0103017. [DOI] [PubMed] [Google Scholar]
- Mechref Y, Novotny M, Krishnan C. Structural characterization of oligosaccharides using MALDI-TOF/TOF tandem mass spectrometry. Anal. Chem. 2003;2003:4895–4903. doi: 10.1021/ac0341968. [DOI] [PubMed] [Google Scholar]
- Shan B, Zhang K, Ma B, Zhang C, Lajoie G. An Algorithm for Determining Glycan Structures from MS/MS Spectra. Proceedings of International Conference on Bioinformatics and its Applications.2004. [Google Scholar]
- Stanley P. Biological consequences of overexpressing or eliminating N-acetylglucosaminyltransferase-TIII in the mouse. Biochim. Biophys. Acta. 2002;1573:363–368. doi: 10.1016/s0304-4165(02)00404-x. [DOI] [PubMed] [Google Scholar]
- Varki A, Cummings R, Esko J, Freeze H, Hart G, Marth J.Essencials of Glycobiology 1999. Cold Spring Harbor Laboratory Press [PubMed] [Google Scholar]
- Zaia J. Mass Spectrometry of oligosaccharides. Mass Spectrometry Reviews. 2004;13:161–227. doi: 10.1002/mas.10073. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Prediction of low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 2004;14:3908–3922. doi: 10.1021/ac049951b. [DOI] [PubMed] [Google Scholar]