

Abstract
Activation of complement on endothelium triggers physiologic changes that promote coagulation, thrombosis and inflammation. Unlike agonists such as cytokines and endotoxin that induce these changes through transcription of many genes, complement, particularly the membrane attack complex, primarily induces release of IL-1α by the endothelial cells; the cytokine may then be removed by normal blood flow or may promote activation of the full range of endothelial cell responses in an autocrine or paracrine manner. We studied the intracellular signaling pathways used by complement to activate IL-1α transcription in cultured endothelial cells. The membrane attack complex and other pore-forming proteins stimulated calcineurin and activated selective transcription of the IL-1α gene. In contrast, the action of cytokines such as IL-1α was not selective and not dependent on calcineurin activity. Transcription of IL-1α, whether stimulated by complement and calcineurin or by “conventional agonists” such as IL-1α independent of calcineurin, proceeded via binding of nuclear factor κB transcriptional activators to the IL-1α gene promoter. These findings define a molecular mechanism through which complement regulates IL-1α production by endothelial cells and explain how blood flow may determine the extent of complement-stimulated inflammation.
Keywords: calcineurin, complement, cyclosporine, endothelium, interleukin-1, membrane attack complex, NF-κB, FK506
Introduction
The endothelial cells lining blood vessels form a highly specialized, metabolically active interface between the blood and underlying tissues. In this capacity, endothelium monitors health and disease in tissues and rapidly integrates signals representing each to induce an appropriate response by blood vessels and circulating cells. For example, under conditions of well being, endothelium expresses thrombomodulin1 and the tissue factor pathway inhibitor,2 which inhibit coagulation; and prostacycline I2,3 which inhibits platelet aggregation; and nitric oxide, which relaxes vascular smooth muscle. This anti-inflammatory and anti-coagulative posture may reflect interactions of endothelial cells with the microenvironment4-6. On the other hand, under conditions of injury, infection, or disease, endothelium is exposed to such agonists as endotoxin, thrombin, and heparan sulfate, and, in response, it expresses tissue factor7,8 and plasminogen activator inhibitor 1 (PAI-1),9 which promote coagulation and E-selectin8; interleukins (IL-610 and IL-1α7) and chemokines (IL-811), which promote inflammation; and thromboxane A2,12 which induces constriction of vascular smooth muscle.
The set of coordinated changes in endothelial function that promote coagulation, inflammation, and vasoconstriction have been called endothelial “perturbation” or “activation”13,14. Endothelial activation was first described in endothelial cells exposed to endotoxin or cytokines such as IL-1β, TNFα or IL-613,15-17. Endotoxin activates endothelial cells by stimulating Toll-like receptor 4 (TLR4)18, and IL-1 stimulates one or more of several IL-1 receptors (IL-1R)19. These receptors, particularly TLR4 and IL-1R, invoke well-known intracellular signaling pathways that among various changes activate NF-κB, a transcriptional regulator that orchestrates expression of pro-inflammatory mediator genes in endothelial cells20.
Activation of endothelium by complement differs in certain respects from activation by endotoxin or cytokines. Endotoxin and cytokines stimulate receptors that initiate well known signaling pathways culminating in translocation of NF-κB. Complement activation generates anaphylotoxins (C3a and C5a) which stimulate G-protein coupled receptors (C3aR and C5aR, respectively) and protein complexes which insert into cell membranes activating the small GTPase Ras21 and mobilizing intracellular calcium22. However, activation of endothelium by C3aR and C5aR is appreciable only after endothelium is first stimulated by IL-1 α23, 24 and rapid inactivation of anaphylotoxins by carboxypeptidase N and carboxypeptidase R25 limit their effect.
Insertion of the membrane attack complex into endothelium induces procoagulant and proinflammatory changes typical of endothelial cell activation; however, it does not do so as a direct response to stimulation. Rather, the membrane attack complex induces transcription, production and secretion by endothelial cells of IL-1α7 and perhaps a few other factors26, but not the range of procoagulant and proinflammatory substances seen with endothelial cell activation. The IL-1α released from endothelial cells can stimulate endothelial cells in an autocrine or paracrine fashion to generate the activated phenotype, or it might be washed away in which case endothelial cells remain quiescent4. We have postulated that autocrine or paracrine activation of endothelial cells by IL-1α following exposure to the membrane attack complex promotes local activation of endothelium when blood flow is low, whereas the washing away of IL-1α prevents activation of endothelium in physiologic conditions4.
We explored the means by which complement or other pore forming proteins induce production of IL-1α by endothelial cells. Our studies revealed that transcriptional activation of IL-1α in response to these proteins requires utilization by NF-κB of specific sequences in the promoter region of the IL-1α gene. Stimulation of endothelial cells by pore-forming proteins like the membrane attack complex stimulates calcineurin-dependent activation and binding of NF-κB to the IL-1α gene promoter, to selectively activate transcription of that gene. The selective activation of IL-1α by complement and calcineurin-dependence of the response contrasts with the typical response of endothelial cells to cytokines and endotoxin, which orchestrate transcription of a broad program of inflammatory gene products20. This finding may help explain endothelial responses to infectious challenges and may offer new avenues for therapy for complement-mediated disease.
Materials and Methods
Materials
Human serum was used as a source of complement7, 27. Recombinant IL-1α and TNF-α were from R&D systems (Minneapolis, MN). Melittin, ionomycin, phorbol-12-myristate-13-acetate (PMA) and FK506 were from Calbiochem (La Jolla, CA).
Cell Cultures
Porcine aortic endothelial cells were cultured and characterized as described27 and were used between the fourth and eighth passage. Human aortic endothelial cells were from Cambrex Bio Science (Santa Rosa, CA) and cultured as suggested by the company. Endothelial cells were transfected on 24 well plates at approximately 50% confluency. For preparation of RNA or nuclear extracts the cells were cultured as confluent monolayers.
Analysis of mRNA by PCR
Total RNA from endothelial cells was isolated by the guanidinium thiocynate method28. Reverse transcriptase-polymerase chain reaction (RT-PCR) was performed as described27. The PCR products were identified by sequencing and the relative levels of various mRNA were compared to GAPDH mRNA7. The sequence of oligonucleotides used for PCR were:
Human IL-1α, CCAGGCGTAGGTCTGGAGTCTCACTTGTCT and TGTTGCGGCAGGAAGGCTTAGGTATTATTC; human GAPDH, CATGCCATCACTGCCACCCAGAAGACTGTG and GAAATGAGCTTGACAAAGTGGTCGTTGAGG; porcine IL-1α, CGGGAAGATTCTGAAGAAGAGACGGTTGAG and TGGGCGGCTGATTTGAAGTAGTCCATATTG; porcine GAPDH, CCGCGTCCCTGAGACACGATGGTGAAGGTC and TTCAAGTGAGCCCCAGCCTTCTCCATGGTC; porcine E-selectin, GCAAAAAGAAGCTCGCCTTGTGCTACACAG and GCTTTACAAACTGGGGCTTGCTGGGTCCAT.
The products were fractionated in 1.2% agarose-TBE gels, stained with ethidium bromide, and quantitated using the Gel Doc 2000 and Quantity One software (Bio-Rad, Hercules, CA).
Cloning of the IL-1α promoter
The porcine IL-1α promoter region was cloned from a porcine liver genomic library prepared in Lambda FIX® II vector (Stratagene, La Jolla, CA). Ten million plaques were screened using porcine IL-1α cDNA (M86730), yielding 3 clones. From these clones we identified a 3.5 kb DNA 5' non-coding region of IL-1α which was sub-cloned in Blusescript KS(+) (Stratagene) for sequencing. The human IL-1α promoter, 156 bp 5' to the human IL-1α transcription start site, was amplified using PCR and genomic DNA obtained from human aortic endothelial cells. The primers used for this reaction were AGTTTTAGCCAGTATCGAGTTGAATGAACATAGAA and AAGCCTGAGTCAGTCTTCTTCGCCTTTTGTAATTG (HGenBank accession no. X03833.1).
Analysis of the IL-1α promoter region
Wild-type or mutated human and porcine IL-1α promoter regions were cloned into pGL3-basic luciferase reporter vector (Promega, Madison, WI). Endothelial cells were transfected with 500 ng of reporter vector using SuperFect (Qiagen, Valencia, CA). The efficiency of transfections was determined by co-transfecting 100 ng of Renilla luciferase reporter vector as an internal control (pRL-TK, Promega). After 40-48 hours, the cells were lysed in 200 μl of Passive Lysis Solution (Promega). Luciferase activity was determined using the Dual-Luciferase Reporter Assay system (Promega) and a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA).
NF-κB translocation analysis
Nuclear translocation of NF-κB was analyzed by electrophoretic mobility shift assay. Nuclear protein was extracted from porcine aortic endothelial cells as described29. Double strand DNA probes were prepared by end-labeling with γ[32P]ATP (Amersham, Piscataway, NJ) and T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and were purified using G-25 Sepahdex Quik Spin Columns (Roche, Indianapolis, IN). The sequences of the oligonucleotide probes were TTCTTCCCTGTAAATTCCCCGTTTTG and TTCTTCCCTGTAAATTCCCCACGA which were derived from the human and porcine IL-1α gene promoters, respectively.
The electrophoretic mobility shift assay was performed as described29. Nuclear extracts (4-5 μg) were incubated with labeled probes (0.25 pmol/reaction) on ice for 15 min. In some experiments 1 μl of antibodies were included in the reactions. Binding reactions (15 μl) contained 2 μg Poly(dI-dC)•Poly(dI-dC) and 15-20 μM non-specific oligonucleotide in binding buffer [30mM HEPES (pH8.0), 0.4mM EDTA (pH8.0), 0.08% Igepal]. The protein-DNA complexes were resolved on 4% non-denaturing polyacrylamide gels and analyzed as described29.
Results
Calcium-mediated induction of IL-1α-transcription
Since complement activates endothelium mainly through insertion of terminal complexes in cell membranes and production and secretion of IL-1α27, we asked how complement induces transcription of IL-1α. Because cellular responses to terminal complement complexes require mobilization of intracellular calcium, we first asked whether changes in intracellular calcium contribute to transcription of IL-1α mRNA. To address this question, porcine and human aortic endothelial cells were stimulated with ionomycin to increase the concentration of intracellular free calcium and changes in the level of IL-1α mRNA was sought by RT-PCR using total cellular RNA and IL-1α-specific primers. As Figure 1 shows, stimulation of endothelial cells with ionomycin dramatically increased expression of IL-1α mRNA within four hours.
Figure 1.

Activation of IL-1α expression in endothelial cells by calcium and complement. Aortic endothelial cells (porcine, PAEC; or human, HAEC) were stimulated for four hours with 1 μM ionomycin to mobilize intracellular calcium, or with 1 ng/ml interleukin 1α (IL-1) or 1 μM phorbol-12-myristate-13-acetate (PMA). In some experiments porcine aortic endothelial cells were exposed to activated complement by incubating the cells with 12% human serum. Total cellular RNA was isolated and the relative amounts of IL-1α and GAPDH mRNA present were determined by RT-PCR and agarose gel electrophoresis. The amount of IL-1α mRNA, relative to GAPDH, was determined by scanning densitometry of gels run following PCR reactions (bar graphs in B and C). Results representative of four experiments are shown.
(A) Ionomycin stimulates IL-1α expression in cultured human and porcine endothelial cells.
(B) Ionomycin-stimulated IL-1α expression is mediated by calcineurin. Porcine aortic endothelial cells were cultured with 1 μM cyclosporine A (CsA) for 2 hours to inhibit calcineurin and then stimulated with ionomycin (In), IL-1α or PMA. Cyclosporine A completely abolished IL-1α expression in response to ionomycin, but did not inhibit IL-1α expression in response to other endothelial cell activators.
(C) Complement-stimulated IL-1α expression requires calcineurin. Porcine aortic endothelial cells were treated with the calcineurin inhibitors FK506 or CsA for 2 hours and then with complement (12% human C) for 4 hours. Calcineruin inhibitors prevented complement-mediated activation of IL-1α expression.
These results demonstrate that calcium mobilization or complement stimulates calcineurin-dependent IL-1α expression.
Since changes in intracellular calcium can activate the calcium-dependant phosphatase calcineurin and the protein kinase C (PKC) serine-threonine protein kinases, we asked whether activation of one or more of these molecules induces transcription of IL-1α. To address that question, porcine aortic endothelial cells were treated with cyclosporine A, an inhibitor of calcineurin, and then with ionomycin and the level of IL-1α transcription was measured. As a positive control, IL-1α transcription was also measured in cells stimulated with recombinant porcine IL-1α, which activates IL-1α transcription, in the presence or absence of cyclosporine A. As shown in Figure 1B, cyclosporine A completely abolished ionomycin-induced IL-1α transcription, but had no effect on the stimulation of IL-1α transcription by IL-1α. These results suggest that mobilization of intracellular calcium activates IL-1α transcription through a calcineurin-dependent mechanism.
Since increases in intracellular calcium activate conventional PKC serine-threonine kinases30, we tested whether these kinases might also contribute to the activation of IL-1α transcription. Porcine aortic endothelial cells were treated with phorbol-12-myristate-13-acetate (PMA) to activate PKC and the amount of IL-1α mRNA was determined four hours later. PMA stimulated IL-1α transcription in endothelial cells to the same degree as treatment with ionomycin or IL-1α, however, in contrast to the response to ionomycin, transcription of IL-1α mRNA induced by PMA was not inhibited by cyclosporine A (Figure 1B). Thus, changes in intracellular calcium stimulate IL-1α gene transcription independent of PKC serine-threonine kinases and through a distinct, calcineurin-dependant pathway.
We next asked whether complement uses calcineurin to induce expression of IL-1α in endothelial cells. To address this question, porcine aortic endothelial cells were incubated with human serum to activate complement in the presence or absence of the cyclosporine A or FK506 and the level of IL-1α transcription was measured. Complement activation on porcine aortic endothelial cells stimulated expression of IL-1α in the cells (Figure 1C). Cells treated with calcineurin inhibitors prior to stimulation with complement expressed significantly less IL-1α mRNA than cells not treated with calcineurin inhibitors (Figure 1C). Thus, complement acts, at least in part through calcineurin to induce expression of IL-1α in endothelial cells. The suppression of IL-1α transcription by calcineurin inhibitors did not reflect “non-specific” toxicity because cells treated with cyclosporine A responded fully to stimulation with IL-1α or PMA (Figure 1B). The concentrations of calcineurin inhibitors used in these experiments represent therapeutic blood concentrations, thus these results suggest that calcineurin inhibitors might find new uses in modifying blood vessel disease caused by complement activation.
Calcineurin-mediated induction of IL-1α-transcription
Endothelial cells respond to endotoxin, IL-1α and other inflammatory stimuli by expressing a number of pro-inflammatory genes and producing corresponding inflammatory gene products, including IL-1α20, 27. Since calcium mobilization by complement activates IL-1α transcription through a distinct, calcineurin-dependant pathway, we asked whether the response of endothelial cells to activation of calcineurin differs from the response to other stimuli. To address this question, we tested whether activated calcineurin itself stimulates transcription of pro-inflammatory genes. We transfected porcine aortic endothelial cells with a vector encoding a constitutively active calcineurin mutant, (ΔCaM-AI)31, and then measured the levels of IL-1α and E-selectin mRNA. Both IL-1α and E-selectin mRNA are found in endothelial cell activated by endotoxin or cytokines but only IL-1α is found as a primary gene product in endothelial cells activated by complement27. Control cells were transfected with a control plasmid with or without added IL-1α. As shown in Figure 2, transfection with the ΔCaM-AI plasmid increased expression of IL-1α mRNA in a dose dependent manner but it did it did not induce expression of E-selectin mRNA (Figure 2) or GAPDH or other genes (not shown). In contrast, treatment of cells with IL-1α induced both IL-1α and E-selectin, as expected. These findings indicate that active calcineurin selectively induces expression of IL-1α in endothelial cells.
Figure 2.
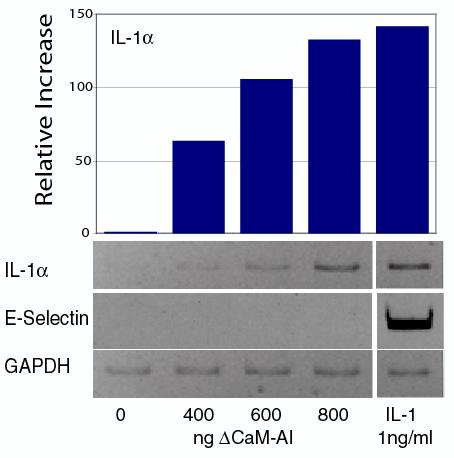
Calcineurin selectively activates IL-1α expression. Porcine aortic endothelial cells were transfected with increasing amounts of a plasmid encoding constitutively-active calcineurin (ΔCaM-AI). Control cells were stimulated with IL-1α. After 12 hours the amounts of IL-1α, Eselectin and GAPDH mRNA were measured by RT-PCR. The results show that calcineurin activity selectively activates IL-1α expression.
Regulation of IL-1α expression
Since mobilization of intracellular calcium and activated calcineurin selectively stimulate expression of IL-1α mRNA, we asked whether the promoter controlling expression of the IL-1α gene in endothelial cells is regulated by calcineurin. To address this question, we cloned the promoter region of the IL-1α gene from porcine aortic endothelial cells and generated luciferase reporter vectors to identify the region controlled by calcineurin. As Figure 3 shows, ionomycin induced luciferase in cells transfected with a vector encoding the full IL-1α promoter and with a vector encoding a 163 base-pair region 5' to the transcription start site (Figures 3A and 3B). Luciferase activity was absent or nearly so in cells transfected with a vector lacking this “calcium-response region”.
Figure 3.

The IL-1α promoter is regulated by calcium and calcineurin. Porcine aortic endothelial cells were transfected with vectors containing a luciferase reporter regulated by various regions of the porcine IL-1α gene promoter. The cells were stimulated with 1 μM ionomycin for 6 hours to mobilize intracellular calcium and activation of the IL-1α gene promoter was measured by luciferase assay of cell lysates.
(A) Ionomycin stimulates porcine IL-1α promoter activity. The calcium-responsive region of the IL-1α gene is an element approximately 163 base-pairs 5' of the IL-1α gene transcriptional start site.
(B) The nucleotide sequence of the calcium-responsive element within the IL-1α gene promoter.
(C) The calcium-responsive element of the IL-1α gene promoter requires calcineurin for activation. Porcine aortic endothelial cells were transfected with a luciferase reporter vector regulated by the calcium-responsive element of the IL-1α gene promoter. Transfected cells were treated for 2 hours with 1 μM cyclosporine A (CsA) or with 100 nM FK506 and then stimulated with ionomycin. Cyclosporine A and FK506 reduced ionomycin-stimulated luciferase production.
These results indicate that the IL-1α gene is regulated by a 163 base-pair calcium- and calcineurin-responsive element near the IL-1α transcriptional start site.
To determine whether the calcium-regulated region of the IL-1α promoter requires calcineurin activity, we transfected endothelial cells with the minimal IL-1α promoter-reporter vector and treated the cells with calcineurin inhibitors and with ionomycin. Cyclosporine A or FK506 treatment inhibited IL-1α promoter responses to ionomycin stimulation (Figure 3C) to the same extent as they had inhibited transcription of endogenous IL-1α mRNA in endothelial cells (Figure 1B). These results show that calcium controls transcription of the IL-1α gene, probably by activation of calcineurin.
To determine whether calcineurin activity directly regulates the IL-1α promoter, we transfected porcine aortic endothelial cells with reporter vectors regulated by either the porcine or the human IL-1α promoter and tested responses to co-transfected ΔCaM-AI. Control cells were cotransfected with the ΔCaM-AI plasmid and with a luciferase reporter vector regulated by the SV40 promoter, or with the IL-1α promoter vector without ΔCaM-AI. The ΔCaM-AI plasmid, at very low concentrations (1:25, ΔCaM-AI:IL-1α) increased porcine promoter activity by 7.2-fold and human IL-1α promoter activity by 8.6-fold compared to controls transfected with the IL-1α promoter luciferase reporter plus a plasmid lacking calcineurin (Figure 4A). Calcineurin activity targets the IL-1α promoter and not other steps in production of luciferase protein, because ΔCaM-AI did not increase the activity of SV40 promoter-driven luciferase production (Figure 4A). Stimulation of the IL-1α promoter by ΔCaM-AI required calcineurin activity, because luciferase production was inhibited by calcineurin inhibitors (Figure 4B). These results show that calcineurin activates transcription from the IL-1α promoter.
Figure 4.

Calcineurin activates a “calcium-regulated” element of the IL-1α gene promoter.
(A) Porcine aortic endothelial cells were transfected with luciferase reporter vectors regulated by porcine or human IL-1α promoters. Control cells were transfected with a luciferase reporter vector controlled by the SV40 promoter. The cells were co-transfected with expression vectors encoding either ΔCaM-AI constitutively active calcineurin or control (empty) vectors and luciferase production was measured 12 hours later. Calcineurin activity specifically stimulated IL-1α promoter activity.
(B) Porcine or human aortic endothelial cells were co-transfected with ΔCaM-AI or control calcineurin expression vectors and with luciferase reporter vectors regulated by porcine (left panel) or human (right panel) IL-1α promoters. Transfected cells were incubated with FK506 for 12 hours, and luciferase production was measured. Calcineurin-activated IL-1α promoter activity was abolished by FK506.
These results show that calcineurin directly stimulates human and porcine IL-1α promoter activity in endothelial cells.
Mapping of the calcineurin response region
To characterize the region of the IL-1α promoter that responds to calcium and calcineurin, we generated deletion and point mutations within the porcine IL-1α promoter in the luciferase reporter construct and tested responsiveness in endothelial cells co-transfected with ΔCaM-AI. Analyses of multiple deletion mutants identified the sequence “CTTCCCTGTAAATTCCCCAC” located ∼ 90 bp upstream of the transcription start site as critical for activation of the promoter by calcineurin. When this sequence was removed, the response of the IL-1α promoter to ΔCaM-AI was significantly decreased (Figure 5). This sequence contains a putative NF-κB binding site (underlined sequences)32 and is identical in the human and the porcine IL-1α promoters. Point mutations of the IL-1α luciferase reporter constructs within “NF-κB sites” were less responsive to calcineurin than were mutants with intact NF-κB binding sites, while point mutations elsewhere responded fully or nearly so to calcineurin (Figure 5). These results suggest that NF-κB activity is essential for induction of IL-1α by calcineurin.
Figure 5.
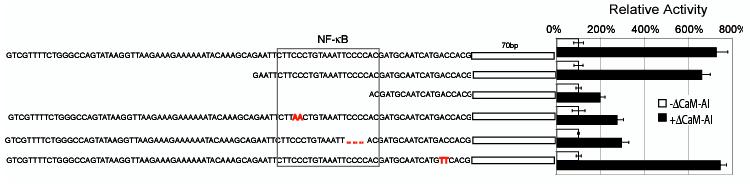
Calcineurin-stimulated IL-1α promoter activity requires NF-κB. Porcine aortic endothelial cells were co-transfected with ΔCaM-AI expression vector and various IL-1α gene promoter reporter vectors. Deletions (dashes in sequence) or mutations (highlighted in red type) within the NF-κB-binding promoter sequence profoundly reduced calcineurin-stimulated IL-1α gene promoter activity.
Identification of NF-κB binding sites in IL-1α promoter
To determine whether the calcineurin responsive region of the IL-1α promoter interacts with NFκB proteins, we performed electrophoretic mobility shift assays using proteins isolated from endothelial cell nuclei and 32P-labled CTTCCCTGTAAATTCCCCAC probes derived from the calcineurin-responsive sequence in the IL-1α gene promoter. Nuclei from human or porcine endothelial cells that had been stimulated with IL-1α contained a protein(s) that formed a complex with the probe (Figure 6A and B). Antibodies specific for NF-κB components p65 and p50 further shifted complexes formed between porcine endothelial cell nuclear proteins and CTTCCCTGTAAATTCCCCACGA derived from the porcine IL-1α promoter, and complexes formed between human endothelial cell nuclear proteins and CTTCCCTGTAAATTCCCCCGTTTG derived from the human IL-1α promoter. Probes with mutated bases in the NF-κB binding consensus site failed to form complexes when incubated with the nuclear extracts (Figure 6B). These findings show that the calcineurin response region of the IL-1α promoter contains a binding site for NF-κB.
Figure 6.

NF-κB complexes form on the IL-1α gene promoter. Human (HAEC) or porcine (PAEC) aortic endothelial cells were stimulated for 4 hours with PBS (Un) or human or porcine IL-1α. Nuclear proteins were extracted and incubated with 32P-labeled oligonucletide probes containing putative NF-κB binding sites found within the human or porcine IL-1α gene promoter. Protein-DNA complexes were detected by polyacrylamide gel electrophoretic mobility shift assay and autoradiography.
(A) IL-1α stimulated nuclear proteins that reacted with human (left) or porcine (right) IL-1α gene promoter probes and with antibodies specific for the p65 and p50 components of NF-κB.
(B) IL-1α-stimulated NF-κB proteins in the nuclei of porcine aortic endothelial cells reacted with wild-type IL-1α gene promoter probe (panel A). Reactivity was reduced (panel B) or eliminated (panel C) with probes containing mutated NF-κB binding sites.
These results demonstrate that the calcium and calcineurin response element in the IL-1α gene promoter binds activated NF-κB proteins.
Role of the NF-κB/calcineurin responsive site in the induction of IL-1α by the membrane attack complex of complement and other pore forming proteins
We next asked whether insertion of pore-forming complexes such as the membrane attack complex generated by complement activation induce NF-κB binding to the calcineurin response site of the IL-1α promoter. To address that question, we incubated porcine endothelial cells with human serum to activate complement or treated the cells with melittin, a pore-forming protein that, like membrane attack complex, mobilizes calcium and induces transcription of IL-1α mRNA in endothelial cells27. Nuclear extracts isolated from endothelial cells exposed to melittin contained proteins that formed specific complexes with the IL-1α promoter probe (Figure 7A). Similar complexes were detected using nuclear extracts from endothelial cells stimulated with complement (Figure 7A). Antibodies specific for NF-κB components p65 and p50 retarded the mobility in gels of the complexes formed between nuclear proteins from complement treated endothelial cells and the probe (Figure 7B). These results indicate that complement and other pore forming proteins stimulate the appearance of NF-κB complexes in endothelial cell nuclei that bind to the IL-1α promoter.
Figure 7.

Complement and other pore-forming peptides stimulate NF-κB-binding to the IL-1α gene promoter. Porcine aortic endothelial cells were incubated for four hours with PBS (Un) or with human complement (Co) or melittin (Mel), polypeptides that form pores in cell membranes, or with IL-1α. Nuclear NF-κB proteins bound to the 32P-labeled porcine IL-1α gene promoter probe was detected by electrophoretic mobility shift assay.
(A) Melittin and complement stimulate NF-κB-DNA binding activity in the nuclei of endothelial cells.
(B) Complement stimulates DNA binding of NF-κB components p65 and p50.
(C) Complement-stimulated NF-κB activity requires calcineurin. FK506 abolished complement-stimulated appearance of NF-κB in cell nuclei.
These results show that complement and other pore-forming polypeptides stimulate calcineurindependant NF-κB appearance in nuclei of endothelial cells.
To determine whether calcineurin modifies NF-κB activation by complement, we treated endothelial cells with FK506 and then with complement and tested whether NF-κB proteins appeared in the nuclei of treated cells. As Figure 7 shows, FK506 completely inhibited complement-stimulated appearance of nuclear proteins reactive with NF-κB-binding IL-1α promoter probe. Since calcineruin inhibitors did not affect the ability of IL-1α to stimulate IL-1α transcription (Figure 1) activation of NFκB in endothelium by complement must proceed through a pathway distinct from that of cytokines and endotoxin.
Discussion
Here we report that activation of endothelial cells by the membrane attack complex of complement and other pore forming proteins proceeds though calcium-dependant activation of calcineurin and activation of NF-κB, which together induce transcription of IL-1α. The combined action of activated calcineurin and NF-κB is highly specific for transcriptional activation of IL-1α and this specificity may explain, in part, how complement, unlike other endothelial cell agonists, focuses responses of endothelial cells on IL-1α production.
Focusing responses of endothelium through regulation of IL-1α transcription may help avoid unwanted activation of endothelium. Thus, full activation of endothelium by complement occurs only in those segments of blood vessels that are directly targeted by complement and injured (thus having reduced blood flow), or those segments immediately downstream (and thus exposed to IL-1α). In contrast, activation of complement without concomitant blood vessel injury and without reduced blood flow, as might occur when complement is activated on the surface of circulating cells, would not perturb endothelium and hence would not induce coagulation and inflammation. Thus, the clearing of dead or neoplastic cells by complement33, 34, and formation of common auto-antibodies such as cold agglutinins35, does not eventuate in widespread coagulation and inflammation since terminal complexes that insert in endothelial cells and activate transcription of IL-1α do not generate full activation of endothelium.
How tissue damage regulates the molecular response to complement activation is an important reflection of regional physiology4. Damage to tissues and particularly damage to blood vessels can eventuate reduction of blood flow through several independent mechanisms. First, endothelial disruption triggers aggregation of platelets which directly occlude blood flow and release thromboxane A2 which stimulates vasoconstriction12. Second, damaged or denuded endothelium provides less nitric oxide, the decrease in availability of which also promotes vasoconstriction. Third, tissue damage causes shedding of heparan sulfate36, 37 the loss of which deprives endothelium of a key anticoagulant38 and thus allows formation of occlusive thrombi. When tissue damage causes vasoconstriction or vaso-occlusion or disruption of blood flow, IL-1α produced in response to complement provokes ongoing coagulation, inflammation and immunity. Such a condition might be advantageous when microorganisms enter injured tissues in which case complement activation may activate endothelium and initiate inflammation and sequestration of microorganisms prior to activation of other immune cells.
Selective stimulation of IL-1α by complement- and calcium-stimulated calcineurin may be restricted to endothelial cells, since constitutively active calcineurin did not activate the IL-1α gene promoter in porcine cardiac smooth muscle cells (unpublished results, G.J. Brunn and J.L. Platt, 2005). The molecular mechanisms that regulate the IL-1α gene and underlie differential expression are complex and only partially understood. McDowell et al.39 recently found that transcription of the human IL-1α gene is constitutively suppressed by a protein(s) bound to a DNA sequence 448 base-pairs 5' to the transcription start site; IL-1α is also positively regulated by the Sp-1 transcription factor, which binds the IL-1α gene promoter sequence 52 base-pairs 5' to the transcriptional start site. These authors did not describe NF-κB activity acting on the IL-1α promoter, as we show here, however Sp-1 has been shown to bind directly to a subset of NF-κB-binding sequences40. The contribution of these positive and negative-regulatory factors, along with tissue-specific epigenetic modifications of the IL-1α chromosomal locus, likely contribute to cell-type specific regulation of the gene.
Activation of endothelium by complement and production of IL-1α may account for systemic inflammatory reactions seen in complement-mediated disease. For example, increased endothelial expression of IL-1α and heightened cardiovascular co-morbidity and mortality have been observed in those with extra-articular rheumatoid arthritis41. Similarly, a single nucleotide polymorphism in the IL-1α gene promoter has been strongly associated with development of end-stage renal disease42. While studies such as ours shed light on molecular mechanisms of immune responses, a full understanding of those responses clearly requires consideration the milieu of the organ or tissue in which the molecular events arise. Thus, in the broadest sense, our results reinforce the concept that complement-stimulated inflammation and disease depends on the microenvironment in which complement-stimulated IL-1α expression occurs.
Acknowledgements
This work was supported by grants from the National Institutes of Health (HL52297 and AI53733).
Footnotes
Subject codes: [138] Cell signaling/signal transduction, [142] Gene expression, [147] Growth factors/cytokines, [95] Endothelium/vascular type/nitric oxide, [155] Physiological and pathological control of gene expression
Conflict of Interest Statement
None.
References
- 1.Esmon CT. The roles of protein c and thrombomodulin in the regulation of blood coagulation. J Biol Chem. 1989;264:4743–4746. [PubMed] [Google Scholar]
- 2.Broze GS., Jr Tissue factor pathway inhibitor. Thromb Haemost. 1995;74:90–93. [PubMed] [Google Scholar]
- 3.Suttorp N, Seeger W, Zinsky S, Bhakdi S. Complement complex C5b-8 induces PGI2 formation in cultured endothelial cells. Am J Physiol. 1987;253:C13–C21. doi: 10.1152/ajpcell.1987.253.1.C13. [DOI] [PubMed] [Google Scholar]
- 4.Saadi S, Wrenshall LE, Platt JL. Regional manifestations and control of the immune system. FASEB J. 2002;16:849–856. doi: 10.1096/fj.01-0690hyp. [DOI] [PubMed] [Google Scholar]
- 5.Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by toll-like receptor 4. J Immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 6.Brunn GJ, Bungum MK, Johnson GB, Platt JL. Conditional signaling by Toll-like receptor 4. FASEB J. 2005;19:872–874. doi: 10.1096/fj.04-3211fje. [DOI] [PubMed] [Google Scholar]
- 7.Saadi S, Holzknecht RA, Patte CP, Stern DM, Platt JL. Complement-mediated regulation of tissue factor activity in endothelium. J Exp Med. 1995;182:1807–1814. doi: 10.1084/jem.182.6.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A. Cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J Exp Med. 1997;185:1619–1627. doi: 10.1084/jem.185.9.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalady MF, Lawson JH, Sorrell RD, Platt JL. Decreased fibrinolytic activity in porcine-to-primate cardiac xenotransplantation. Mol Med. 1998;4:629–637. [PMC free article] [PubMed] [Google Scholar]
- 10.Kishimoto T. The biology of interleukin-6. Blood. 1989;74:1–10. [PubMed] [Google Scholar]
- 11.Huber AR, Kunkel SL, Todd RF, III, Weiss SJ. Regulation of transendothelial neutrophil migration by endogenous interleukin-8. Science. 1991;254:99–102. doi: 10.1126/science.1718038. [DOI] [PubMed] [Google Scholar]
- 12.Bustos M, Coffman TM, Saadi S, Platt JL. Modulation of eicosanoid metabolism in endothelial cells in a xenograft model: role of cyclooxygenase-2. J Clin Invest. 1997;100:1150–1158. doi: 10.1172/JCI119626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stern D, Nawroth P, Handley D, Kisiel W. An endothelial cell-dependent pathway of coagulation. Proc Natl Acad Sci USA. 1985;82:2523–2527. doi: 10.1073/pnas.82.8.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA., Jr Interleukin-1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am J Pathol. 1985;121:394–403. [PMC free article] [PubMed] [Google Scholar]
- 15.Nawroth PP, Stern DM. Modulation of endothelial cell hemostatic properties by tumor necrosis factor. J Exp Med. 1986;163:740–745. doi: 10.1084/jem.163.3.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pober JS, LaPierre LA, Stolpen AH, Brock TA, Springer TA, Fiers W, Bevilacqua MP, Mendrick DL, Gimbrone MA., Jr. Activation of cultured human endothelial cells by recombinant lymphotoxin: comparison with tumor necrosis factor and interleukin 1 species. J Immunol. 1987;138:3319–3324. [PubMed] [Google Scholar]
- 17.Strieter RM, Kunkel SL, Showell HJ, Remick DG, Phan SH, Ward PA, Marks RM. Endothelial cell gene expression of a neutrophil chemotactic factor by TNF-alpha, LPS, and IL-1 beta. Science. 1989;243:1467–1469. doi: 10.1126/science.2648570. [DOI] [PubMed] [Google Scholar]
- 18.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 19.Warner SJC, Auger KP, Libby P. Interleukin 1 induces interleukin 1. II. Recombinant human interleukin 1 induces interleukin 1 production by adult human vascular endothelial cells. J Immunol. 1987;139:1911–1917. [PubMed] [Google Scholar]
- 20.Kempe S, Kestler H, Lasar A, Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33:5308–5319. doi: 10.1093/nar/gki836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niculescu F, Rus H, van Biesen T, Shin ML. Activation of Ras and mitogen-activated protein kinase pathway by terminal complement complexes is G protein dependent. J Immunol. 1997;158:4405–4412. [PubMed] [Google Scholar]
- 22.Hattori R, Hamilton KK, McEver RP, Sims PJ. Complement proteins C5b-9 induce secretion of high molecular weight multimers of endothelial von Willebrand factor and translocation of granule membrane protein GMP-140 to the cell surface. J Biol Chem. 1989;264:9053–9060. [PubMed] [Google Scholar]
- 23.DiScipio RG, Daffern PJ, Jagels MA, Broide DH, Sriramarao P. A comparison of C3a and C5a-mediated stable adhesion of rolling eosinophils in postcapillary venules and transendothelial migration in vitro and in vivo. J Immunol. 1999;162:1127–1136. [PubMed] [Google Scholar]
- 24.Coulpier M, Andreev S, Lemercier C, Dauchel H, Lees O, Fontaine M, Ripoche J. Activation of the endothelium by IL-1 alpha and glucocorticoids results in major increase of complement C3 and factor B production and generation of C3a. Clin Exp Immunol. 1995;101:142–149. doi: 10.1111/j.1365-2249.1995.tb02290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell WD, Lazoura E, Okada N, Okada H. Inactivation of C3a and C5a octapeptides by carboxypeptidase R and carboxypeptidase N. Microbiol Immunol. 2002;46:131–134. doi: 10.1111/j.1348-0421.2002.tb02669.x. [DOI] [PubMed] [Google Scholar]
- 26.Selvan RS, Kapadia HB, Platt JL. Complement-induced expression of chemokine genes in endothelium: regulation by IL-1-dependent and -independent mechanisms. J Immunol. 1998;161:4388–4395. [PubMed] [Google Scholar]
- 27.Saadi S, Holzknecht RA, Patte CP, Platt JL. Endothelial cell activation by pore forming structures: pivotal role for IL-1α. Circulation. 2000;101:1867–1873. doi: 10.1161/01.cir.101.15.1867. [DOI] [PubMed] [Google Scholar]
- 28.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 29.Brunn GJ, Falls EL, Nilson AE, Abraham RT. Protein-tyrosine kinase-dependent activation of STAT transcription factors in interleukin-2- or interleukin-4-stimulated T lymphocytes. J Biol Chem. 1995;270:11628–11635. doi: 10.1074/jbc.270.19.11628. [DOI] [PubMed] [Google Scholar]
- 30.Parker PJ, Murray-Rust J. PKC at a glance. J Cell Sci. 2004;117:131–132. doi: 10.1242/jcs.00982. [DOI] [PubMed] [Google Scholar]
- 31.O'Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O'Neill EA. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- 32.Kunsch C, Ruben SM, Rosen CA. Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol Cell Biol. 1992;12:4412–4421. doi: 10.1128/mcb.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rus HG, Niculescu FI, Shin ML. Role of the C5b-9 complement complex in cell cycle and apoptosis. Immunol Rev. 2001;180:49–55. doi: 10.1034/j.1600-065x.2001.1800104.x. [DOI] [PubMed] [Google Scholar]
- 34.Ramos-Casals M, Brito-Zeron P, Yague J, Akasbi M, Bautista R, Ruano M, Claver G, Gil V, Font J. Hypocomplementaemia as an immunological marker of morbidity and mortality in patients with primary Sjogren's syndrome. Rheumatology (Oxford) 2005;44:89–94. doi: 10.1093/rheumatology/keh407. [DOI] [PubMed] [Google Scholar]
- 35.Schollkopf C, Kjeldsen L, Bjerrum OW, Mourits-Andersen HT, Nielsen JL, Christensen BE, Jensen BA, Pedersen BB, Taaning EB, Klausen TW, Birgens H. Rituximab in chronic cold agglutinin disease: a prospective study of 20 patients. Leuk Lymphoma. 2006;47:253–260. doi: 10.1080/10428190500286481. [DOI] [PubMed] [Google Scholar]
- 36.Key NS, Platt JL, Vercellotti GM. Vascular endothelial cell proteoglycans are susceptible to cleavage by neutrophils. ArterioThromb. 1992;12:836–842. doi: 10.1161/01.atv.12.7.836. [DOI] [PubMed] [Google Scholar]
- 37.Ihrcke NS, Platt JL. Shedding of heparan sulfate proteoglycan by stimulated endothelial cells: evidence for proteolysis of cell surface molecules. J Cell Physiol. 1996;168:625–637. doi: 10.1002/(SICI)1097-4652(199609)168:3<625::AID-JCP15>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 38.Marcum JA, Atha DH, Fritze LM, Nawroth P, Stern D, Rosenberg RD. Cloned bovine aortic endothelial cells synthesize anticoagulantly active heparan sulfate proteoglycan. J Biol Chem. 1986;261:7507–7517. [PubMed] [Google Scholar]
- 39.McDowell TL, Symons JA, Duff GW. Human interleukin-1 alpha gene expression is regulated by Sp1 and a transcriptional repressor. Cytokine. 2005;30:141–153. doi: 10.1016/j.cyto.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 40.Hirano F, Tanaka H, Hirano Y, Hiramoto M, Handa H, Makino I, Scheidereit C. Functional interference of Sp1 and NF-kappaB through the same DNA binding site. Mol Cell Biol. 1998;18:1266–1274. doi: 10.1128/mcb.18.3.1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turesson C, Englund P, Jacobsson LT, Sturfelt G, Truedsson L, Nennesmo I, Lundberg IE. Increased endothelial expression of HLA-DQ and interleukin 1alpha in extra-articular rheumatoid arthritis. Results from immunohistochemical studies of skeletal muscle. Rheumatology (Oxford) 2001;40:1346–1354. doi: 10.1093/rheumatology/40.12.1346. [DOI] [PubMed] [Google Scholar]
- 42.Wetmore JB, Hung AM, Lovett DH, Sen S, Quershy O, Johansen KL. Interleukin-1 gene cluster polymorphisms predict risk of ESRD. Kidney Int. 2005;68:278–284. doi: 10.1111/j.1523-1755.2005.00403.x. [DOI] [PubMed] [Google Scholar]