

Abstract
Single metallic bowtie nanoantennas provide a controllable environment for surface-enhanced Raman scattering (SERS) of adsorbed molecules. Bowties have experimentally measured electromagnetic enhancements, enabling estimation of chemical enhancement for both the bulk and the few-molecule regime. Strong fluctuations of selected Raman lines imply that a small number of p-mercaptoaniline molecules on a single bowtie show chemical enhancement >107, much larger than previously believed, likely due to charge transfer between the Au surface and the molecule. This chemical sensitivity of SERS has significant implications for ultra-sensitive detection of single molecules.
Rapid and accurate detection and identification of trace amounts of chemical species is of the utmost importance in biology, chemistry, medicine, and defense. Recent advances in fluorescence spectroscopy methods offer exquisite sensitivity, enabling the ultimate in analytical detection: single molecules.1 However, fluorescence studies require specially engineered labels, a limitation that places constraints on potential applications. Raman spectroscopy does not suffer from this limitation, since most molecules display a unique set of molecular vibrations that give rise to a distinctive chemical fingerprint, especially attractive for ultra-selective analysis.
Because Raman transitions are incredibly weak, this technique was not generally believed to offer the potential sensitivity afforded by fluorescence. However, over 30 years ago, it was first observed that the Raman signal of pyridine dramatically increases when adsorbed on a roughened Ag electrode,2,3 and the detailed origins of surface-enhanced Raman scattering (SERS) arising from nanostructured metals have remained a topic of debate. Researchers linked SERS signals to a combination of two effects,4 electromagnetic (EM) enhancement, where illumination intensity is enhanced due to sharp metal edges or plasmon effects, and chemical enhancement (CE), where the Raman cross-section of adsorbed molecules is increased above the solution value,5 with EM enhancement dominant. Detailed SERS experiments performed on roughened metal films in electrochemical cells revealed the importance of CE due to the applied potential dependence of SERS spectra,6,7 but measured values of either enhancement were not available.
Interest in the SERS mechanism blossomed with the recent observation of Raman lines apparently arising from single molecules adsorbed onto colloidal Ag and Au particles.4,8,9 To obtain the 14-order of magnitude enhancement required to make Raman signals competitive with fluorescence, it was believed that an enormous EM enhancement at “hot spots” was combined with a modest CE in these systems. Detailed investigation revealed signals are greatest at junctions between plasmonically coupled metal nanoparticles, usually formed when two or more colloidal particles of ∼100 nm diameter happen to aggregate,4,10 or within fractal island films.11,12
Michaels et al.4 suggested that coupled colloidal particles locally enhance the incident laser pump intensity, proportional to |E|2 where E is the electric field, by as much as 105. The same factor enhances the probability of detecting Raman emission as in the case of a transmitting antenna by another |E|2 factor yielding a total EM enhancement potentially as high as 1010.
The search for large and controllable EM enhancements has renewed interest in studying near-field coupling between metal particles, because optimized structures should readily produce SERS-active substrates. Scattering experiments have explored the resonant wavelength of subwavelength-sized particles,13,14 and have been accompanied by important theoretical analysis15,16 aiding nanostructure design. Because of their ability to increase incident and received signals of molecules in their gap, enhanced lithographically fabricated metallic nanostructures have been dubbed “nanoantennas” and experimentally produced large local E-fields.17,18
However, there is still uncertainty about the relative importance of the chemical effect, which has been estimated to enhance SERS signals by factors anywhere from 10 to as high as 106.19,20 Here, we describe the first use of single resonant “bowtie” nanoantennas consisting of two metallic triangles oriented tip to tip designed to facilitate Raman spectroscopy, measuring SERS from chemically bonded p-mercaptoaniline (pMA) molecules. Since these structures provide a coupled plasmon system with lithographically controllable gap and known (measured) EM enhancement,17 this strategy allows the first direct exploration of the role of CE in SERS.
Au bowties were fabricated with electron-beam lithography producing gap sizes [see Fig. 1(B) inset], and coated with pMA.21 Single bowties were pumped at their resonance (830 nm) using a linearly polarized cw laser diode, and imaged in a confocal microscope, whose collection path was equipped with a grating spectrometer and cooled Si CCD detector to acquire Raman spectra. pMA was chosen because the thiol moiety forms a self-assembled monolayer (SAM) upon binding to Au, it is nonabsorbing at 830 nm, and several bulk SERS studies were previously performed on pMA in electrochemical cells6,7 and on metallic nanoshells.22
FIG. 1.
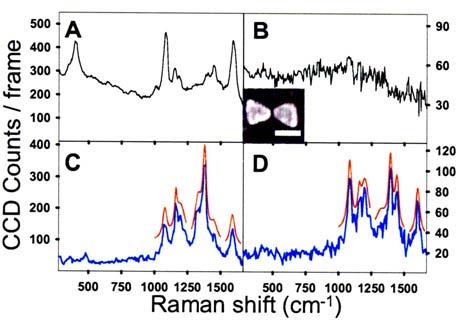
(Color online) (A) Bulk SERS spectrum of pMA on roughened Au (10 s integration); (B) fluorescence continuum from a bowtie without adsorbed pMA. A representative SEM image, inset (scale bar=100 nm, 17 nm gap); (C) SERS for pMA on a bowtie (2 s integration) at t=326 s; (D) SERS from the same bowtie at t=441 s. Spectra (C) and (D) differ in observed SERS modes and total intensity. Red curves in (C) and (D) are Lorentzian fits for modes between 1000 and 1600 cm−1, offset for clarity. I=155 kW/cm2.
For verification of SAM preparation, a 35 nm thick roughened Au film was thermally evaporated, incubated in pMA,21 and its Raman spectrum [Fig. 1(A)] shows the peaks expected for pMA SERS.6,21 Prior to pMA incubation, single bowtie spectra showed only broad continuum emission [Fig. 1(B)], which scales linearly with power below the bowtie damage threshold, and is attributed to 1-photon photoluminescence excited in Au by the high optical fields near the bowtie gap.23 Continuum is also present in the pMA Raman spectra on bowties and the Au film. Figures 1(C) and 1(D) show spectra (2 s integration) taken at t=326 s and t=441 s for the same bowtie revealing most of the characteristic pMA lines seen in Fig. 1(A). In the fingerprint region (1000–1600 cm−1), 6–7 lines appear with similar Raman shift but radically different amplitudes. For bowties, both continuum and SERS signals decrease to undetectable levels (2 s integration) when excitation light is polarized perpendicular to the bowtie long axis.
Using the known monolayer packing area of pMA (19 A2/molecule)21 and the surface area of the bowtie experiencing enhanced E-fields (3000 nm2) as predicted by electromagnetic calculations,16 the SERS signal is expected to arise from N∼104 molecules near the tips of the two triangles forming the bowtie gap. We estimate total SERS enhancement (TE) for pMA on the bowtie using: TE =S/(N σRaman Φ I), where S is the integrated Raman signal with continuum fluorescence subtracted (S=5 ×104 photons/s), σRaman =10−29 cm2, Φ the collection efficiency (0.01), and I=6.5×1023 photons/s cm2. Thus in Fig. 1(D), TE=7×107. The TE value is often broken into EM and CE contributions in the following manner: , where E0 is the incident electric field. In our bowties, is measured17 to be ∼1500|E0|2. Assuming that is also ∼1500|E0|2, reasonable for Stokes radiation considering the fairly broad antenna resonance,14 a value of CE∼30 is obtained, consistent with previous estimates.21
Closer examination of the spectra shown in Fig. 1(C) and 1(D) reveals that, while the spectra are similar to the bulk spectrum [Fig. 1(A)], there are intriguing dynamics; particular lines “blink” and their mode frequencies shift discontinuously, even though an entire monolayer of pMA covers the bowtie. Behavior like this has been reported previously,4,8,9,19 but only at concentrations of a few molecules per nanoparticle. For example, in Fig. 1(C) there is an intense band at 1380 cm−1 that dramatically decreases in panel 1(D), where the spectrum has a lower total intensity and resembles the bulk pMA spectrum.
To display the typical spectral dynamics we see, Fig. 2(A) shows a “waterfall” plot, where Raman shift, Δν, is plotted as a function of time, and SERS intensity is displayed as a colorscale. First we consider the very intense (several thousands of photons/s) broadband flash of emission, as seen at t∼150 s in Fig. 2(A) (yellow arrow). This brief flash of broad infrared fluorescence is possibly due to the creation and subsequent destruction of a metastable Au cluster of ∼30 atoms, previously shown to be fluorescent.24,25 Because these events are rare, and smaller, bluer emitting clusters would not be resonant with the bowties, we do not consider cluster formation to be central to the SERS effects reported here.
FIG. 2.
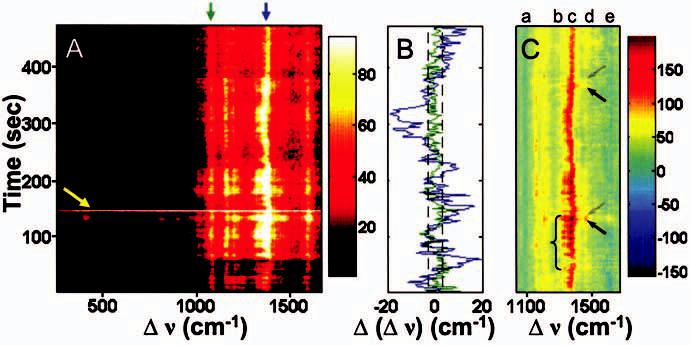
(Color) (A) Waterfall plot showing the evolution of SERS spectra with time (2 s integration). I=11 kW/cm2 for t=0–57 s, and I=38 kW/cm2 for t=57–460 s. Notice the intense flash of continuum fluorescence at t∼150 s (yellow arrow), the relatively stable modes at 1077 and 1590 cm−1, and fluctuations of both frequency and amplitude for the 1160, 1195, 1325, 1380, and 1450 cm−1 Raman modes. (B) Mode center frequency fluctuations for 1077 (green) and 1380 cm−1 (blue) lines, with error bar shown as dashed lines.21 Only modes with b2 symmetry fluctuate significantly. (C) A waterfall plot of difference spectra showing discrete intensity fluctuations evident for b2 modes. Note the blinking in the 1325 cm−1 mode (bracket, mode “b”) and “on” (black arrows) “off” (gray arrows) switching for mode “d,” 1450 cm−1. Mode “c” (1380 cm−1) also fluctuates. Modes (“a,” 1077 cm−1 and “e,” 1590 cm−1) having a1 symmetry are suppressed by the subtraction which is produced in two steps: first, intensity for the non-fluctuating fiducial 1077 cm−1 mode for each Raman spectrum in Fig. 2(A) is used to scale the observed bulk SERS spectrum of pMA on a rough Au film [Fig. 1(A)], and this scaled bulk SERS spectrum is subtracted from the measured spectrum in A.
Immediately obvious is that the intensity and frequency of some Raman lines are stable, but vary significantly for other modes. Using previous pMA mode assignments,6 modes at 1077 and 1590 cm−1, assigned to a1 symmetry,21 are stable in their frequency, and their intensity varies by less than a factor of 2 over the entire spectral series. For this reason, we will use the a1 modes as key reference modes in our analysis below. Conversely, peaks at 1160, 1195, 1325, 1380, and 1450 cm−1, which have b2 symmetry and have been interpreted as charge transfer (CT) modes for pMA,6,7 display large frequency and intensity fluctuations. For frequency fluctuations, Fig. 2(B) displays the center frequency time trajectory of each of two modes, determined by fitting the peaks to Lorentzian profiles.21 The 1077 cm−1 (a1 symmetry, C–S stretch) reference mode (green) displays little change, while the 1380 cm−1 (b2 symmetry, combination C–H bend and C–C stretch) mode (blue) varies significantly, often “jumping” as much as ±20 cm−1 from its expected value.
We assume that the Raman spectra include contributions from a large number of molecules with the small enhancement calculated above, plus a contribution from a small number of molecules with large (possibly huge) enhancement. To highlight the latter contribution, we use the strength of the 1077 cm−1 reference mode to scale the bulk SERS spectrum [Fig. 1(A)] and subtract it from the observed spectra, generating the difference spectra in Fig. 2(C). Note that scaling by the 1077 cm−1 mode automatically makes the other a1 mode at 1590 cm−1 disappear. (It should be noted that a single blinking event is observed for mode “e” at t ∼110 s, because a weak b2 symmetry mode exists at 1575 cm−1.) Figure 2(C) shows strong on/off amplitude fluctuations in several b2 modes (designated “b” and “d”), for example at the arrows and bracket. The discrete fluctuations of the b2 modes suggests a dynamic CT process is occurring and that the SERS signal is quite sensitive to CT.
Due to the difficulty in synchronizing even a small number of molecules, discrete intensity fluctuations (blinking) and spectral jumping are two spectroscopic signatures of single molecules.1 We note that blinking alone is not necessarily a fair indicator of single-molecule SERS, because the EM enhancement could rapidly change, leading to temporary fluctuations of SERS signals for all molecules near the gap. The stability of the reference a1 modes shows that large EM changes are not occurring. Because the frequency and/or amplitude of the b2 modes often discontinuously “jump,” and in any one 2 s spectrum, a single frequency is observed (movie in Ref. 21), this observation implies that there is either a synchronization of all 104 molecules near the gap, very unlikely, or that a very small subset of molecules is responsible for the discontinuous changes, with the possibility that this SERS signature is from a single molecule. Therefore, the varying frequencies [Fig. 2(B)] and amplitudes [Fig. 2(C)] of the CT b2 modes suggest that selected molecule(s) are subject to a huge variation in their CE, from ∼30 to as much as ∼107. Apparently, signal from a small subset is comparable, and often exceeds (by more than an order of magnitude) that of the remaining 104 molecules near the bowtie gap, even though both are subject to the same EM value, not an unreasonable possibility.26
Because we are able to distinguish EM and CE enhancement components, and EM is not varying, it is clear that fluctuating CE is of primary importance in producing the dynamic SERS behavior, a result that should help elucidate the elusive SERS mechanism. Since this idea is quite new, it is not possible to produce a precise mechanism. However, huge local CE is only present for selected low symmetry Raman modes of the benzene ring long associated with CT processes. How can a huge change in CT select for one (or a few) molecules? We suggest a few reasonable possibilities based on previous CT theory that could produce the observed effects, in order to stimulate further experimental and theoretical exploration of CE mechanisms.
Very small Ag clusters [O(10 atoms)] have shown surprisingly large Raman signals in the absence of significant EM enhancement possibly due to extremely large molecule-cluster couplings.27-29 Indeed, one could imagine that an Au cluster could form in the bowtie gap, and due to its small surface area, accommodate a few molecules that would be strongly enhanced, but we rarely observe cluster fluorescence or huge a1 mode changes and thus rule out clusters as the mechanism. Helpfully, CT effects have been characterized in pMA SERS experiments on rough Ag/Au films in electro-chemical cells for over a decade,6,7 where b2 modes were found to be very sensitive to applied potential, and attributed to CT. To explain this, Osawa et al.6 borrow from the comprehensive theory of Lombardi et al.30 who describe the Herzberg-Teller effect for CT. In short, for a molecule adsorbed onto a metal, vibronic mixing between the electronic and vibrational wave functions of adsorbed molecules and those of the metal leads to electron transfer, strongly increasing Raman scattering associated with mixed modes by inducing an effect analogous to resonant Raman.21 A simple physical picture is presented in Fig. 3 that could produce these effects. Normally, pMA molecules should orient perpendicular to the surface in a close-packed SAM, making it difficult for a molecule to access the metal surface. However, at a defect, which could be light-induced, the SAM could be interrupted, allowing a single or a few pMA molecules to lie flat on the surface and undergo CT. In this position, benzene ring π-orbitals better couple to the Au surface, dramatically increasing vibronic mixing for selected benzene modes while leaving mixing for other modes, such as the C–S stretch, unchanged, strongly enhancing CE for selected modes only.26 The transient “on”/“off” switching shown for b2 modes in Fig. 2(C) (especially “b” and “d” lines) is consistent with this picture.
FIG. 3.
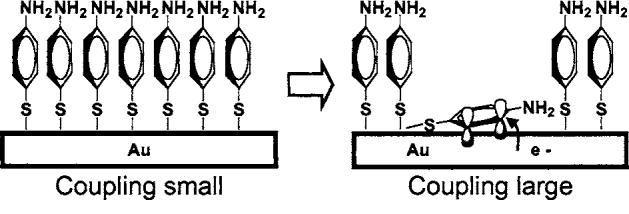
Proposed CT mechanism. At a defect site, a pMA molecule can adsorb flat on the surface, enabling π-electrons in the benzene ring to more strongly interact with the Au surface, increasing vibronic coupling for selected modes, increasing SERS.
In summary, SERS is observed for pMA adsorbed on lithographically produced single bowtie nanoantennas with known local field enhancement, yielding the expected Raman lines on top of a fluorescent continuum originating in the Au. Despite the fact that ∼104 molecules are present near the bowtie gap, the Raman spectra display discrete blinking and spectral shifting events, indicating that a few or possibly one molecule produces a SERS signal comparable to that of the remaining molecules. This means some molecules experience a larger CE than previously believed possible, as large as 107. This is possibly understood using existing CT theory, in which we suggest that an increase in wave function mixing between π-orbitals in the benzene ring and Au occurs when a pMA molecule lies down at the surface. Due to the extreme sensitivity of SERS enhancement to the properties of adsorbed chemical species, the design and production of nanoantenna-based SERS substrates holds great promise in performing trace chemical detection, but achieving the proper orientation for large CE will be a challenge.
Footnotes
This work was supported in part by the National Institutes of Health Grant No. 1R21-GM065331, and the Stanford Nanofabrication Facility (a member of the National Nanotechnology Infrastructure Network), supported by the National Science Foundation Grant No. ECS-9731293.
References
- 1.Moerner WE, Orrit M. Science. 1999;283:1670. doi: 10.1126/science.283.5408.1670. [DOI] [PubMed] [Google Scholar]
- 2.Fleischmann M, Hendra PJ, McQuillan AJ. Chem. Phys. Lett. 1974;26:163. [Google Scholar]
- 3.Jeanmaire DL, Van Duyne RP. J. Electroanal. Chem. 1977;84:1. [Google Scholar]
- 4.Michaels AM, Jiang J, Brus L. J. Phys. Chem. B. 2000;104:11965. [Google Scholar]
- 5.Moskovits M. Rev. Mod. Phys. 1985;57:783. [Google Scholar]
- 6.Osawa M, Matsuda N, Yoshii K, Uchida I. J. Phys. Chem. 1994;98:12702. [Google Scholar]
- 7.Hill W, Wehling B. J. Phys. Chem. 1993;97:9451. [Google Scholar]
- 8.Kneipp K, Wang Y, Kneipp H, Perelman LT, Itzkan I, Dasari RR, Feld MS. Phys. Rev. Lett. 1997;78:1667. doi: 10.1103/PhysRevLett.76.2444. [DOI] [PubMed] [Google Scholar]
- 9.Nie S, Emory SR. Science. 1997;275:1102. doi: 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- 10.Xu H, Kaell M. ChemPhysChem. 2003;4:1001. doi: 10.1002/cphc.200200544. [DOI] [PubMed] [Google Scholar]
- 11.Gresillon S, Aigouy L, Boccara AC, Rivoal JC, Quelin X, Desmarest C, Gadenne P, Shubin VA, Sarychev AK, Shalaev VM. Phys. Rev. Lett. 1999;82:4520. [Google Scholar]
- 12.Stockman MI, Pandey LN, Muratov LS, George TF. Phys. Rev. Lett. 1994;72:2486. doi: 10.1103/PhysRevLett.72.2486. [DOI] [PubMed] [Google Scholar]
- 13.Haynes CL, McFarland AD, Zhao L, Van Duyne RP, Schatz GC, Gunnarson L, Prikulis J, Kasemo B, Kaell M. J. Phys. Chem. B. 2003;107:7337. [Google Scholar]
- 14.Fromm DP, Sundaramurthy A, Schuck PJ, Kino GS, Moerner WE. Nano Lett. 2004;4:957. doi: 10.1021/nl052322c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hao E, Schatz GC. J. Chem. Phys. 2004;120:357. doi: 10.1063/1.1629280. [DOI] [PubMed] [Google Scholar]
- 16.Sundaramurthy A, Fromm DP, Crozier KB, Schuck PJ, Moerner WE, Kino GS. Phys. Rev. B. 2005;72:165409. doi: 10.1103/PhysRevLett.94.017402. [DOI] [PubMed] [Google Scholar]
- 17.Schuck PJ, Fromm DP, Sundaramurthy A, Kino GS, Moerner WE. Phys. Rev. Lett. 2005;94:017402. doi: 10.1103/PhysRevLett.94.017402. [DOI] [PubMed] [Google Scholar]
- 18.Muhlschlegel P, Eisler HJ, Martin OJF, Hecht B, Pohl DW. Science. 2005;308:1607. doi: 10.1126/science.1111886. [DOI] [PubMed] [Google Scholar]
- 19.Haran G. Isr. J. Chem. 2004;44:385. [Google Scholar]
- 20.Otto A. J. Raman Spectrosc. 2002;33:593. [Google Scholar]
- 21.See EPAPS Document No. E-JCPSA6-124-009604 for a movie of time-dependent SERS spectra, details regarding experimental conditions and statistical methods employed, and for detailed discussion. This document can be reached via a direct link in the online article's HTML reference section or via the EPAPS homepage (http://www.aip.org/pubservs/epaps.html).
- 22.Jackson JB, Halas NJ. Proc. Nat. Acad. Sci. USA. 2004;101:17930. doi: 10.1073/pnas.0408319102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beversluis MR, Bouhelier A, Novotny L. Phys. Rev. B. 2003;68:115433. doi: 10.1103/PhysRevLett.90.013903. [DOI] [PubMed] [Google Scholar]
- 24.Link S, Beeby A, FitzGerald S, El-Sayed MA, Schaaff TG, Whetten RL. J. Phys. Chem. B. 2002;106:3410. [Google Scholar]
- 25.Zheng J, Zhang C, Dickson RM. Phys. Rev. Lett. 2004;93:077402. doi: 10.1103/PhysRevLett.93.077402. [DOI] [PubMed] [Google Scholar]
- 26.Wang Z, Rothberg LJ. J. Phys. Chem. B. 2005;109:3387. doi: 10.1021/jp0460947. [DOI] [PubMed] [Google Scholar]
- 27.Jacobson ML, Rowlen KL. Chem. Phys. Lett. 2005;401:52. [Google Scholar]
- 28.Monti OLA, Fourkas JT, Nesbitt DJ. J. Phys. Chem. B. 2004;108:1604. [Google Scholar]
- 29.Capadona LP, Zheng J, Gonzalex JI, Lee TH, Patel SA, Dickson RM. Phys. Rev. Lett. 2005;94:058301. doi: 10.1103/PhysRevLett.94.058301. [DOI] [PubMed] [Google Scholar]
- 30.Lombardi JR, Birke R, Lu LT, Xu J. J. Chem. Phys. 1986;84:4174. [Google Scholar]