

Abstract
The Saccharomyces cerevisiae Rad24 and Rad17 checkpoint proteins are part of an early response to DNA damage in a signal transduction pathway leading to cell cycle arrest. Rad24 interacts with the four small subunits of replication factor C (RFC) to form the RFC-Rad24 complex. Rad17 forms a complex with Mec3 and Ddc1 (Rad17/3/1) and shows structural similarities with the replication clamp PCNA. This parallelism with a clamp-clamp loader system that functions in DNA replication has led to the hypothesis that a similar clamp-clamp loader relationship exists for the DNA damage response system. We have purified the putative checkpoint clamp loader RFC-Rad24 and the putative clamp Rad17/3/1 from a yeast overexpression system. Here, we provide experimental evidence that, indeed, the RFC-Rad24 clamp loader loads the Rad17/3/1 clamp around partial duplex DNA in an ATP-dependent process. Furthermore, upon ATP hydrolysis, the Rad17/3/1 clamp is released from the clamp loader and can slide across more than 1 kb of duplex DNA, a process which may be well suited for a search for damage. Rad17/3/1 showed no detectable exonuclease activity.
DNA damage in eukaryotic cells provokes a range of cellular responses which includes DNA repair, apoptosis, and cell-cycle arrest. The DNA damage checkpoint response arrests cells at appropriate points in the cell cycle to allow recovery of the integrity of the DNA before reentering the cell cycle (1). The later steps of the DNA damage checkpoint that ultimately lead to inhibition of the cdk kinases that drive the cell cycle are relatively well understood (recently reviewed in refs. 2 and 3). However, molecular details about the initial steps of damage recognition that activate the checkpoint response are still lacking. Two distinct complexes independently localize to sites of DNA damage in Saccharomyces cerevisiae, but the presence of both complexes is required for proper checkpoint function (4–6). The Mec1 protein kinase forms a complex with Ddc2 and may function in both DNA damage recognition and signal transduction (6, 7). Another set of proteins, Rad24, Rad17, Mec3, and Ddc1, which may primarily function in DNA damage recognition and processing, shows sequence similarities with replication factor C (RFC) and proliferating cell nuclear antigen (PCNA), the eukaryotic clamp loader-clamp system that is central to the structure of the replication fork. RFC is a heteropentameric complex, consisting of a large subunit, Rfc1, and the four small Rfc2-5 subunits, that clamps PCNA around the DNA at primer/template junctions in an ATP-dependent process (8). PCNA is the processivity factor of DNA polymerase δ and many other proteins involved in various aspects of DNA metabolism (9).
Rad24 (Rad17 in human and Schizosaccharomyces pombe) shows sequence similarity with the Rfc1 subunit of RFC and interacts with the small subunits of RFC (10–12). A heteropentameric complex, consisting of Rad24 and the Rfc2-5 subunits, has been purified from yeast, and the analogous human complex has been purified from overproduction systems (13–15). Protein-threading algorithms have predicted a PCNA-like fold for Rad17, Mec3, and Ddc1 (16). These three S. cerevisiae proteins interact in the cell and also show genetic interactions with the putative clamp loader RFC-Rad24 (17, 18). Biochemical studies with the analogous putative human clamp complex (Rad9/Rad1/Hus1) show its subunit structure to be that of a heterotrimer (14, 15, 19).
Electron microscopic studies of the two human checkpoint complexes are consistent with that of a clamp and a clamp loader (15, 19). However, despite all of these structural similarities with the well characterized RFC-PCNA pair, evidence that the checkpoint system constitutes that of a clamp-clamp loader pair is still missing. Indeed, one study on the human system failed to detect positive proof for this current hypothesis (14). To understand the initial DNA damage-induced events that generate the cell cycle checkpoint, we have studied the two complexes from S. cerevisiae. Here, we provide evidence that the RFC-Rad24 clamp loader interacts with the Rad17/3/1 clamp in an ATP-dependent fashion and loads the clamp onto partial duplex DNA. Hydrolysis of ATP is required for release of the clamp loader and to initiate sliding of the clamp across double-stranded DNA.
Materials and Methods
Plasmids and DNA.
All checkpoint genes were cloned in multicopy yeast shuttle vectors under control of the bidirectional GAL1–10 promoter for high-level galactose-inducible expression in yeast. PBL766 (2 μM ori URA3 GST-RAD24) was made by first amplifying the RAD24 ORF in vitro, cloning it in-frame downstream of the GST gene in pGEX-6P-1 (Amersham Pharmacia), then reamplifying the fusion gene and cloning it into vector pRS426-GAL. The GST gene is separated form the RAD24 gene by a protease site. Treatment with prescission protease (Amersham Pharmacia) generates the full-length Rad24 protein, together with a 5-aa (GPLGS) N-terminal extension. PBL422 (2 μM ori TRP1 RFC2 RFC3 RFC4 RFC5), with the four small subunit genes under control of the GAL1-10 cassette, has been described (20). Plasmid pBL764 (2 μM ori URA3 GST-MEC3 RAD17) contains both GST-MEC3, obtained analogously to GST-RAD24, and RAD17, divergently transcribed from the GAL1-10 cassette; pBL760 (2 μM ori TRP1 DDC1) contains the DDC1 gene under similar control. The correct sequence of all genes was confirmed by sequencing of the constructs.
The oligonucleotides used in the SPR analysis and ATPase assays were A9 (5′-ccagtgaattcgagctcggtacccgctagcggggatcctcta), and complementary oligos A2 (5′-tagaggatccccgctagcgggtaccgagctcgaattcactgg), 5′-20 (5′-accgagctcgaattcactgg), and 3′-20 (5′-tagaggatccccgctagcgg). For attachment to SPR chips, the A9 oligo was biotinylated either at the 3′ or 5′ terminus.
Expression and Purification of Protein Complexes.
S. cerevisiae strain BJ2168 (MATa: ura3-52, trp1-289, leu2-3,112, prb1-1122, prc1-407, pep4-3) was used as a host for protein overproduction. For RFC-Rad24 expression, yeast cells were cotransformed with plasmids pBL766 and pBL422, grown and induced in a 35-liter fermenter by using conditions and media described (21). After a 3-h induction with galactose, methylmethane sulfonate was added to a final concentration of 0.01%, followed by a further 2 h of growth. For Rad17/3/1 expression, yeast cells were cotransformed with plasmids pBL760 and pBL764 and grown and induced similarly.
RFC-Rad24 was purified by adapting a published protocol (13). Crude protein extract, prepared from 150 g of yeast in 150 ml of 2× buffer A (buffer A: 50 mM Hepes, pH 7.5/150 mM KCl/10% glycerol/3 mM DTT/0.1% Tween 20/0.01% Nonidet P-40/1 mM EDTA/1 mM EGTA/1 mM PMSF/2 μM pepstatin A/2 μM leupeptin/10 mM NaHSO3/1 mM benzamidine/0.2 mM Na3VO4/1 mM α-naphtyl phosphate/5 mM Na-pyrophosphate/2 mM β-glycerophosphate) by blending with dry ice, followed by a clearing step at 35,000 rpm for 1 h, was loaded onto an 80-ml heparin agarose column equilibrated with buffer A (21). The column was washed with 500 ml of buffer A250 (containing 250 mM KCl), and RFC-Rad24 was eluted with 100 ml of buffer A500. The enzyme was diluted with an equal volume of buffer A0 and loaded onto a 3-ml glutathione-Sepharose column over a period of 3 h. The column was washed with 50 ml of buffer A250, and RFC-Rad24 was eluted with 10 ml of buffer A250 containing 20 mM glutathione (reduced form) and digested overnight at 4°C with 50 units of Prescission protease (Amersham Pharmacia). The enzymatic digest was diluted with an equal volume of buffer A0 and loaded onto a 1-ml MonoQ column equilibrated with buffer B125 (as buffer A, but without α-naphtyl phosphate, pepstatin, and leupeptin). The column was eluted with a 30-ml linear gradient of 125–1,000 mM KCl in buffer B; RFC-Rad24 eluted at ≈300 mM KCl. The same overall procedure was used for Rad17/3/1 purification, except that the heparin agarose column was loaded in 100 mM KCl and eluted with 250 mM KCl. The Rad17/3/1 complex eluted from the MonoQ column at 250 mM KCl and was passed over a final glutathione-Sepharose column to remove trace impurities of GST.
Analysis of the Checkpoint Complexes and Their Interactions.
The RFC-Rad24 and Rad17/3/1 complexes (0.1–5 μg) were analyzed by SDS/10% PAGE and stained with either Coomassie blue or Sypro red (Molecular Dynamics), and relative bands intensities were determined either by photographing the gel with a charge coupled-device camera and analyzing with GELIMAGE software or by using PhosphorImager and IMAGEQUANT software, respectively.
A (6%) native polyacrylamide gel was used to separate the complexes under nondenaturating conditions. The electrophoresis was carried out in 25 nM Tris⋅Base, 192 mM glycine buffer. Gels were stained with Coomassie blue, or the bands were cut out and analyzed by SDS/12% PAGE. Twenty micrograms of Rad17/3/1 was used in the assay.
Gel filtration analysis was performed at 15°C by using a SMART chromatography system (Amersham Pharmacia) with a Superose 12 PC3.2/30 column equilibrated in buffer C (25 mM Hepes, pH 7.8/100 mM NaCl/5 mM MgCl2/1 mM DTT). The column was run at a flow rate of 40 μl/min and 3 μg of each protein complex was loaded in 25 μl of the running buffer. ATP or ATPγS were used at a concentration of 100 μM when present. Thyroglobulin (85 Å), ferritin (61 Å), BSA (35.5 Å), and cytochrome C (16.5 Å) were used for column calibration.
Glycerol gradient centrifugation (25 μg of each complex) was performed in a 5-ml 10–30% glycerol gradient in buffer C, with 50 μM of ATP or ATPγS when present, in an SW65 rotor (Beckman) at 4°C for 20 h at 50,000 rpm. The protein in 180-μl fractions were precipitated with 10% (vol/vol) trichloroacetic acid and analyzed by SDS/10% or 12% PAGE. Carbonic anhydrase (2.8 S), BSA (4.3 S), alcohol dehydrogenase (7.4 S), β-amylase (8.9 S), catalase (11.3 S), and ferritin (17.6 S) were used as sedimentation markers.
ATPase Assays.
Twenty-five microliter assays were performed in buffer C supplemented with 50 μg/ml BSA. The assays contained 50 nM RFC-Rad24, 100 nM Rad17/3/1 or PCNA, 50 μM [α-32P]ATP, and 1 μM effector DNA. After 2.5, 5, 7.5, and 10 min at 30°C, 5-μl aliquots were removed from the reaction and quenched with 2 μl of 50 mM EDTA, 1% (wt/vol) SDS, and 20 mM each ADP and ATP. Two microliters were spotted on a polyethyleneimine cellulose sheet and dried. The sheet was washed in water for 10 min, rinsed in ethanol, dried and developed in 0.5 M LiCl/1M formic acid. The sheet was dried and subjected to PhosphorImager analysis (Molecular Dynamics).
Bio-Gel A15m Filtration of Complexes.
Complexes were formed in 200 μl of buffer C supplemented with 10 μg/ml BSA. The reactions contained 50 nM RFC-Rad24, 50 nM RFC, 100 nM Rad17/3/1, 100 nM PCNA, 1.5 μg RPA, 50 μM ATP or ATPγS, and 2 μg DNA (5 nM circles if pBluescript SKII (+) DNA). The mixtures were incubated at room temperature for 2 min, cooled on ice, and immediately filtered through a 5-ml Bio-Gel A15m (Bio-Rad) column. Six-drop fractions were collected. The DNA eluted predominantly in fractions 8–10, and free protein eluted in 12–20. Fractions 8–10 were combined, protein-precipitated with 10% trichloroacetic acid, and analyzed by SDS/10- or 12% PAGE. Quantitation of the protein/DNA ratio in the BioGel fractions was made based on the assumption that 100% of the DNA was recovered in the fractions, and 100% of the protein was recovered after acid precipitation. Gapped DNA (95 nucleotide gap) was prepared by cutting SKII DNA with SacI and XhoI and reannealing, by boiling/cooling, to single-stranded circular SKII DNA prepared by phagemid technology (22). The gapped DNA was purified on a 0.8% agarose gel to separate it from other DNA forms in the annealing mixture, including contaminating helper-phage DNA. The gapped DNA was digested with ScaI to generate the linearized gapped form.
DNA–Protein Interaction Analysis.
Surface plasmon resonance was performed in a BIAcore X apparatus. Buffer C containing 100 μg/ml BSA was the running buffer used in the analysis. DNA-chips were prepared as described and contained either ≈20 or ≈50 resonance units of biotinylated DNA attached via streptavidin to a CM5 chip (23).
The interaction between RFC-Rad24 (30 nM) and Rad17/3/1 (30 nM) or PCNA (30 nM) with DNA was monitored at 20°C by injecting 80 μl of the factors at the indicated concentration in the presence of 100 μM ATP or ATPγS where indicated over a DNA-chip at a flow rate of 30 μl/min.
Results and Discussion
Purification of the RFC-Rad24 and Rad17/3/1 Complexes.
We overproduced the five-subunit RFC-Rad24 complex in yeast from multicopy plasmids with each gene placed downstream of the galactose-inducible GAL1-10 promoter. A cleavable GST tag was added to the RAD24 gene to aid in purification. A similar strategy, with the GST tag fused to the MEC3 gene was used to overproduce and purify the Rad17/3/1 complex (see Materials and Methods for details). Copurification on the GST-affinity column of Rfc2-5 with GST-Rad24, and of Rad17 and Ddc1 with Mec3, indicates that the two complexes are stable. After the glutathione-affinity column, the GST-tag was proteolytically cleaved, and the remaining complex was purified further by HPLC ion-exchange chromatography. Typical isolation procedures yielded ≈5 mg of the RFC-Rad24 complex and ≈7 mg of the Rad17/3/1 from 100 g of yeast.
An electrophoretic analysis of the stoichiometry of each of the subunits in the two complexes revealed an average molar ratio of 1:0.9 for Rad24:Rfc2-5 and 0.9:1:1.4 for Mec3:Ddc1:Rad17 (see Fig. 4, lanes 7 and 8). The slight excess of Rad17 protein in the preparations may reflect the presence of Rad17 homodimers, which possibility was investigated by performing a native polyacrylamide electrophoretic analysis (24). This analysis showed three distinct bands (Fig. 1A). SDS/PAGE analysis of the predominant species (≈80%) showed it to be the Rad17/3/1 complex in a 1:1:1 ratio, whereas one of the minor bands (≈15%) consisted of a complex of Rad17 and Mec3 in an ≈2:1 ratio; the third band (≈5%) is a higher oligomer of Rad17/3/1 (Fig. 1B).
Figure 4.

Rad17/3/1 slides across double-stranded DNA. Complexes were formed in 200 μl and contained, when present, 50 nM RFC-Rad24, 100 nM Rad17/3/1, 1.5 μg of RPA, 50 μM ATP or ATPγS, and 2 μg of DNA (5 nM double-stranded or gapped double-stranded, 10 nM single-stranded). The mixtures were incubated at room temperature for 2 min, cooled on ice, and immediately filtered through a 5-ml Bio-Gel A15m column. Six drop fractions were collected, protein-precipitated with 10% trichloroacetic acid, and analyzed by SDS/10% PAGE. Lanes 7 and 8 are purified RFC-Rad24 and Rad17/3/1, respectively. The arrows in lanes 10 and 12 indicate the large subunit of RPA. In lanes 1–6, the molar ratio of Rad17/3/1:DNA is indicated at the bottom. Lanes 1–8 and 9–12 are from independent experiments.
Figure 1.

Native gel analysis of the Rad17/3/1 complex. Twenty micrograms of the complex was separated by 6% nondenaturating PAGE and stained with Coomassie blue (A), or the bands were cut out and analyzed by SDS/12% PAGE (B). Lane 4 is purified Rad17/3/1 complex.
A determination of the molecular weights by gel filtration and velocity centrifugation yielded Stokes radii of 48 and 54 Å, and S values of 9.5 and 6.5S, for RFC-Rad24 and Rad17/3/1, respectively, with calculated molecular weights of 192,000 and 147,000, respectively (25). Together with the determination of the relative subunit compositions by SDS/PAGE, these data are indicative of monomeric complexes with all subunits present at equimolar ratios in each complex. These data also suggest that the Rad17/3/1 complex is moderately asymmetric, with a frictional ratio f/fo of 1.55. However, electron microscopy studies have suggested a very symmetric donut-like shape for the human clamp (14, 15, 19). Modeling studies have shown that the donut-like structure could be made up by contributions of only approximately half of the Rad17, Mec3, and Ddc1 polypeptides (16). Possibly those portions of the proteins which do not participate in the formation of the central core structure may have extended conformations, which are not easily visualized by electron microscopy, but could contribute substantially to the observed solution properties of the complex.
ATP-Dependent Complex Formation Between RFC-Rad24 and Rad17/3/1.
Gel filtration analysis of the Rad17/3/1 complex without nucleotide present gave a single peak (Fig. 2A), whereas analysis of RFC-Rad24 gave two peaks, the one at the void volume of the column representing aggregated material. In the presence of ATP or the nonhydrolyzable analog ATPγS, RFC-Rad24 was much less aggregated, as indicated by the decrease in material eluting at the void volume (Fig. 2B).
Figure 2.

ATP-driven complex formation between RFC-Rad24 and Rad17/3/1. (A and B) Superose 12 elution profiles of RFC-Rad24 (black lines), of Rad17/3/1 (blue lines), and of the mixture of both (red lines), without nucleotide (A) or with prior incubation (10 min at 0°C) with 100 μM ATPγS (B). Vo = excluded volume. (C–E) Fractions were collected of the three gel filtration experiments in B, precipitated with trichloroacetic acid, and analyzed by SDS/10% PAGE. (C) RFC-Rad24 with ATPγS. (D) Rad17/3/1 with ATPγS. (E) RFC-Rad24+Rad17/3/1 with ATPγS. (F–H) Glycerol gradient sedimentation of RFC-Rad24+Rad17/3/1 without nucleotide (F), of RFC-Rad24+Rad17/3/1 with ATPγS (G), and of RFC-Rad24+PCNA with ATPγS (H). Fractions were collected from the bottom of the gradient, precipitated with 10% trichloroacetic acid, and analyzed by SDS/10% PAGE. The faint doublet at ≈65 kDa in all gels (indicated by *) is a gel-staining artifact resulting from the acid precipitation procedure. See Materials and Methods for further details.
Gel filtration analysis of a mixture of RFC-Rad24 and Rad17/3/1 in the absence of nucleotide yielded an elution pattern equal to the sum of those of the individual complexes, indicative of a lack of interaction (Fig. 2A). However, upon addition of ATP or the nonhydrolyzable analog ATPγS, a faster migrating complex was detected (shown for ATPγS in Fig. 2B), whereas the elution positions of the separate complexes remained unaffected by the addition of nucleotide. Analysis of the fractions showed that the faster migrating complex consisted of both RFC-Rad24 and Rad17/3/1, with all subunits present in approximately equimolar ratios (Fig. 2E).
Velocity sedimentation analysis of the factors yielded analogous results. In the presence of ATP or ATPγS a faster sedimenting complex consisting of equimolar amounts of all subunits of RFC-Rad24 and Rad17/3/1 complexes was detected (shown for ATPγS in Fig. 2G), whereas no interaction was observed in the absence of nucleotide (Fig. 2F). These results mirror those with RFC and PCNA, i.e., formation of a stable complex required ATP binding, but not its hydrolysis (20).
No stable complex between RFC-Rad24 and PCNA could be identified by gel filtration or sedimentation analysis, either in the presence of ATP or ATPγS (Fig. 2H). Therefore, RFC uniquely forms a stable ATP-dependent complex with PCNA, and RFC-Rad24 forms a stable ATP-dependent complex with Rad17/3/1.
ATPase Activity of RFC-Rad24.
Four of the five subunits of RFC bind ATP, and ATPase activity is not only observed in RFC but also in the four-subunit Rfc2-5 complex (23, 26). Therefore, the observation that RFC-Rad24 also exhibited an ATPase activity was not unexpected (Table 1). Surprisingly, this ATPase was strongly stimulated by PCNA. However, as no stable complex between RFC-Rad24 and PCNA could be detected, the increased ATPase must result from transient interactions between the two factors. These transient interactions could not be stabilized in the presence of ATPγS, a nonhydrolyzable analog of ATP (Fig. 2H). In contrast to the stimulation of the ATPase observed with PCNA, the ATPase of RFC-Rad24 was actually significantly inhibited in the presence of the checkpoint clamp Rad17/3/1, similar to that observed with the Escherichia coli clamp-clamp loader system (27).
Table 1.
ATPase activity of RFC-Rad24
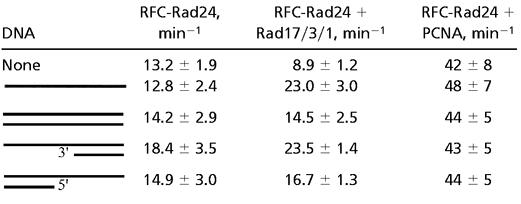 |
The assays contained 50 nM RFC-Rad24, 100 nM Rad17/3/1 or PCNA, 50 μM [α-32P]ATP, and 1 μM effector DNA (SS = A9, DS = A9/A2, 3′-recessed = A9/3′-20, 5′-recessed = A9/5′-20; see Materials and Methods). Percentage of ADP formed was quantitated by PhosphorImager analysis and used to calculate rates (expressed in turnover number per min). The data are averages of at least three independent experiments.
The ATPase activity of the Rad17/3/1⋅RFC-Rad24 complex was stimulated 2- to 3-fold by the addition of DNA. The strongest stimulation was measured for single-stranded DNA and 3′-junction DNA. The activity of RFC-Rad24 with PCNA, however, was not stimulated further by DNA. The regulatory effect of Rad17/3/1 on the ATPase of RFC-Rad24 required the Rad24 subunit. The ATPase activity of RFC or of the four-subunit Rfc2-5 complex was not altered by the addition of Rad17/3/1, with or without DNA cofactor (data not shown).
Interaction of RFC-Rad24 and Rad17/3/1 with DNA.
Genetic evidence has laid a firm foundation that RFC-Rad24 and Rad17/3/1 act together in the DNA damage checkpoint pathway. Given the structural similarities of RFC-Rad24 and Rad17/3/1 to RFC and PCNA, respectively, it is reasonable to hypothesize that a similar clamp loader-clamp relationship exists during DNA damage response (16). To investigate this problem, we have used surface plasmon resonance (SPR) to quantitate combinatorial interactions of the checkpoint factors with DNA. The DNA oligonucleotides were the same as those used in the ATPase studies (see Materials and Methods) and were attached to a sensor chip by means of a biotin linkage. Proteins of interest were flowed across the chip, and binding and dissociation were measured in real time.
When RFC-Rad24 and ATPγS were flowed across a chip charged with partially double-stranded DNA with a 3′-recessed junction, a weak but significant signal was observed, indicative of nucleotide-dependent binding of the clamp loader to DNA (Fig. 3A). This signal was not observed in the presence of ATP or without nucleotide present. These results suggest that ATP binding to RFC-Rad24 mediates binding to DNA, whereas its hydrolysis dissociates the clamp loader from the DNA, analogously to that previously observed with RFC (23, 28). No binding of Rad17/3/1 was observed with or without nucleotide present. However, when a mixture of RFC-Rad24, Rad17/3/1, and ATP was flowed across the chip, a strong signal was observed. Whether this signal is indicative of binding or clamp loading cannot be determined by this technique. However, with ATPγS present, a stronger signal was observed, and the dissociation phase was much slower than with ATP. These data are suggestive of a mechanism in which ATP binding allows loading of Rad17/3/1 by RFC-Rad24, but its hydrolysis is required to release RFC-Rad24. Therefore, the long-lived signal with ATPγS may represent stalled complexes, as observed previously by SPR in an analysis of PCNA loading by RFC (23). No SPR signal suggestive of PCNA loading by RFC-Rad24 was detected (Fig. 3A).
Figure 3.

Surface plasmon resonance analysis of checkpoint factor binding to DNA. DNA chips were prepared by using standard methodology and contained either ≈50 (A) or ≈20 (B–D) resonance units of biotinylated DNA. The DNA was either partially double-stranded 3′-recessed DNA A9/3′-20 (A and D), fully double-stranded DNA A9/A2 (B), or partially double-stranded 5′-recessed DNA A9/5′-20 (C) (see Materials and Methods). All proteins were injected at a concentration of 30 nM with 100 μM ATP or ATPγS, where indicated. No signal was observed without ATP or ATPγS present in B–D.
To determine the preferred DNA substrate for interaction with RFC-Rad24 and Rad17/3/1, chips with three distinct DNA substrates were tested for binding. To make a proper comparison possible and minimize artifacts inherent in high-density chips, very low and approximately equal levels of the DNA substrates were attached to the chips (29). With double-stranded DNA, a rapid-on and rapid-off signal was observed, suggesting transient binding of RFC-Rad24⋅Rad17/3/1 to the double-stranded break (Fig. 3B). This transient binding required either ATP or ATPγS, because in its absence, no signal was observed (data not shown). Similar rapid kinetics were observed with partially double-stranded DNA with a 5′-primer/template junction (Fig. 3C). In contrast, very long-lived complexes were observed with 3′-junction effector DNA and ATPγS, suggesting the presence of an RFC-Rad24⋅Rad17/3/1 complex stalled in loading at the 3′-primer/template junction (Fig. 3D). These data suggest that, as with RFC and PCNA, a normal primer/template is the preferred effector for the checkpoint proteins.
The Rad17/3/1 Clamp, Loaded by RFC-Rad24 onto Partial Duplex DNA, Is Released from RFC-Rad24 upon ATP Hydrolysis and Slides Across Double-Stranded DNA.
We carried out loading reactions onto DNA substrates derived from pBluescript SKII (+) to determine the nature and stoichiometry of the complexes actually loaded onto DNA as schematically shown in Fig. 4. We found that only RFC-Rad24 was recovered in the DNA-containing fractions when gapped DNA was incubated with a mixture of RFC-Rad24 and Rad17/3/1 without nucleotide (Fig. 4, lane 1). Loading of Rad17/3/1 alone onto either gapped circular or linear DNA was also not observed without the clamp loader, with or without nucleotide present (data not shown, but see Fig. 5, lanes 3 and 4).
Figure 5.

Rad17/3/1 and RFC-Rad24 constitute a specific clamp-clamp loader pair. Experiments were performed as described in Fig. 4. The assays contained 50 nM RFC-Rad24, 50 nM RFC [lacking the ligase-homology domain (amino acid 1–272) of Rfc1 (23)], 100 nM Rad17/3/1, 100 nM PCNA, 50 μM ATP, and 5 nM DNA (see Fig. 4). The fractions were analyzed by SDS/12% PAGE. Lanes 7–10 are RFC, PCNA, RFC-Rad24, and Rad17/3/1, respectively. Lanes 1–2, 3–6, and 7–10 are from independent experiments.
Loading of Rad17/3/1 required the presence of both the clamp loader and ATP (Fig. 4, lanes 2 and 11). Remarkably, the molar ratio of Rad17/3/1 to RFC-Rad24 in the DNA-bound fraction was ≈3–4:1, and that of Rad17/3/1 to DNA was ≈2–3:1 (three independent experiments). These results suggest that RFC-Rad24, after having loaded Rad17/3/1, released the clamp and then proceeded to load additional Rad17/3/1 clamps. If the Rad17/3/1 clamp released by the loader were free to slide across double-stranded DNA, then linearization of the DNA opposite the gap should allow the checkpoint clamp to slide off the DNA, thereby causing a reduction in signal. This type of experiment was first carried out to demonstrate that the E. coli β-subunit of DNA polymerase III holoenzyme is a sliding clamp (30). Indeed, the occupancy of Rad17/3/1 was reduced to that equimolar with RFC-Rad24 when the gapped DNA was linearized (Fig. 4, lane 3).
As a control experiment, PCNA, which is known to slide across DNA, was loaded by RFC and ATP onto the circular-gapped and the linear-gapped DNA under the same conditions as the checkpoint complexes (31). About 15-fold more PCNA was bound to the circular than to the linear DNA, and essentially, no RFC was detected (Fig. 5, lanes 5 and 6). These results show that Rad17/3/1, like PCNA, is a sliding clamp which can traverse over 1 kb of double-stranded DNA. The presence of significant residual Rad17/3/1 bound to the linear DNA indicates either that release of the clamp from the clamp loader is a slow process or that sliding of the clamp itself proceeds at a slow rate.
The same loading reactions with ATPγS as cofactor gave distinctively different results. Rad17/3/1 and RFC-Rad24 bound DNA to the same extent, whether the gapped DNA had been linearized or not (Fig. 4, lanes 5 and 6). Consistent with the data in Fig. 3D, these experiments suggest that clamps loaded with ATPγS are frozen onto the DNA, because hydrolysis of bound ATP is required to release the clamp from the clamp loader. The same mechanism has been proposed for RFC and PCNA (26, 32).
We observed very inefficient loading of Rad17/3/1 onto single-stranded DNA (Fig. 4, lane 9). Furthermore, addition of the yeast single-stranded DNA-binding protein RPA suppressed this loading, presumably by melting secondary structures that could resemble template/primer-loading sites (Fig. 4, lanes 10 and 12). Interestingly, RPA did not only increase the association of RFC-Rad24 with DNA, it also reduced the excess Rad17/3/1 loaded onto gapped DNA, perhaps through stabilization of an RPA⋅RFC-Rad24⋅Rad17/3/1 complex at the site of loading (Fig. 4, lanes 11 and 12). We did not observe any clamp loading onto circular double-stranded DNA (Fig. 4, lane 4) nor on damaged DNA. We damaged covalently closed circular double-stranded DNA by UV irradiation (1,500 J/m2) to the extent of about 4 dimers per plasmid molecule (as judged by the generation of T4 UV-endonuclease V-sensitive sites), but no loading of Rad17/3/1 onto this damaged DNA was observed (data not shown).
Rad17/3/1 and RFC-Rad24 Constitute a Specific Clamp-Clamp Loader Pair.
The interaction studies in Fig. 2 already indicated that RFC-Rad24 forms stable interactions with Rad17/3/1, but not with PCNA. To investigate whether the transient interactions between the checkpoint clamp loader and PCNA inferred from the ATPase data might promote loading of PCNA by RFC-Rad24, we carried out additional loading experiments on gapped DNA (Fig. 5). We observed loading only for the pairs Rad17/3/1–RFC-Rad24 (Fig. 4) and PCNA–RFC (Fig. 5, lanes 5 and 6). No PCNA loading by RFC-Rad24 was observed by SPR analysis (Fig. 3A), nor by product analysis (Fig. 5, lanes 1 and 2), nor by a functional DNA replication analysis, in which loading of PCNA is detected indirectly by stimulation of DNA synthesis by DNA polymerase δ (data not shown). Likewise, no loading of Rad17/3/1 by RFC onto gapped DNA was detected (Fig. 5, lanes 3 and 4). These data suggest that the two pairs of complexes, PCNA–RFC and Rad17/3/1–RFC-Rad24, define nonexchangeable clamp-clamp loader systems which are active during DNA replication and the checkpoint response, respectively.
Studies with the Rec1 checkpoint protein from Ustilago maydis, which is the homologue of Rad17, have revealed an intrinsic 3′-5′-exonuclease activity. Similar activities have been reported for the human homologues of Rad17 and Ddc1 (33–36). We detected no nuclease activity in our Rad17/3/1 preparations, when they were measured on oligonucleotide substrates that gave high activity with Rec1 (36). Genetic studies suggest that RAD24 may regulate the activity of a downstream 3′-5′-exonuclease (37). Possibly, a cryptic exonuclease is present in Rad17/3/1, but a more specialized DNA structure may be needed to elicit its activity. Alternatively, the clamp may provide a loading site for a downstream exonuclease that functions in DNA degradation.
Our biochemical studies of the DNA damage checkpoint system not only present experimental evidence for the hypothesis that indeed a clamp-clamp loader system is involved in the checkpoint response, but also provides important new insights into substrate recognition and processing. Although the major clamp in our preparations consisted of a Rad17/Mec3/Ddc1 heterotrimer, we also identified a complex consisting of Rad17 and Mec3, perhaps in the form of a Rad17/Rad17/Mec3 trimer (Fig. 1). The possibility of alternative checkpoint clamps may provide diversity to the response and is in agreement with a study that identified Rad17–Rad17 interactions in response to damage, and with genetic studies ascribing overlapping but not identical functions to the three components of the checkpoint clamp (38, 39). From our biochemical studies, it seems that the clamp loader does not directly load the clamp onto sites of damage, but rather on regular 3′-primer/template junctions, which may be generated during repair of DNA damage or which preexist at stalled replication forks (Fig. 3D). However, we also detected interactions with 5′-primer/template junctions and even double-stranded DNA ends (Fig. 3 B and C), suggesting that the range of substrates recognized by the checkpoint clamp loading system may be broader than the replicational clamp loading system. Finally, the ability of Rad17/3/1 to slide across more than 1 kb of duplex DNA could provide a mechanism by which the checkpoint clamp, either alone or in a complex with associated damage recognition factors, could search for DNA damage after having been loaded at normal primer/template junctions.
Acknowledgments
We thank Kim Gerik and Carrie Welch for technical assistance, and John Majors and Anita Niedziela-Majka for critical discussions during the course of this work. This work was supported in part by National Institutes of Health Grant GM32431.
Abbreviations
- RFC
replication factor C
- PCNA
proliferating cell nuclear antigen
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Hartwell L H, Weinert T A. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- 2.Rouse J, Jackson S P. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 3.Melo J, Toczyski D. Curr Opin Cell Biol. 2002;14:237–245. doi: 10.1016/s0955-0674(02)00312-5. [DOI] [PubMed] [Google Scholar]
- 4.Kondo T, Wakayama T, Naiki T, Matsumoto K, Sugimoto K. Science. 2001;294:867–870. doi: 10.1126/science.1063827. [DOI] [PubMed] [Google Scholar]
- 5.Melo J A, Cohen J, Toczyski D P. Genes Dev. 2001;15:2809–2821. doi: 10.1101/gad.903501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rouse J, Jackson S P. Mol Cell. 2002;9:857–869. doi: 10.1016/s1097-2765(02)00507-5. [DOI] [PubMed] [Google Scholar]
- 7.Sun Z, Fay D S, Marini F, Foiani M, Stern D F. Genes Dev. 1996;10:395–406. doi: 10.1101/gad.10.4.395. [DOI] [PubMed] [Google Scholar]
- 8.Ellison V, Stillman B. Cell. 2001;106:655–660. doi: 10.1016/s0092-8674(01)00498-6. [DOI] [PubMed] [Google Scholar]
- 9.Warbrick E. BioEssays. 2000;22:997–1006. doi: 10.1002/1521-1878(200011)22:11<997::AID-BIES6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 10.Griffiths D J, Barbet N C, McCready S, Lehmann A R, Carr A M. EMBO J. 1995;14:5812–5823. doi: 10.1002/j.1460-2075.1995.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lydall D, Weinert T. Mol Gen Genet. 1997;256:638–651. doi: 10.1007/s004380050612. [DOI] [PubMed] [Google Scholar]
- 12.Shimomura T, Ando S, Matsumoto K, Sugimoto K. Mol Cell Biol. 1998;18:5485–5491. doi: 10.1128/mcb.18.9.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green C M, Erdjument-Bromage H, Tempst P, Lowndes N F. Curr Biol. 2000;10:39–42. doi: 10.1016/s0960-9822(99)00263-8. [DOI] [PubMed] [Google Scholar]
- 14.Lindsey-Boltz L A, Bermudez V P, Hurwitz J, Sancar A. Proc Natl Acad Sci USA. 2001;98:11236–11241. doi: 10.1073/pnas.201373498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffith J D, Lindsey-Boltz L A, Sancar A. J Biol Chem. 2002;277:15233–15236. doi: 10.1074/jbc.C200129200. [DOI] [PubMed] [Google Scholar]
- 16.Venclovas C, Thelen M P. Nucleic Acids Res. 2000;28:2481–2493. doi: 10.1093/nar/28.13.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paciotti V, Lucchini G, Plevani P, Longhese M P. EMBO J. 1998;17:4199–4209. doi: 10.1093/emboj/17.14.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondo T, Matsumoto K, Sugimoto K. Mol Cell Biol. 1999;19:1136–1143. doi: 10.1128/mcb.19.2.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiomi Y, Shinozaki A, Nakada D, Sugimoto K, Usukura J, Obuse C, Tsurimoto T. Genes Cells. 2002;7:861–868. doi: 10.1046/j.1365-2443.2002.00566.x. [DOI] [PubMed] [Google Scholar]
- 20.Gerik K J, Gary S L, Burgers P M. J Biol Chem. 1997;272:1256–1262. doi: 10.1074/jbc.272.2.1256. [DOI] [PubMed] [Google Scholar]
- 21.Burgers P M. Methods. 1999;18:349–355. doi: 10.1006/meth.1999.0796. [DOI] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 23.Gomes X V, Burgers P M. J Biol Chem. 2001;276:34768–34775. doi: 10.1074/jbc.M011631200. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Zhu Z, Vidanes G, Mbangkollo D, Liu Y, Siede W. J Biol Chem. 2001;276:26715–26723. doi: 10.1074/jbc.M103682200. [DOI] [PubMed] [Google Scholar]
- 25.Siegel L M, Monty K J. Biochim Biophys Acta. 1966;112:346–362. doi: 10.1016/0926-6585(66)90333-5. [DOI] [PubMed] [Google Scholar]
- 26.Gomes X V, Schmidt S L, Burgers P M. J Biol Chem. 2001;276:34776–34783. doi: 10.1074/jbc.M011743200. [DOI] [PubMed] [Google Scholar]
- 27.Hingorani M M, O'Donnell M. J Biol Chem. 1998;273:24550–24563. doi: 10.1074/jbc.273.38.24550. [DOI] [PubMed] [Google Scholar]
- 28.Tsurimoto T, Stillman B. J Biol Chem. 1991;266:1950–1960. [PubMed] [Google Scholar]
- 29.Schuck P. Curr Opin Biotechnol. 1997;8:498–502. doi: 10.1016/s0958-1669(97)80074-2. [DOI] [PubMed] [Google Scholar]
- 30.Stukenberg P T, Studwell-Vaughan P S, O'Donnell M. J Biol Chem. 1991;266:11328–11334. [PubMed] [Google Scholar]
- 31.Burgers P M J, Yoder B L. J Biol Chem. 1993;268:19923–19936. [PubMed] [Google Scholar]
- 32.Waga S, Stillman B. Mol Cell Biol. 1998;18:4177–4187. doi: 10.1128/mcb.18.7.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Onel K, Koff A, Bennett R L, Unrau P, Holloman W K. Genetics. 1996;143:165–174. doi: 10.1093/genetics/143.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parker A E, Van de Weyer I, Laus M C, Verhasselt P, Luyten W H. J Biol Chem. 1998;273:18340–18346. doi: 10.1074/jbc.273.29.18340. [DOI] [PubMed] [Google Scholar]
- 35.Bessho T, Sancar A. J Biol Chem. 2000;275:7451–7454. doi: 10.1074/jbc.275.11.7451. [DOI] [PubMed] [Google Scholar]
- 36.Naureckiene S, Holloman W K. Biochemistry. 1999;38:14379–14386. doi: 10.1021/bi991495b. [DOI] [PubMed] [Google Scholar]
- 37.Lydall D, Weinert T. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- 38.Paulovich A G, Armour C D, Hartwell L H. Genetics. 1998;150:75–93. doi: 10.1093/genetics/150.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H, Zhu Z, Vidanes G, Mbangkollo D, Liu Y, Siede W. J Biol Chem. 2001;276:26715–26723. doi: 10.1074/jbc.M103682200. [DOI] [PubMed] [Google Scholar]