

Abstract
p53 ubiquitination catalysed by MDM2 (murine double minute clone 2 oncoprotein) provides a biochemical assay to dissect stages in E3-ubiquitin-ligase-catalysed ubiquitination of a conformationally flexible protein. A mutant form of p53 (p53F270A) containing a mutation in the second MDM2-docking site in the DNA-binding domain of p53 (F270A) is susceptible to modification of long-lived and high-molecular-mass covalent adducts in vivo. Mutant F270A is hyperubiquitinated in cells as defined by immunoprecipitation and immunoblotting with an anti-ubiquitin antibody. Transfection of His-tagged ubiquitin along with p53R175H or p53F270A also results in selective hyperubiquitination in cells under conditions where wild-type p53 is refractory to covalent modification. The extent of mutant p53R175H or p53F270A unfolding in cells as defined by exposure of the DO-12 epitope correlates with the extent of hyperubiquitination, suggesting a link between substrate conformation and E3 ligase function. The p53F270A:6KR chimaeric mutant (where 6KR refers to the simultaneous mutation of lysine residues at positions 370, 372, 373, 381, 382 and 386 to arginine) maintains the high-molecular-mass covalent adducts and is modified in an MDM2-dependent manner. Using an in vitro ubiquitination system, mutant p53F270A and the p53F270A:6KR chimaeric mutant is also subject to hyperubiquitination outwith the C-terminal domain, indicating direct recognition of the mutant p53 conformation by (a) factor(s) in the cell-free ubiquitination system. These data identify an in vitro and in vivo assay with which to dissect how oligomeric protein conformational alterations are linked to substrate ubiquitination in cells. This has implications for understanding the recognition of misfolded proteins during aging and in human diseases such as cancer.
Keywords: chaperone, murine double minute clone 2 oncoprotein (MDM2), p53, protein folding, ubiquitin
Abbreviations: 6KR, simultaneous mutation of lysine residues at positions 370, 372, 373, 381, 382 and 386 to arginine; Ada-Ahx3-Leu3-vinyl sulphone, adamantane acetyl-(6-aminohexanoyl)3-(leucyl)3-vinyl(methyl) sulphone; ALLN, N-acetyl-L-leucyl-L-leucylnorleucinal; ARNIP, androgen receptor N-terminal-interacting protein; CDK2, cyclin-dependent kinase 2; CHIP, C-terminus of Hsc70 (heat-shock cognate 70)-interacting protein; DMEM, Dulbecco's modified Eagle's medium; DTT, dithiothreitol; EGS, ethylene glycol bis(succinimidyl succinate); FBS, foetal bovine serum; HBSS, Hanks balanced salt solution; HRP, horseradish peroxidase; Hsp90, 90 kDa heat-shock protein; IHD, interferon-binding homology domain; M5A, McCoy's 5A; MDM2, murine double minute clone 2 oncoprotein; NEDD8, neural precursor cell expressed, developmentally down-regulated gene 8; RLU, relative light units; RNAi, RNA interference; SiRNA, small interfering RNA; TMB, tetramethylbenzidine
INTRODUCTION
The p53 protein is a tumour suppressor that functions as a transcription factor at a key nodal point in the response of cells to environmental change. p53 protein is active as a transcription factor in undamaged cells towards a subset of growth-responsive gene products, but insults like DNA damage, hypoxia or oxidant stress recruits p53 towards a distinct set of promoters that mediate the stress response [1]. Such a response leads either to growth arrest, repair and cell survival or apoptosis. Two dominating pathways control p53 activity. Negative control of p53 function is mediated by four distinct proteins with E3 ubiquitin ligase activity that mediate ubiquitination of p53, namely MDM2 (murine double minute clone 2 oncoprotein) [2–4], PrihH2 [5], COP-1 [6] and CHIP [C-terminus of Hsc70 (heat-shock cognate 70)-interacting protein] [7]. Positive control of p53-dependent tumour suppression is mediated by the transcriptional co-activator protein p300, which is essential for p53 protein stabilization in response to DNA damage [8]. The balance between p300 co-activation of transcription and E3-ligase-catalysed ubiquitination controls the transcriptional flux of the p53 pathway [9].
p300 and MDM2 both bind to the N-terminal LXXLL transactivation domain of p53. This provides one form of p53 regulation through mass action that comes about by competition of MDM2 or p300 for the same binding site on p53. In this model, MDM2 can act as a repressor of p53-dependent transcription by occluding the binding of transcription components by the direct binding of MDM2 to DNA-bound p53 [10,11]. MDM2 also binds to two distinct regions of p53 with differing effects on the p53 ubiquitination. The original MDM2-binding site was defined within the LXXLL N-terminal activation domain of p53 [12–14], the binding of which by MDM2 can repress p53-dependent transcription by competition with p300 and other basal factors that can bind to the LXXLL activation motif. A second biochemical effect of MDM2 binding to the LXXLL region of p53 is mono-ubiquitination in the C-terminal regulatory domain of p53 [15]. Multiple sites of mono-ubiquitination in the C-terminal domain of p53 appear to play a role in the first stage in p53-protein degradation by the proteasome [16]. The N-terminal domain in MDM2 contains a hydrophobic pocket that binds with the LXXLL activation domain of p53 and is the target of drug-design programmes aimed at inhibiting the MDM2–p53 complex, thus activating the p53 pathway [17,18]. Mutation of the Phe19 residue in p53 flanking the LXXLL activation domain can inhibit MDM2-dependent ubiquitination of p53, further suggesting that MDM2 binding to the N-terminus can mediate p53 ubiquitination in the C-terminus [19]. However, p53 substrate conformation is critical in MDM2-dependent ubiquitination, since monomeric forms of p53 are not ubiquitinated by MDM2, despite having an intact N-terminal MDM2-binding site [20,21].
A second MDM2-binding site was identified within the S10 β-sheet in the central DNA-binding domain of p53 that plays a role in anchoring MDM2 and which can modulate p53 ubiquitination in vitro and in vivo [22,23]. The relationship between the N-terminal MDM2-binding site on p53 and the DNA-domain-localized MDM2-binding site on p53 with respect to ubiquitination is not yet defined. However, this second binding site of MDM2 is within a conformationally flexible motif known to be misfolded in human cancers [24], suggesting a link between p53 conformation and MDM2 binding. A chaperoning role for MDM2 in altering the folding of p53 might be required to assist in ubiquitin transfer to the substrate by the E2–ubiquitin complex. In fact, p53 protein conformation can be altered in vitro through the action of an MDM2–Hsp90–CHIP protein chaperone complex (Hsp90 is 90 kDa heat-shock protein) [23], and CHIP-mediated ubiquitination of p53 can occur in cells [7]. Although the precise mechanism whereby MDM2 embraces tetrameric p53 either at the LXXLL site or at the central DNA-binding domain remains undefined, this second binding site gives support to the idea that the alternative MDM2-docking site may mediate p53 ubiquitination by changing the substrate folding status. In the present study we evaluated the role of p53 substrate conformation in controlling MDM2-dependent ubiquitination of p53 and identified a link between substrate misfolding and susceptibility to ubiquitination.
EXPERIMENTAL
Chemicals
All reagents were supplied by Sigma unless otherwise stated. Restriction enzymes were supplied by New England Biolabs. Oligonucleotides were synthesized and desalted or HPLC- purified by Sigma–Genosys. Ada-Ahx3-Leu3-vinyl sulphone [adamantane-acetyl-(6-aminohexanoyl)3-(leucyl)3-vinyl(methyl) sulphone] was from BIOMOL International. All siRNA (small interfering RNA) sequences were obtained from and synthesized by Dharmacom. FBS (foetal bovine serum), DMEM (Dulbecco's modified Eagle's medium), M5A (McCoy's 5A) medium, HBSS (Hanks balanced salt solution), trypsin/EDTA solution and Lipofectamine™ 2000 were supplied by Invitrogen. Cycloheximide was obtained from Supelco. Microlite 2 96-well ELISA plates were supplied by Dynex. Streptavidin was supplied by Vector Laboratories. TMB (tetramethylbenzidine) was supplied by Kirkegaard & Perry Laboratories. Precast 4–12% (w/v) NuPAGE® Bis-Tris gel and Mops buffer were provided by Invitrogen. Hybond-C nylon membrane for immunoblotting, Protein G–Sepharose beads, and ECL® (enhanced chemiluminescence) Hyperfilm were supplied by Amersham Pharmacia Biotech. Synthetic peptides were synthesized by Chiron Mimotopes. The HiTrap-SP column was obtained from Amersham Biotech. Antibodies used include anti-MDM2 (2A10), anti-MDM2 (4B2), anti-MDM2 (SMP14), anti-p53 (DO-1), anti-p53 (DO-11), anti-p53 (DO-12), anti-p53 (19.1), anti-p53 (ICA-9), anti-p53 (240), anti-p53 (1620), anti-p53 (421) and anti-p53 (CM1). Anti-p21 (Ab-1) was supplied by Calbiochem. HRP (horseradish peroxidase)-conjugated secondary antibodies were supplied by Dako. pcDNA3.1-p53175H, pcDNA3.1-p53341A, the expression vector for NEDP1 (a deNEDDylating cysteine protease) and expression constructs for His–ubiquitin (ubiquitin with six histidine residues at its N-terminus) were provided by Dr Dimitris Xirodimas (School of Life Sciences Research Biocentre, University of Dundee, Dundee, Scotland, U.K.). pCMV-hMDM2 plasmid was a gift from Dr Bert Vogelstein (Johns Hopkins Oncology Center, Boston, MA, U.S.A.). pcDNA3.1-p53-6KR was a gift from Professor Ronald T. Hay (Centre for Biomolecular Sciences, University of St. Andrews, St. Andrews, Scotland, U.K.) and was previously used to demonstrate defects in p53-mediated and p300-co-activated chromatin interactions [25].
Site-specific mutagenesis
QuikChange™ Site-Directed Mutagenesis Kit (Stratagene) was used to create p53 mutants. Primers were designed according to the manufacturer's protocol. The mutagenesis primers were as follows:
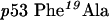 |
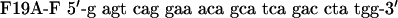 |
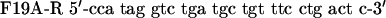 |
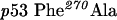 |
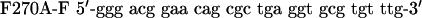 |
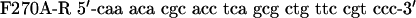 |
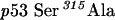 |
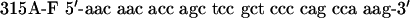 |
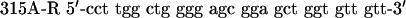 |
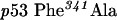 |
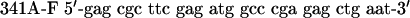 |
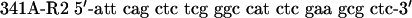 |
Immunoblotting
The resolved proteins were transferred on to Hybond-C nitrocellulose membrane in transfer buffer [0.192 M glycine, 25 mM Tris and 20% (v/v) methanol, pH 8.3] at 100 mA for 2 h, or, alternatively, at 20 mA overnight. Following transfer, the membrane was stained with black Indian ink to confirm even protein transfer and loading. Non-specific antibody binding was blocked by incubating the membrane for 1 h in 3% milk-PBST [3% (w/v) dried skimmed milk and 0.1% (v/v) Tween 20 in PBS] and then incubated with primary antibody at the dilution recommended by the suppliers in 3% milk-PBST for 1 h. The blot was then washed twice for 10 min in PBST [0.1% (v/v) Tween 20 in PBS] before incubating for 1 h in HRP-coupled secondary antibody in 3% milk-PBST to detect specific antibody binding. Finally, the blot was washed six times with PBST for 10 min and then incubated with ECL® solution, and specific bands were detected by being exposed to ECL® hyperfilm.
ELISA
All procedures were carried out at 4 °C unless mentioned otherwise. The antibodies used and the exact quantity of protein employed is stated for each Figure. Wells were coated with pure monoclonal antibodies (DO-1, D-12, PAb240, PAb421 and PAb1620 for p53, and 4B2 for MDM2) at 40–100 ng/well in 50 μl of 0.1 M sodium borate, pH 9.0, overnight, and washed three times with 180 μl of PBST (0.02%, v/v) to remove non-binding antibodies and sodium borate. To prevent non-specific binding, non-reactive sites were blocked with 180 μl of 3% (w/v) BSA/PBST and incubated for 1 h. p53 or MDM2 was resuspended in 50 μl of 3% BSA/PBST and added to the antibody-coated ELISA wells and incubated for 1 h. Unbound proteins were washed away with 180 μl of PBST (0.02%, v/v) three times. For p53 conformation detection using anti-p53 monoclonal antibodies, p53 protein captured by anti-p53 monoclonal antibodies were incubated with anti-p53 polyclonal antibody CM1 for 1 h in 50 μl of 3% BSA/PBST. Unbound antibodies were washed away with 180 μl of PBST (0.02%, v/v) three times, and 50 μl of HRP-coupled anti-rabbit Ig in 3% BST/PBST were added to the wells for a 1 h incubation. The amount of CM1 captured on the ELISA plate was detected by the TMB method or ECL® method. In the TMB method, 50 μl of TMB was added per well to initiate the colour reaction, the reaction being stopped by the addition of 50 μl of 0.5 M H2SO4 per well. The intensity of the colour was read at 450 nm in a microplate reader (Dynex Laboratories). In the ECL® method, 50 μl of ECL® solution was added to each well to develop chemiluminescence and the luminescence produced was immediately detected with a luminometer.
Cell maintenance
H1299 cells were incubated in RPMI 1640 medium, and SAOS-2 cells and A375 cells were incubated in DMEM. Both media were supplemented with 10% (w/v) FBS. Cells were cultured at 37 °C under a 5%-CO2 humidified atmosphere. Cells were maintained in 175 cm2 flasks and subcultured as required. Cells were rinsed with 5 ml of PBS for 1 min, followed by the addition of 1 ml of trypsin/EDTA solution/175 cm2 flask and incubated at 37 °C for 5 min. Trypsinized cells were diluted 10-fold and centrifuged at 1000 g for 3 min, and the collected cells were resuspended in fresh medium in a fresh flask. Cells were plated in 10-cm-diameter tissue-culture dishes and incubated for 24 h. All transfections were performed using the liposome-mediated method with Lipofectamine™ 2000, with the manufacturer's recommendations being followed. The exact quantity of transfected DNA is indicated for each experiment and, where necessary, carrier DNA (pcDNA3.1) was transfected to keep the same quantity of DNA consistent in each transfection. Unless stated otherwise, cells were harvested 24 h post-transfection.
In vitro and in vivo ubiquitination assay
H1299 cells were transfected with wild-type and mutant p53 with or without co-transfection of MDM2 (the exact quantity of DNA transfected is indicated for each experiment) and, where necessary, carrier DNA (pcDNA3.1) was incorporated to keep the quantity of DNA consistent in each transfection. All cells were treated (except the cells treated with cycloheximide) with 50 μM of the proteasome inhibitor ALLN (N-acetyl-L-leucyl-L-leucylnorleucinal) for 2 h before harvest. At 24 h post-transfection, cells were washed twice with ice-cold PBS and transferred to a microcentrifuge tube. The harvested cells were lysed in 50 μl of lysis buffer A [100 mM Tris/HCl, pH 8.0, 100 mM dithiothreitol (DTT) and Complete™ protease inhibitor mixture (Roche)] by gentle pipetting, incubated on ice for 15 min and centrifuged at 11000 g for 10 min. The supernatant was transferred to a fresh microcentrifuge tube before freezing in liquid nitrogen. The protein concentration was determined using the Bradford assay and the same quantity of total protein from each cell lysate was examined for immunoblot analysis. In vitro p53 ubiquitination assays using recombinant MDM2 protein purified from Escherichia coli were carried out as described previously [23]. However, instead of using recombinant p53 purified from Sf9 (Spodoptera frugiperda 9 insect cells) or E. coli cells, the p53 protein was synthesized in rabbit reticulocyte lysates (Promega) from the indicated p53 allele as described previously [26–28]. In order to monitor in vitro-co-translation-linked ubiquitination, 0.75 μg of wild-type p53, p53F270A, p53F19A or p53F19A/F270A (double mutant) were in vitro-translated with 0.75 μg of either empty pCDNA vector or MDM2. After a 90 min incubation at 30 °C, the translated proteins were resolved on a 4–12%-polyacrylamide/Bis-Tris gel and probed with DO-1 antibody. To examine the effect of NEDP1 on wild-type-p53 ubiquitination, H1299 cells were co-transfected at 70% confluency with wild-type p53 (1 μg) and MDM2 (3 μg) and incremental amounts (1–8 μg) of pcDNA encoding the deNEDDylating enzyme NEDP1. For mutant p53 ubiquitination, since significant ubiquitination occurs without transfected MDM2, H1299 cells were co-transfected at 70% confluency with p53F270A (1 μg) and incremental amounts (1–8 μg) of pcDNA encoding the deNEDDylating enzyme NEDP1. Cells were lysed 24 h after transfection in Triton X-100 lysis buffer [50 mM Tris/HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% (w/v) Triton X-100 and 10% (w/v) glycerol] and Complete™ protease inhibitor mixture. Samples were run on a 4–12% Bis-Tris gel and probed with DO-1 antibody.
His-ubiquitin conjugates pull-down assay
Cells transfected with pCMV-His-Ub plasmid for 24 h were incubated with 50 μM ALLN for 2 h and then harvested and washed in PBS before the addition of 6 ml of HUBA (His–ubiquitin buffer A; 6 M guanidinium chloride, 0.1 M Na2HPO4/NaH2PO4, pH 8.0, 10 mM Tris/HCl and 10 mM 2-mercaptoethanol), including 5 mM imidazole, to half of the pellet. The lysate was then homogenized using a 24-gauge syringe needle before adding 75 μl of Ni2+-nitrilotriacetate–agarose beads and rotating at room temperature (21 °C) for 4 h. The beads were then centrifuged at 2000 g for 5 min and the supernatant was discarded before washing with 750 μl of HUBA, HUBB (His–ubiquitin buffer B; 8 M urea, 0.1 M Na2HPO4/NaH2PO4, pH 8.0, 10 mM Tris/HCl and 10 mM 2-mercaptoethanol), HUBC (His–ubiquitin buffer C; 8 M urea, 0.1 M Na2HPO4/NaH2PO4, pH 6.3, 10 mM Tris/HCl and 10 mM 2-mercaptoethanol) and 0.2% (v/v) Triton X-100, HUBC, and HUBC and 0.1% (v/v) Triton X-100. To the washed beads, 75 μl of HUEB [His–ubiquitin elution buffer; 0.2 M imidazole, 5% (w/v) SDS, 0.15 M Tris/HCl, pH 6.7, 10% (v/v) glycerol and 0.72 M 2-mercaptoethanol] was added and incubated for 20 min at room temperature. Finally, 75 μl of 2× SDS sample buffer buffer was added to the eluted ubiquitin conjugates and subject to Western blot analysis. The other half of the pellet was lysed in Reporter Lysis Buffer (Promega) and the whole-cell lysate was analysed by Western blotting with anti-p53 antibody (DO-1).
Ablation of endogenous MDM2 by siRNA
All siRNAs were obtained from Dharmacom and consist of 21-nucleotide siRNA duplexes with 3′-dTdT overhangs. The sequences of MDM2 siRNA and negative control siRNA (non-specific control duplex II) are concealed by Dharmacom. PARC siRNA sequence was adopted from [29]; 21-nucleotide siRNA duplex with 3′-dTdT overhangs corresponding to PARC mRNA (AAGCUUUCCUCGAGAUCCAGG). About 40% confluent H1299 cells in six-well tissue-culture plates were transfected with 200 pmol of each siRNA (non-specific control, MDM2 or PARC siRNA), which is 100 nM in 2 ml of medium, using 5 μl of Lipofectamine™. At 24 h after transfection, medium was exchanged with fresh medium after cells had been washed once with PBS, and the second round of siRNA transfection was carried out with wild-type p53 or p53R175H expression vectors. A 200 pmol sample of each siRNA was co-transfected with 1 μg of wild-type p53 or p53R175H expression vectors using 5 μl of Lipofectamine™, and the cells were incubated for a further 22 h. At 46 h after the first transfection, ALLN was added to a final concentration of 50 μM to preserve any ubiquitinated p53 products in the cells, and cells were incubated for 2 h before harvesting. The harvested cells were lysed in 50 μl of lysis buffer A (100 mM Tris/HCl, pH 8.0, 100 mM DTT and protease inhibitor mixture) on ice for 15 min by gentle pipetting, and spun at 11000 g for 10 min, and the supernatant was transferred to a fresh microcentrifuge tube before freezing in liquid nitrogen. Protein concentration was determined using the Bradford assay and the same quantity of total protein from each cell lysate was subjected to immunoblot analysis. Cell lysis buffer contained Complete™ protease inhibitor mixture (1×PIM) (2 mM Pefabloc, 42 μM leupeptin, 0.16 μM aprotinin, 3 μM pepstatin, 1 mM EDTA, 0.5 μM soyabean trypsin inhibitor and 1 mM benzamidine). Lysis buffer A contained 100 mM Tris/HCl, pH 8.0, 100 mM DTT and 1×PIM. Lysis buffer B contained 1% Igepal, 50 mM Hepes, pH 7.6, 0.4 M NaCl, 0.05% Triton X-100, 10 mM NaF, 1 mM β-phosphoglycerate, 20 mM okadaic acid, 2 mM DTT and 1×PIM.
RESULTS
An S10 β-sheet mutation in p53 partially restores ubiquitination to the p53F19A allele of p53
Mutation of the MDM2-binding site in the central DNA-binding domain of p53 can enhance p53 ubiquitination in vivo [22]. However, the mechanism was not defined. Here we extend this observation by determining whether: (i) this ubiquitination reaction is affected by p53 conformation; (ii) this p53 ubiquitination reaction is MDM2-dependent; and (iii) this p53 ubiquitination reaction occurs at the known C-terminal ubiquitination sites on p53. As reported previously, the p53F270A allele of p53 has enhanced ubiquitination in cells compared with wild-type p53 [22], as determined by the high-molecular-mass ladder of p53-associated bands (Figure 1A, lane 4 versus lane 2), despite the fact that the p53F270A allele cannot induce MDM2 induction (Figure 2, lane 4 versus lane 2). Further, the transfection of MDM2 into cells resulted in enhanced ubiquitination of wild-type p53 to a level similar to ubiquitination of p53F270A (Figures 1B and 1C, lanes 4 versus lanes 2). As a control, p53F19A mutant protein is not ubiquitinated well in the absence or presence of transfected MDM2 (Figure 1, lanes 3 versus lanes 1), even though the p53F19A protein induces MDM2 protein (Figure 2, lane 3 versus lane 2). These data are consistent with previous models suggesting that MDM2 binding to the N-terminus is required to mediate p53 ubiquitination. However, introducing the F270A missense mutation into the p53F19A mutant protein partially restored mono-ubiquitination and high-molecular-mass bands after MDM2 transfection (Figure 1, lanes 7 versus lanes 3). Since the p53F270A protein is better than wild-type p53 in binding stably to MDM2 [22], these data indicate that mutation of the MDM2-binding site in the DNA-binding domain of p53 can partially restore MDM2-mediated ubiquitination, even with the F19A mutation in the N-terminal domain of p53.
Figure 1. Monomeric p53 encoded by the F341A allele is not ubiquitinated by MDM2.

Expression vectors encoding wild-type p53, p53F19A, p53F270A, p53S315A, p53F341A, p53F19A/F270A and p53F270A/S315A (1 μg of DNA as indicated) were co-transfected with pCMV-MDM2 [0 (A), 1 (B) or 3 (C) μg] into H1299 cells, and the expressed proteins were examined for changes in their steady-state levels of p53 with the amount of ubiquitination products determined by immunoblotting with anti-p53 antibody (DO-1).
Figure 2. Transactivation function of p53 MDM2-binding site mutants.

Expression vectors encoding wild-type p53, p53F19A, p53F270A and p53F19A/F270A (1 μg of DNA) were transfected into H1299 cells, and the expressed proteins were examined for changes in their steady-state levels of p53 with the amount of ubiquitination products determined by immunoblotting with anti-p53 antibody (DO-1, top panel), anti-MDM2 antibody (middle panel) or anti-p21 antibody (bottom panel) as indicated.
We examined the contribution of another binding region in p53, implicated in its degradation, to the MDM2-dependent ubiquitination reaction. The CDK2 (cyclin-dependent kinase 2) phosphorylation site of p53 was previously shown to contain a determinant that mediates p53 degradation when fused to a carrier protein [30]. The mutant form of p53 encoded by the F270A allele was mutated at the CDK2 phosphorylation sites to determine whether this residue can modulate the mutant protein ubiquitination. Compared with the ubiquitination of p53F270A in the absence of transfected MDM2 (Figure 1A, lane 4), the p53F270A/S315A double mutant gave rise to a similar pattern of hyperubiquitination (Figure 1A, lane 8). Further, mutating the CDK2 site alone did not alter basal (Figure 1A, lane 5 versus lane 2) and MDM2-mediated ubiquitination of p53 (Figure 1B and 1C, lanes 5 versus lanes 2). The most significant clue into the possible nature of mutant p53F270A ubiquitination under these conditions is that monomeric p53 encoded by the F341A allele cannot be ubiquitinated well after transfection of MDM2 (Figure 1, lanes 6 versus lanes 2). These data suggest that the conformation of tetrameric p53 may be an important recognition determinant in p53 ubiquitination and we evaluated whether the hyperubiquitination we see in p53F270A could be explained in part by alterations in p53 conformation induced by the mutation.
Unfolding of p53 correlates with enhanced ubiquitination
A growing concept in the protein-folding field is the realization that many regulatory proteins are proving to be thermodynamically unstable or intrinsically disordered in their ‘native’ state [31]. The binding of regulatory partner protein or covalent modifications can stabilize such subdomains by an induced-fit mechanism and drive functional structural elements. This model explains why many human diseases can be attributed to conformational defects or the production of temperature-sensitive alleles of a polypeptide and provides the rationale for restoring polypeptide function with therapeutic benefit. p53 protein fits into this class of regulatory proteins that are intrinsically unfolded in their native conformation [32]. For example, the mechanism of p53 latency with respect to DNA binding appears to be driven by the ability of the C-terminal domain to destabilize the folding of the core DNA-binding domain in cis and reduce the specific activity as a sequence-specific DNA-binding protein [33]. Phosphorylation or deletion of the C-terminal regulatory domain of p53 neutralizes this domain and permits folding and enhanced thermostability of the core DNA-binding domain [34].
Furthermore, many inactivating mutant forms of p53 naturally occurring in human cancers are thermodynamically unstable [35], but the conformation can be restored by lowering temperatures or using chemical solvents, such as glycerol, that can stabilize polypeptide conformation [36]. The extent of folding or unfolding of wild-type or mutant p53 can be quantified by the use of monoclonal antibodies specific for epitopes confined to each respective conformation (Figure 3). Since the F270A mutation exists ina conformationally sensitive epitope (Figure 3) and the monomeric F341A mutant allele of p53 cannot be ubiquitinated well in cells (Figure 1), these data suggest that the conformation of tetrameric p53 may affect its ubiquitination. Therefore we examined whether p53 encoded by the F270A allele is unfolded and whether the most commonly mutated and most unfolded p53 protein in cancers (encoded by the R175H allele) is also hyperubiquitinated.
Figure 3. The DNA-binding domain, MDM2-binding site and two cryptic epitopes of p53.

(A) Antibodies recognize cryptic epitopes which are exposed in unfolded/denatured p53. The grey ribbon depicts p53 protein and the white ribbons highlight two cryptic epitopes of p53 (recognized by monoclonal antibodies PAb240 and DO-12) (as indicated by arrows). The DO-12 epitope contains the S9-S10 linker and the S10 β-sheet containing the MDM2 interface in the core domain of p53. (B) p53 conformation and conformation-sensitive antibodies. PAb1620 is able to detect only the native conformation of p53, whereas the other three antibodies recognize cryptic epitopes that are hidden in the folded conformation of p53.
To quantify the levels of unfolded or folded p53 in cells grown at 37 °C, lysates from H1299 p53-null cells transfected with wild-type or mutant p53 alleles were incubated in ELISA wells coated with the monoclonal antibodies PAb1620 or PAb240 specific for folded and unfolded p53, respectively. Following the capture of the respective conformational variants of p53, the total p53 was detected using polyclonal antibody to the captured p53 protein. The transfection of wild-type p53 into cells produces a pool of p53 that is predominantly in the PAb1620 conformation (Figure 4A, black bar) with a minor, but statistically significant, pool of wild-type p53 in the unfolded conformation (Figure 4A, white bar). The ratio of folded to unfolded wild-type p53 is approx. 9:1, which indicates that only 10% of the wild-type p53 is in the unfolded conformation. The transfection of the mutant p53 allele known to be unfolded, p53R175H, into cells produces a pool of mutant p53 that is unfolded or folded in approximately equal levels (Figure 4C). The ratio of folded to unfolded wild-type p53 is approx. 1:1, which indicates that approx. 50% of the mutant p53 is in the unfolded conformation. By contrast, the transfection of p53F270A into cells produces a pool of mutant p53 that is predominantly unfolded (Figure 4B, white bar). The ratio of folded to unfolded wild-type p53 is approx. 1:4, which indicates that over 75% of the mutant p53 is in the unfolded conformation. These data indicate that p53F270A is significantly unfolded in cells and this unfolding may play a role in substrate ubiquitination. The oligomeric status of the wild-type p53 and mutant p53F270A is relatively similar, as defined by in vivo cross-linking with EGS [ethylene glycol bis(succinimidyl succinate); Pierce], as similar higher-molecular-mass species is observed in cells (Figure 4D versus Figure 4E). A higher-resolution gel of the differences is shown in Figure 4(F). We therefore examined whether oligomeric p53 encoded by the R175H allele was also hyperubiquitinated in cells and, if so, whether p53F270A was more hyperubiquitinated than p53R175H.
Figure 4. Extent of mutant p53F270A and p53R175H unfolding in cells.

Antibody-capture ELISA using conformation-sensitive antibodies revealed that most of the p53F270A mutant shows an unfolded conformation. Expression vector encoding wild-type p53 (A), or p53F270A (B) and p53R175H (C) (1 μg of DNA) was transfected into cells, and the conformation of p53 protein was examined by antibody-capture ELISA. Cell lysates were prepared in a non-detergent-containing buffer (the same buffer as in vivo ubiquitination; see the Experimental section). p53 proteins in the lysates were captured by monoclonal antibodies PAb1620 or PAb240 coated on to ELISA wells, followed by polyclonal antibody CM1 incubation and ECL® detection. The data are represented as luminescence in RLU (relative light units) as a function of the monoclonal antibody used to capture p53. (D–F) Cross-linking of p53 in vivo. The indicated p53 genes were transfected into cells and treated with EGS for 1 h prior to cell harvesting, lysis and immunoblotting on a (D and E) 12% polyacrylamide gel or (F) an 8% polyacrylamide gel. The asterisks highlight the cross-linked higher-molecular-mass oligomeric forms of p53.
To determine whether the two unfolded mutant forms of p53 are more susceptible to ubiquitination, His–ubiquitin was used as a reagent. The transfection of a gene encoding p53R175H into cells without His–ubiquitin (Figure 5A, lane 3 versus lane 1) or with His–ubiquitin (Figure 5C, lane 3 versus lane 1) results in more ubiquitination of the mutant protein, relative to wild-type p53. The extent of high-molecular-mass ubiquitination of p53R175H is not as pronounced as that seen with p53F270A (Figure 5A, lane 6 versus lane 3) and the lower-molecular-mass product is also only seen with p53F270A (Figures 5A and 5B, lanes 6 versus lanes 3). Since p53F270A is more unfolded than p53R175H (Figure 4), these data provide a correlation between the extent of unfolding of p53 and hyperubiquitination. The transfection of MDM2 with His–ubiquitin (Figure 5D) eliminated this selective effect of the mutant protein ubiquitination, and similar patterns of ubiquitination were observed (Figure 5D, lanes 6 and 3 versus lane 1). These data suggest that endogenous and transfected pools of ubiquitin may be differentially affecting the ubiquitination pattern of the mutant and that transfected MDM2 may not be a good tool with which to modulate mutant p53 ubiquitination. However, there is selective mutant p53 ubiquitination without transfected MDM2 and with His–ubiquitin (Figure 5C, lanes 3 and 6 versus lane 1). We finally tried the transfection by the classic protocol, involving His pull-down and immunoblotting of ubiquitinated p53 in order to evaluate whether this method can be used to study selective ubiquitination of mutant p53. When this assay was employed, there was no selective MDM2-dependent ubiquitination or cleavage/degradation of mutant p53 observed, relative to wild-type p53 (Figure 5E, right panel, lanes 4 and 5 versus lane 1). These data indicate that the assays we use to study mutant p53 ubiquitination cannot employ transfected MDM2 and His-tagged ubiquitin.
Figure 5. Mutant p53 encoded by the R175H allele is hyperubiquitinated in vivo.

H1299 cells were transfected with: (A) wild-type or mutant pcDNA-p53 (0.5 μg); (B) wild-type or mutant pcDNA-p53 (0.5 μg) and pCMV-MDM2 (1.5 μg); (C) wild-type or mutant pcDNA-p53 (0.5 μg) and His–ubiquitin (His-Ub) (1.5 μg); (D) wild-type or mutant pcDNA-p53 (0.5 μg) and His–ubiquitin (1.5 μg) and pCMV-MDM2 (1.5 μg). The expressed proteins were examined for changes in their amount of post-translational modification by immunoblotting with an anti-p53 antibody (DO-1). (E) His pull-down assay using His-tagged ubiquitin. H1299 cells were transfected with or without p53 (0.5 μg), and pCMV-MDM2 (1.5 μg) and His-tagged ubiquitin (1.5 μg). Harvested cell lysates were incubated with Ni2+-nitrilotriacetate–agarose to purify the ubiquitinated proteins and blotted with anti-p53 monoclonal antibody (DO-1).
Transfection of genes encoding wild-type p53 and mutant p53F270A followed by precipitation with the monoclonal antibody DO-1 and immunoblotting with the anti-ubiquitin antibody also demonstrated enhanced ubiquitination of p53F270A in the presence of MG132 (Figure 6A, lane 3 versus lane 4). The transfection of MDM2 has an impact on the ubiquitin-adduct formation on wild-type p53 (Figure 6A, lane 8 versus lane 4; and as observed in Figure 5B, lane 1 versus Figure 5A, lane 1). These data indicate that a large proportion of high-molecular-mass adducts linked to mutant p53 are ubiquitin, but they do not address whether the lower-molecular-mass pattern of p53 adducts stem from cleaved fragments of p53 or whether they stem from other adducts such as the recently identified NEDD8 (neural precursor cell expressed, developmentally down-regulated gene 8) conjugation. As such, we evaluated whether protease manipulation alters the production of these covalent adducts on p53. First, the NEDP1 protease was co-transfected into the cell with either wild-type p53 (and MDM2) or mutant p53 (Figure 6B). As reported previously [37], a proportion of wild-type p53 adducts is reduced upon deconjugation by NEDP1 (Figure 6B, lanes 2–5 versus lane 1). By contrast, mutant p53 adducts remain unchanged when co-transfected with NEDP1 (Figure 6B, lanes 7–10 versus lane 6), indicating that lower- and higher-molecular-mass adducts do not stem from NEDD8 conjugation. We examined whether the lower-molecular-mass pattern of bands stemmed from the action of proteases; for example, intermediates in the polyubiquitin-coupled proteolysis would produce laddering of fragments similar to that seen in the mutant p53 immunoblots. When cells were transfected with mutant p53 and incubated with increasing amounts of the proteasome inhibitor Ada-(Ahx)3-(Leu)3-vinyl sulphone results in the accumulation of higher-molecular-mass adducts (Figure 6C, lane 3 versus lane 4) and also changes in the accumulation of lower-molecular-mass adducts (arrows, Figure 6C, lanes 1–3 versus lane 4). These lower-molecular-mass species are similar to that seen with mutant p53 and suggest together that proteasomal cleavage events can modulate the extent of banding patterns observed on the mutant and wild-type p53. The predominance of these lower-molecular-mass banding patterns on mutant p53 might be due to its enhanced susceptibility for proteolysis owing to its unfolded nature (Figure 4).
Figure 6. Immunoprecipitation of the p53F270A mutant in a hyperubiquitinated state.

(A) Mutant p53 ubiquitination. H1299 cells were transfected with either wild-type or mutant pcDNA-p53 (0.5 μg) or pCMV-MDM2 (1.5 μg) as indicated. The cells were treated as indicated with MG132 and examined for changes in p53 ubiquitination after immunoprecipitation with the anti-p53 monoclonal antibody DO-1 and immunoblotting with an anti-ubiquitin antibody. (B) Mutant p53 ubiquitin adducts are not cleaved by NEDP1 protease. The indicated p53 allele (wild-type p53+MDM2, lanes 1–5; or F270A, lanes 6–10) were transfected into cells alone or with increasing amounts of NEDP1 (1–8 μg, as indicated). After 24 h, cells were harvested and immunoblotted with an anti-p53 antibody. (C). Accumulation of lower- and higher-molecular-mass adducts on mutant p53 using a broad-range proteasome inhibitor. The mutant-p53-specific ubiquitination system was assembled in vivo as described above using transfected genes, followed by addition of increasing amounts of Ada-(Ahx)3-(Leu)3-vinyl sulphone. Lysates were immunoblotted to quantify changes in mutant p53 adducts, and higher-molecular-mass accumulation and production of lower-molecular-mass adducts are highlighted with arrows.
Ubiquitination of mutant p53 is MDM2-dependent and occurs outwith the C-terminal acetylation sites
Having established that hyperubiquitination of mutant p53 correlates with the unfolded state of p53, we examined whether the adduct formation mapped to the location of the previous ubiquitination sites using the in vivo ubiquitination assay. Previous studies have indicated that MDM2-dependent ubiquitination of p53 occurs at lysine residues in the C-terminus that are also acetylated by p300. This was most specifically shown using p536KR, a mutant form of p53 that can enter the nucleus similarly to wild-type p53 and which has all six C-terminal lysine residues (at positions 370, 372, 373, 381, 382 and 386) replaced by arginine [38]. The F270A and F19A mutations were introduced into this multiple-arginine-mutant background and assessed for in vivo ubiquitination without transfected MDM2 (Figure 7A, from the left, lanes 1–6) or with transfected MDM2 (Figure 7A, from the left, lanes 7–12). The extent of p53F270A hyperubiquitination was similar to that observed with p53F270A/6KR (Figure 7A, lane 6 versus lane 3). Further, transfection of the MDM2 gene similarly stimulated p53F270A hyperubiquitination and p53F270A/6KR hyperubiquitination (Figure 7A, lane 12 versus lane 9). Surprisingly, the control wild-type p53 or p536KR was ubiquitinated to similar extents in the presence of transfected MDM2 (Figure 7A, lane 10 versus lane 7). There was clear attenuation of two adducts on p53 (Figure 7A, lane 9 versus lane 7, see the asterisks), suggesting that there can in fact be some C-terminal ubiquitination of p53 by MDM2.
Figure 7. The mutant p53F270A/6KR protein is ubiquitinated in vivo.

(A) p536KR mutant is ubiquitinated in an MDM2-dependent manner. H1299 cells were co-transfected with wild-type or mutant pcDNA-p53 (0.5 μg) and with or without pCMV-MDM2 (1.5 μg). (B) Comparison of wild-type p53, p53F270A and p536KR mutant ubiquitination using anti-p53 antibodies. The same cell lysates from (A) was examined with different anti-p53 antibodies. (C) Map positions of the monoclonal antibodies DO-1, DO-12, PAb421 and ICA-9.
The wild-type p536KR gene is defective in recruiting p300 to the p21 promoter, indicating that the gene product can be defective in acetylation-dependent transactivation [25,39], suggesting that the ubiquitination observed with wild-type p536KR is not due to general loss of a mutant phenotype. Further, using antibodies to various epitopes on p53 (Figure 7C), we immunoblotted lysates expressing wild-type p53, p53F270A and wild-type p536KR to confirm that p536KR is indeed mutant in the C-terminus (Figure 7B; in addition to confirmation by DNA sequencing). Two high-affinity monoclonal antibodies, including DO-12 and DO-1, show similar patterns of mutant p53 hyperubiquitination in the absence of transfected MDM2 (Figure 7B, from the left, lanes 1–6). However, with the antibody PAb421, which requires contact with key lysine residues in the 6KR mutant epitope (Figure 7C; [40]), only the p536KR mutant protein failed to bind (Figure 7B, from the left, lane 9 versus lane 7 and 8), thus indicating that p536KR is indeed mutant in that region. Further, the monoclonal antibody that has an epitope at the penultimate amino acid in the C-terminus of p53 outside the 6KR region (ICA-9; [41]) can bind well to p536KR (Figure 7B, from the left, lane 12 versus lane 10), indicating that the C-terminus of p536KR is not truncated or cleaved in vivo. The differences in the levels of p53 and p53F270A using ICA-9 and PAb421 antibodies may be because these two sites are differentially phosphorylated, a modification that can block the epitopes [41]. Together these data indicate that the majority of mutant p53 ubiquitination occurs outwith the C-terminal acetylation sites.
The unusual feature of mutant p53 hyperubiquitination is that it can occur in the absence of transfected MDM2 (Figure 5), whereas wild-type p53 exhibits a requirement for transfection of the MDM2 gene to be maximally ubiquitinated. Further, the ubiquitinated p53F270A protein has a very long half-life in cells [22], suggesting that this pool may be forming inclusion aggregates, as it is not being recognized by the proteasome. These data could indicate that mutant p53 hyperubiquitination is MDM2-independent. However, mutant p53 hyperubiquitination appears to be MDM2-dependent, since transfecting the MDM2 gene into cells can stimulate ubiquitination of the p53F270A/F19A mutant protein in cells (Figure 1). Nevertheless, MDM2-dependence in mutant p53 ubiquitination needs to be evaluated by an independent method. siRNA was used as a method to determine whether mutant p53 ubiquitination was MDM2-dependent. H1299 cells were co-transfected with vector only or genes encoding either wild-type p53 or p53R175H and with or without siRNA towards MDM2. As a positive control, cells were also treated with siRNA towards PARC, a protein that was identified, interestingly, as specifically binding to mutant p53 encoded by the R175H allele, but which is reported to be a negative regulator of wild-type p53 [29]. Wild-type-p53-transfected cells treated with MDM2 siRNA, compared with the control, have substantially lower levels of ubiquitination (Figure 8A, lane 5 versus lane 2), along with substantial increases in endogenous p21WAF1 (Figure 8B, lane 5 versus lane 2). These data are consistent with MDM2 being a key negative regulator of the p53 pathway. The treatment of cells with control PARC siRNA marginally attenuated wild-type p53 ubiquitination (Figure 8A, lane 8 versus lane 2) and induced slight increases in p21WAF1 (Figure 8B, lane 8 versus lane 2), though not as much as siRNA to MDM2. Such marginal increases in p21 protein using siRNA to PARC were similar to that reported previously [29].
Figure 8. Ubiquitination of unfolded p53 is attenuated using siRNA to MDM2.

Empty vector or genes encoding either wild-type p53 or p53R175H (1 μg) were co-transfected with control siRNA, MDM2 siRNA or PARC siRNA into H1299 cells (see the Experimental section). The cells were transfected with 100 nM siRNA twice at an interval of 24 h before harvesting. ALLN was added into the cells at 46 h post-transfection for 2 h. The expressed proteins were examined for changes in their amount of ubiquitination by immunoblotting with: (A) anti-p53 antibody (DO-1); (B) anti-p21 antibody (AB-1); (C) anti-MDM2 antibody (2A10). (D). MDM2-stimulated ubiquitination of p53R175H is attenuated by the IHD region of p300. Vectors encoding p53R175H (1 μg) were co-transfected with control DNA (lane 1), MDM2 (1 μg, lane 2) or IHD (1 and 2 μg, lanes 3 and 4) expression constructs into H1299 cells (see the Experimental section). ALLN was added to the cells at 24 h post-transfection for 2 h. The expressed proteins were examined for changes in their amount of ubiquitination by immunoblotting with anti-p53 antibody (DO-1). (E) PARC attenuates ubiquitination of p53R175H. Vectors encoding p53R175H (1 μg) were co-transfected with control DNA (lane 1) and FLAG-PARC (2 and 5 μg, lanes 2 and 3) expression constructs into H1299 cells (see the Experimental section). ALLN was added into the cells at 24 h post-transfection for 2 h. The expressed proteins were examined for changes in their amount of ubiquitination by immunoblotting with anti-p53 antibody (DO-1).
In contrast with wild-type p53, both MDM2 RNAi (RNA interference) and PARC RNAi affected mutant p53 hyperubiquitination differently. Cells transfected with mutant p53 and treated with MDM2 siRNA had lower amounts of mutant p53 ubiquitination (Figure 8A, lane 6 versus lane 3) without substantial increases in endogenous p21WAF1 protein (Figure 8B, lane 6 versus lane 3). The reason the mutant p53 ubiquitinated protein was not ablated using siRNA to MDM2 like wild-type p53 is presumably due to the very long half-life of the mutant p53 ubiquitinated product [22]. As a control, the treatment of cells with PARC siRNA induced significant stimulation of mutant p53 ubiquitination and degradation/cleavage fragments (Figure 8A, lane 9 versus lanes 6 and 3). Further, this siRNA treatment actually resulted in a marginal increase in steady-state levels of endogenous MDM2 (Figure 8C, lane 9 versus lanes 7 and 6). It is interesting that PARC was identified as a protein that bound specifically to mutant p53R175H [29] and, together, these data suggest that PARC is a negative regulator of the MDM2-dependent mutant p53 ubiquitination pathway. Transfection of PARC into cells confirms this effect, since a decrease in mutant p53R175H ubiquitination is observed upon PARC transfection, relative to MDM2 (Figure 8E, lanes 2 and 3 versus lane 1). As a control, the MDM2-mediated stimulation of p53R175H ubiquitination (Figure 8D, lanes 2 versus lane 1) is attenuated by the dominant-negative minidomain of p300, termed IHD (interferon-binding homology domain) [18], which binds to the LXXLL domain of p53 (Figure 8D, lanes 3 and 4 versus lane 2). The stimulation or attenuation of mutant p53 ubiquitination by MDM2 and PARC respectively (Figures 8D and 8E) is consistent with the siRNA approach that led to attenuation or stimulation of mutant p53 adduct formation be targeting MDM2 or PARC respectively (Figure 8A).
These studies, linking p53 conformation to ubiquitination rates, are limiting in that the assays are based in vivo and, as such, the link between mutant p53 conformation and its ubiquitination might be indirect. For example, the mutant unfolded p53 might be in equilibrium with an intracellular compartment different from that obtaining for wild-type p53, leading to an enhanced ubiquitination in cells that is actually unrelated directly to their conformational differences. More direct evidence linking p53 protein unfolding to ubiquitination is required. As such, we set up the in vitro protein-synthesis system previously described for p53 that resolves p53 into the folded or unfolded conformational variants [26–28]. Wild-type p53 synthesized in reticulocyte lysates can adopt the PAb1620-positive (i.e. folded) conformation (Figure 9A) relative to the p53F270A protein, which is largely in the DO-12-reactive (i.e. unfolded) conformation (Figure 9B). These data confirm that the reticulocyte-lysate protein-synthesis system faithfully reflects the p53 conformational changes that occur in cells. When the reticulocyte lysate containing wild-type p53 was added to ubiquitination assays containing E1, E2 and required conjugation substrates, relatively little ubiquitination was observed (Figure 9C, lane 10). However, the addition of MDM2 protein stimulated mono-ubiquitination (Figure 9C, lane 9 versus lane 10). Similarly to what is observed in cells, p53 encoded by the F270A allele exhibits enhanced ubiquitination without the addition of exogenous MDM2 (Figure 9C, lanes 8 and 7). Similarly, using 35S-labelled p53, p53 encoded by the F270A allele exhibits enhanced ubiquitination without the addition of exogenous MDM2 compared with wild-type p53 (Figure 9C, lane 11 versus lane 12). The addition of small peptide inhibitors to MDM2 can block mutant p53 ubiquitination (results not shown), suggesting that rabbit MDM2 in the lysate has a high specificity towards mutant p53. The gene encoding p53F270A/6KR suppressed basal ubiquitination (Figure 9C, lane 2), but the addition of human MDM2 restores the ubiquitination ladders representative of the mutant protein (Figure 9C, lane 2 versus lane 1). In place of adding recombinant bacterially expressed MDM2, the MDM2 protein was synthesized by co-translation in vitro with various p53 isoforms (Figure 9D). Both wild-type p53 and mutant p53 demonstrated enhanced high-molecular-mass adduct formation after co-translation with MDM2 (Figure 9D, lanes 2 and 4 versus lanes 1 and 3). Furthermore, although p53F19A was not ubiquitinated by MDM2 (Figure 9D, lane 6 versus lane 5), re-introducing the F270A mutation into the F19A background induced the high-molecular-mass adduct that was in turn stimulated by co-translated MDM2 in vitro (Figure 9D, lane 7 versus lane 8). Together these data suggest that the there are factors that can directly discriminate between the wild-type and mutant conformations of p53 in vitro and provide a model system from which to purify factors that control MDM2-dependent mutant protein ubiquitination.
Figure 9. Mutant p53 protein is more sensitive to ubiquitination in vitro.

(A and B) Antibody-capture ELISA using conformation-sensitive antibodies towards in vitro-synthesized p53 protein. Expression vectors encoding wild-type p53 or p53F270A as indicated were added to transcription-coupled translation reticulocyte-lysate systems to produce p53 protein. An aliquot of the synthesized p53 protein was titrated on to ELISA wells precoated with the indicated monoclonal antibodies, followed by polyclonal antibody CM1 incubation and ECL® detection. The results are represented as luminescence in RLU as a function of the monoclonal antibody used to capture p53. (C). In vitro ubiquitination of mutant p53. Reticulocyte lysates containing the p53 protein isoform (as indicated, lanes 1–10) were added to ubiquitination reactions without MDM2 (lanes 2, 4, 6, 8 and 10) or with bacterially expressed MDM2 (100 ng; lanes 1, 3, 5, 7 and 9) as described previously [23]. After 10 min, the reactions were quenched and the material processed for immunoblotting to reveal p53 protein (arrow) and modifications of the indicated p53 protein isoforms (as highlighted by the brackets). For radioactive p53 production, reticulocyte lysates containing the p53 protein isoform (as indicated, lanes 11 and 12) was added to ubiquitination reactions and, after 10 min, the reactions were quenched and the material processed for phosphorimaging to reveal p53 protein (arrow) and higher-molecular-mass species. (D) Co-translation of MDM2 with mutant p53 in reticulocyte lysates stimulates ubiquitination in vitro. Reticulocyte lysates containing co-translated MDM2 (+) or vector control (−) and p53 protein isoform (as indicated) were added to the ubiquitination reaction mixtures. After 10 min the reactions were quenched and the material was processed for immunoblotting to reveal p53 protein (arrow) and high-molecular-mass modifications of the indicated p53 protein isoforms.
DISCUSSION
The tumour suppressor protein p53 is a conformationally flexible and allosterically regulated protein that functions as a transcription factor in a homotetramer form. The activity of p53 is tightly regulated by protein–protein interactions, and post-translational modifications, such as phosphorylation, acetylation, sumoylation and ubiquitination [32]. MDM2 is the most well-studied negative regulator of p53 and inhibits the activity of p53 activity by catalysing a ubiquitination that results in the 26 S-proteasome-dependent degradation of p53 [42]. MDM2 itself is a conformationally flexible protein also regulated in its binding to partner proteins by additional cofactors such as zinc and RNA [22,23,43].
The mechanisms of MDM2-mediated ubiquitination of p53 are largely undefined, although our current focus is on testing the hypothesis that alteration in the folding of the p53 DNA-binding domain by MDM2 (plus associated cofactors) may play a role in controlling ubiquitination. In general it is thought that MDM2 binding to the N-terminal domain of p53 catalyses p53 mono-ubiquitination, since mutation at a key phenylalanine residue in the N-terminus residue blocks this ubiquitination. However, wild-type p53 in a monomeric, rather than tetrameric, conformation is not ubiquitinated by MDM2, suggesting that another binding interface or a complex tertiary conformation exists for MDM2 to catalyse the ubiquitination of p53 [20]. Previous work had highlighted the location of a second binding site for MDM2 in the DNA-binding domain of p53 the mutation of which can stabilize MDM2–p53 complex formation in vitro and stimulate p53 ubiquitination in cells [22]. Previously, Dawson et al. [44] determined whether the binding constant of full-length p53 for human MDM2 is similar to that of N-terminal part of p53 (amino acids 1–93) by using fluorescence spectroscopy. A lower KD value for the interaction between full-length p53 and MDM2 should be observed if the secondary MDM2-binding site is significant. The KD values were 4 μM for the N-terminal part of p53 for MDM2 binding and 0.3 μM for the full-length p53 for MDM2 binding [44]. The binding of MDM2 to full-length p53 is therefore approx. 10-fold stronger than the binding to the N-terminal part of p53, a finding which supports the notion that there is an additional MDM2-binding site outside the p53 N-terminus. In the present study we used the p53 mutant in this second MDM2-binding site to explore the contribution of substrate conformation to mutant p53 ubiquitination for two reasons. First, previous work had shown that MDM2 is an Hsp90-binding protein functioning as a co-chaperone in altering the folding of the p53 tetramer [23] and mutant misfolded p53 can form a stable complex with Hsp90 and MDM2 in cancer cells [45]. In fact, in a series of primary breast cancers with mutant unfolded p53, high-level expression of MDM2 can be detected, suggesting that MDM2 may play a role in p53 unfolding. Secondly, monomeric p53 is surprisingly not ubiquitinated by MDM2 [20], indicating that there are other determinants that drive MDM2-mediated ubiquitination of p53.
Three observations from the present study now suggest that the unfolded p53 ubiquitination is MDM2-dependent: (1) MDM2-dependent p53F19A mutant ubiquitination is enhanced by re-introduction of the F270A mutation in vitro and in vivo; (2) MDM2 siRNA reduces the mutant p53 ubiquitination in cells; and (3) p53F270A/6KR exhibits enhanced MDM2-dependent ubiquitination when synthesized in vitro or in vivo, providing direct evidence that factors can recognize the altered conformational p53 variant. However, these observations would not completely exclude the possibility that mutant p53 hyperubiquitination is partially regulated by other p53-specific ubiquitin E3 ligases. Previously, ubiquitin ligases Pirh2, COP-1 and CHIP were reported to bind to and/or ubiquitinate p53 [5,6]. Pirh2 and COP-1 are also induced by p53, in a similar fashion to MDM2, and the induction of Pirh2 or COP-1 promotes p53 degradation. Therefore it is proposed that Pirh2 or COP-1 also participates in an autoregulatory feedback loop with p53, although it is not yet clear whether MDM2 and Pirh2 or COP-1 function co-operatively in the same pathway or are independent degraders of p53. Interestingly, an independent function of Pirh2 protein was first published under a different name, ARNIP (androgen receptor N-terminal-interacting protein), which binds to the androgen receptor in an androgen-dependent manner and co-localizes to the nucleus [46], suggesting that a hormone-dependent signalling pathway could negatively regulate p53 and determine the substrate specificity of ARNIP/Pirh2. Further, given the role of the E3 ligase CHIP in mediating ubiquitination of mutant proteins [47] and the observation that CHIP can stimulate MDM2-dependent misfolding of p53 in vitro [23] and p53 ubiquitination in vivo [7], there may prove to be co-operation between the CHIP-dependent ubiquitination machinery, p53 unfolding and the p53-specific E3 ligases.
One surprising result to emerge from this work in characterizing whether mutant p53 ubiquitination is MDM2-dependent was the data acquired using the p536KR mutant as a substrate for ubiquitination. Our observations showing that ubiquitination of wild-type p536KR mutant occurs suggest that p53-ubiquitination sites are outwith the C-terminal region classically defined as sites of ubiquitination. This observation is consistent with independent data showing that the wild-type p536KR mutant can be ubiquitinated outwith the C-terminus by human-papillomavirus E6 protein/E6-associated protein [48]. The data demonstrating that the level of mutant p53F270A/6KR ubiquitination is not different in cells from that of the p53F270A mutant suggests that the major ubiquitination sites are also outwith the C-terminus. This result can be reconstituted in vitro, although the mutations in the C-terminus of the p53F270A/6KR attenuate basal ubiquitination in the absence of MDM2 (Figure 9C). In a key report showing the sites of p53 ubiquitination that occur in the C-terminal domain of p53 [38], His-tagged ubiquitin and associated pull-downs were used to show the C-terminus has multiple lysine residues as the ubiquitination sites, and we subsequently tested whether His-tagged ubiquitin and endogenous ubiquitin may cause different patterns of ubiquitination of p53. When we compare transfected mutant-p53 ubiquitination (with endogenous MDM2 and endogenous ubiquitin), ubiquitination patterns are observed that are different from those we obtain if we examine mutant ubiquitination with endogenous MDM2 and transfected His–ubiquitin (Figure 5) or transfected MDM2 and transfected His–ubiquitin (Figure 5). These data suggest that mutant p53 ubiquitination is operating along a control pathway different from that followed by wild-type p53 and that transfected ubiquitin and MDM2 cannot be employed necessarily to dissect out mutant p53 ubiquitination mechanisms. Furthermore, the fact that substantial ubiquitination can occur on wild-type p536KR indicates that other major sites of ubiquitination exist, despite the fact that, under certain transfection conditions, the C-terminus can be the major site of ubiquitination. As a potential candidate site for p53 N-terminal ubiquitination site, one possibility is the very-N-terminal methionine residue. Ubiquitin generally attaches covalently to the ϵ-amino group of a lysine residue of the substrate protein. However, studies have revealed that ubiquitination can occur at the α-amino group of the N-terminal residue instead of internal lysine residues. Replacement of all lysine residues in MyoD [49], Epstein–Barr-virus protein LMP1 (The latent membrane protein 1) [50] and E7 human papillomavirus oncoprotein [51] has no significant effect on their ubiquitin conjugation and degradation pathway. These ubiquitin substrate proteins were stabilized by the addition of a few amino acids to their N-terminus, but not to the C-terminal residue, and the same effect was observed on the deletion of the N-terminus. Therefore these findings suggest that lysine-independent ubiquitination occurs whereby the first ubiquitin moiety is attached linearly to the free N-terminal residue of the ubiquitin recipients.
Apart from the fact that major sites of ubiquitination are occurring outwith the C-terminal domain of mutant p53, on the basis of the in vitro sensitivity of the mutant p53 to ubiquitination (Figure 9), the ubiquitination of mutant p53 may also be operating under a sensing system in the cell different from that used by wild-type p53. This alternative pathway shows three notable differences from wild-type p53 folding and ubiquitination: (1) the mutant p53 protein is largely unfolded in vitro and in vivo; (2) the mutant p53 ubiquitinated adduct has a very long half-life in cells and is MDM2-dependent in vitro and in vivo (Figures 8 and 9B, lanes 1 versus lanes 2); and (3) the mutant p53 MDM2-mediated ubiquitination can occur without exogenous or transfected MDM2 in vitro and in vivo, suggesting a sensitivity to small amounts of MDM2. Together these data highlight the possibility that mutant p53 is processed by the ubiquitination machinery in a manner distinct from that by which the wild-type p53 protein is processed. This difference in processing may be related to the unfolded nature of mutant p53 and the cellular detection system that exists to discriminate between folded and unfolded proteins.
Mechanisms of protein ubiquitination and degradation are though to be linked by the chaperone protein folding system. The protein triage hypothesis [52] argues that the decision for a protein being translated to assemble into its pathway or to be degraded depends on the unfolding state at the ribosome [53]. Some proteins that are intrinsically unstable may be unfolded more and undergo more rapid ubiquitination and/or degradation [54]. Mutant p53 protein forms a unique model to begin to explore links between substrate conformation, chaperone flux and associated ubiquitination, mainly because immunochemical probes exist that can monitor the unfolding of the protein in vitro and in vivo. The observation that MDM2, Hsp90 and CHIP can co-operate to alter the folding of p53 and ubiquitinate p53 [7,23,45] opens the door to evaluate how unfolded protein conformation is linked to ubiquitination flux and the chaperone-mediated degradation programme. Further, the ability of the acidic domain of MDM2 to alter the conformation of p53 at the second MDM2-binding site with the Phe270-containing motif in the DNA-binding domain of p53, as defined by NMR [55], provides independent evidence that an MDM2 minidomain has a chaperone-like activity in its ability to distort the conformation of the DNA-binding domain.
Acknowledgments
This work was supported by a Programme Grant from Cancer Research UK (to T. R. H., p53 Signal Transduction Group), a Cancer Research UK Translational Programme Grant to the Edinburgh Cancer Centre, and the Medical Research Council.
References
- 1.Vogelstein B., Kinzler K. W. Cancer genes and the pathways they control. Nat. Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 2.Honda R., Tanaka H., Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 3.Kubbutat M. H., Jones S. N., Vousden K. H. Regulation of p53 stability by Mdm2. Nature (London) 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 4.Haupt Y., Maya R., Kazaz A., Oren M. Mdm2 promotes the rapid degradation of p53. Nature (London) 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 5.Leng R. P., Lin Y., Ma W., Wu H., Lemmers B., Chung S., Parant J. M., Lozano G., Hakem R., Benchimol S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–791. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 6.Dornan D., Wertz I., Shimizu H., Arnott D., Frantz G. D., Dowd P., O'Rourke K., Koeppen H., Dixit V. M. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature (London) 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- 7.Esser C., Scheffner M., Hohfeld J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 2005;280:27443–27448. doi: 10.1074/jbc.M501574200. [DOI] [PubMed] [Google Scholar]
- 8.Iyer N. G., Chin S. F., Ozdag H., Daigo Y., Hu D. E., Cariati M., Brindle K., Aparicio S., Caldas C. p300 regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7386–7391. doi: 10.1073/pnas.0401002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shimizu H., Hupp T. R. Intrasteric regulation of MDM2. Trends Biochem. Sci. 2003;28:346–349. doi: 10.1016/S0968-0004(03)00140-3. [DOI] [PubMed] [Google Scholar]
- 10.Knights C. D., Liu Y., Appella E., Kulesz-Martin M. Defective p53 post-translational modification required for wild type p53 inactivation in malignant epithelial cells with mdm2 gene amplification. J. Biol. Chem. 2003;278:52890–52900. doi: 10.1074/jbc.M300279200. [DOI] [PubMed] [Google Scholar]
- 11.Thut C. J., Goodrich J. A., Tjian R. Repression of p53-mediated transcription by MDM2: a dual mechanism. Genes Dev. 1997;11:1974–1986. doi: 10.1101/gad.11.15.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin J., Chen J., Elenbaas B., Levine A. J. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes Dev. 1994;8:1235–1246. doi: 10.1101/gad.8.10.1235. [DOI] [PubMed] [Google Scholar]
- 13.Kussie P. H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A. J., Pavletich N. P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 14.Picksley S. M., Vojtesek B., Sparks A., Lane D. P. Immunochemical analysis of the interaction of p53 with MDM2: fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene. 1994;9:2523–2529. [PubMed] [Google Scholar]
- 15.Lai Z., Ferry K. V., Diamond M. A., Wee K. E., Kim Y. B., Ma J., Yang T., Benfield P. A., Copeland R. A., Auger K. R. Human mdm2 mediates multiple mono-ubiquitination of p53 by a mechanism requiring enzyme isomerization. J. Biol. Chem. 2001;276:31357–31367. doi: 10.1074/jbc.M011517200. [DOI] [PubMed] [Google Scholar]
- 16.Shmueli A., Oren M. Regulation of p53 by Mdm2: fate is in the numbers. Mol. Cell. 2004;13:4–5. doi: 10.1016/s1097-2765(03)00529-x. [DOI] [PubMed] [Google Scholar]
- 17.Lai Z., Yang T., Kim Y. B., Sielecki T. M., Diamond M. A., Strack P., Rolfe M., Caligiuri M., Benfield P. A., Auger K. R., Copeland R. A. Differentiation of Hdm2-mediated p53 ubiquitination and Hdm2 autoubiquitination activity by small molecular weight inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14734–14739. doi: 10.1073/pnas.212428599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vassilev L. T. Small-molecule antagonists of p53-MDM2 binding: research tools and potential therapeutics. Cell Cycle. 2004;3:419–421. [PubMed] [Google Scholar]
- 19.Kubbutat M. H., Ludwig R. L., Ashcroft M., Vousden K. H. Regulation of Mdm2-directed degradation by the C terminus of p53. Mol. Cell. Biol. 1998;18:5690–5698. doi: 10.1128/mcb.18.10.5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marston N. J., Jenkins J. R., Vousden K. H. Oligomerisation of full length p53 contributes to the interaction with mdm2 but not HPV E6. Oncogene. 1995;10:1709–1715. [PubMed] [Google Scholar]
- 21.Maki C. G. Oligomerization is required for p53 to be efficiently ubiquitinated by MDM2. J. Biol. Chem. 1999;274:16531–16535. doi: 10.1074/jbc.274.23.16531. [DOI] [PubMed] [Google Scholar]
- 22.Shimizu H., Burch L. R., Smith A. J., Dornan D., Wallace M., Ball K. L., Hupp T. R. The conformationally flexible S9-S10 linker region in the core domain of p53 contains a novel MDM2 binding site whose mutation increases ubiquitination of p53 in vivo. J. Biol. Chem. 2002;277:28446–28458. doi: 10.1074/jbc.M202296200. [DOI] [PubMed] [Google Scholar]
- 23.Burch L., Shimizu H., Smith A., Patterson C., Hupp T. R. Expansion of protein interaction maps by phage peptide display using MDM2 as a prototypical conformationally flexible target protein. J. Mol. Biol. 2004;337:129–145. doi: 10.1016/j.jmb.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Vojtesek B., Dolezalova H., Lauerova L., Svitakova M., Havlis P., Kovarik J., Midgley C. A., Lane D. P. Conformational changes in p53 analysed using new antibodies to the core DNA binding domain of the protein. Oncogene. 1995;10:389–393. [PubMed] [Google Scholar]
- 25.Dornan D., Shimizu H., Burch L., Smith A. J., Hupp T. R. The proline repeat domain of p53 binds directly to the transcriptional coactivator p300 and allosterically controls DNA-dependent acetylation of p53. Mol. Cell. Biol. 2003;23:8846–8861. doi: 10.1128/MCB.23.23.8846-8861.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milner J., Medcalf E. A. Temperature-dependent switching between “wild-type” and “mutant” forms of p53-Val135. J. Mol. Biol. 1990;216:481–484. doi: 10.1016/0022-2836(90)90371-R. [DOI] [PubMed] [Google Scholar]
- 27.Milner J., Medcalf E. A. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell. 1991;65:765–774. doi: 10.1016/0092-8674(91)90384-b. [DOI] [PubMed] [Google Scholar]
- 28.Milner J., Medcalf E. A., Cook A. C. Tumor suppressor p53: analysis of wild-type and mutant p53 complexes. Mol. Cell. Biol. 1991;11:12–19. doi: 10.1128/mcb.11.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikolaev A. Y., Li M., Puskas N., Qin J., Gu W. Parc: a cytoplasmic anchor for p53. Cell. 2003;112:29–40. doi: 10.1016/s0092-8674(02)01255-2. [DOI] [PubMed] [Google Scholar]
- 30.Lin W. C., Desiderio S. Regulation of V(D)J recombination activator protein RAG-2 by phosphorylation. Science. 1993;260:953–959. doi: 10.1126/science.8493533. [DOI] [PubMed] [Google Scholar]
- 31.Dyson H. J., Wright P. E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 32.Lane D. P., Hupp T. R. Drug discovery and p53. Drug Discov. Today. 2003;8:347–355. doi: 10.1016/s1359-6446(03)02669-2. [DOI] [PubMed] [Google Scholar]
- 33.Bell S., Klein C., Muller L., Hansen S., Buchner J. p53 contains large unstructured regions in its native state. J. Mol. Biol. 2002;322:917–927. doi: 10.1016/s0022-2836(02)00848-3. [DOI] [PubMed] [Google Scholar]
- 34.Nichols N. M., Matthews K. S. Human p53 phosphorylation mimic, S392E, increases nonspecific DNA affinity and thermal stability. Biochemistry. 2002;41:170–178. doi: 10.1021/bi011736r. [DOI] [PubMed] [Google Scholar]
- 35.Bullock A. N., Fersht A. R. Rescuing the function of mutant p53. Nat. Rev. Cancer. 2001;1:68–76. doi: 10.1038/35094077. [DOI] [PubMed] [Google Scholar]
- 36.Brown C. R., Hong-Brown L. Q., Welch W. J. Correcting temperature-sensitive protein folding defects. J. Clin. Invest. 1997;99:1432–1444. doi: 10.1172/JCI119302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xirodimas D. P., Saville M. K., Bourdon J. C., Hay R. T., Lane D. P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118:83–97. doi: 10.1016/j.cell.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez M. S., Desterro J. M., Lain S., Lane D. P., Hay R. T. Multiple C-terminal lysine residues target p53 for ubiquitin–proteasome-mediated degradation. Mol. Cell. Biol. 2000;20:8458–8467. doi: 10.1128/mcb.20.22.8458-8467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dornan D., Eckert M., Wallace M., Shimizu H., Ramsay E., Hupp T. R., Ball K. L. Interferon regulatory factor 1 binding to p300 stimulates DNA-dependent acetylation of p53. Mol. Cell. Biol. 2004;24:10083–10098. doi: 10.1128/MCB.24.22.10083-10098.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephen C. W., Helminen P., Lane D. P. Characterisation of epitopes on human p53 using phage-displayed peptide libraries: insights into antibody–peptide interactions. J. Mol. Biol. 1995;248:58–78. doi: 10.1006/jmbi.1995.0202. [DOI] [PubMed] [Google Scholar]
- 41.Hupp T. R., Lane D. P. Allosteric activation of latent p53 tetramers. Curr. Biol. 1994;4:865–875. doi: 10.1016/s0960-9822(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 42.Vousden K. H., Lu X. Live or let die: the cell's response to p53. Nat. Rev. Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 43.Linares L. K., Scheffner M. The ubiquitin–protein ligase activity of Hdm2 is inhibited by nucleic acids. FEBS Lett. 2003;554:73–76. doi: 10.1016/s0014-5793(03)01108-6. [DOI] [PubMed] [Google Scholar]
- 44.Dawson R., Muller L., Dehner A., Klein C., Kessler H., Buchner J. The N-terminal domain of p53 is natively unfolded. J. Mol. Biol. 2003;332:1131–1141. doi: 10.1016/j.jmb.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Peng Y., Chen L., Li C., Lu W., Chen J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J. Biol. Chem. 2001;276:40583–40590. doi: 10.1074/jbc.M102817200. [DOI] [PubMed] [Google Scholar]
- 46.Beitel L. K., Elhaji Y. A., Lumbroso R., Wing S. S., Panet-Raymond V., Gottlieb B., Pinsky L., Trifiro M. A. Cloning and characterization of an androgen receptor N-terminal-interacting protein with ubiquitin–protein ligase activity. J. Mol. Endocrinol. 2002;29:41–60. doi: 10.1677/jme.0.0290041. [DOI] [PubMed] [Google Scholar]
- 47.McDonough H., Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones. 2003;8:303–308. doi: 10.1379/1466-1268(2003)008<0303:calbtc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Camus S., Higgins M., Lane D. P., Lain S. Differences in the ubiquitination of p53 by Mdm2 and the HPV protein E6. FEBS Lett. 2003;536:220–224. doi: 10.1016/s0014-5793(03)00054-1. [DOI] [PubMed] [Google Scholar]
- 49.Breitschopf K., Bengal E., Ziv T., Admon A., Ciechanover A. A novel site for ubiquitination: the N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 1998;17:5964–5973. doi: 10.1093/emboj/17.20.5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aviel S., Winberg G., Massucci M., Ciechanover A. Degradation of the Epstein–Barr virus latent membrane protein 1 (LMP1) by the ubiquitin-proteasome pathway. Targeting via ubiquitination of the N-terminal residue. J. Biol. Chem. 2000;275:23491–23499. doi: 10.1074/jbc.M002052200. [DOI] [PubMed] [Google Scholar]
- 51.Reinstein E., Scheffner M., Oren M., Ciechanover A., Schwartz A. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: targeting via ubiquitination of the N-terminal residue. Oncogene. 2000;19:5944–5950. doi: 10.1038/sj.onc.1203989. [DOI] [PubMed] [Google Scholar]
- 52.Connell P., Ballinger C. A., Jiang J., Wu Y., Thompson L. J., Hohfeld J., Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat. Cell. Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 53.Cyr D. M., Hohfeld J., Patterson C. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem. Sci. 2002;27:368–375. doi: 10.1016/s0968-0004(02)02125-4. [DOI] [PubMed] [Google Scholar]
- 54.Meacham G. C., Patterson C., Zhang W., Younger J. M., Cyr D. M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001;3:100–105. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- 55.Yu G., Rudiger S., Veprintsev D., Freund S., Fernandez-Fernandez M., Fersht A. R. The central region of HDM2 provides a second binding site for p53. Proc. Natl. Acad. Sci. U.S.A. 2006;103:1227–1232. doi: 10.1073/pnas.0510343103. [DOI] [PMC free article] [PubMed] [Google Scholar]