

Abstract
Interleukin-6 (IL-6) has been implicated as an autocrine factor involved in growth of several human cancers, such as tumors arising from the biliary tract or cholangiocarcinoma. In malignant biliary tract epithelia, IL-6 activates the p38 MAPK pathway, which mediates a dominant survival signaling pathway. Serum and glucocorticoid-stimulated kinase (SGK) has been implicated as a survival kinase, but its role in survival signaling by IL-6 is unknown. After IL-6 stimulation, p38 MAPK activation preceded phosphorylation of SGK at Ser78. Pretreatment with the pharmacological inhibitors of p38 MAPK SB-203580 or SB-202190 blocked IL-6-induced SGK phosphorylation at Ser78 and SGK activation. Overexpression of p38α increased constitutive SGK phosphorylation at Ser78, whereas dominant negative p38α MAPK blocked IL-6-induced SGK phosphorylation and nuclear translocation. Interestingly, in addition to stimulating SGK phosphorylation, both IL-6 stimulation and p38α MAPK overexpression increased SGK mRNA and protein expression. An increase in p38 MAPK and SGK occurred following enforced expression of IL-6 in vivo. Furthermore, inhibition of SGK expression by siRNA increased toxicity due to chemotherapeutic drugs. Taken together, these data identify SGK as both a downstream kinase substrate as well as a transcriptionally regulated gene target of p38 MAPK in response to IL-6 and support a role of SGK during survival signaling by IL-6 in human cancers, such as cholangiocarcinoma.
Keywords: cytokines, intracellular kinases, cancer
serum and glucocorticoid-inducible kinase (SGK) is a serine/threonine kinase that is highly expressed in the liver and mediates cell survival in response to diverse environmental stresses (28). SGK is a member of the AGC subfamily that includes cAMP and the cGMP-dependent kinases PKC and Akt (2, 13, 31, 44). Although it was initially identified as being under acute transcriptional control by serum and glucocorticoids, SGK has subsequently been shown to be stimulated by diverse stimuli, such as osmotic and oxidative stress, growth factors, or hormones (1, 5, 28, 30, 32, 46). Stimulation of SGK can occur in response to cell surface nuclear receptors and intracellular stress pathways. Furthermore, SGK can be regulated at multiple levels, including transcriptional regulation, modulation of enzymatic activity, and subcellular localization. The diversity of stimulatory mechanisms and complexity of regulation of SGK support a role for SGK as an intracellular modulator of cellular responses to distinctive environmental signals. Several different biological functions have been described in response to SGK signaling, including cell survival, proliferation, and osmoregulation (8, 13, 47). Survival signaling by SGK has been shown to involve downstream phosphorylation of Bad as well as the forkhead transcription factor FKHRL1 (FOXO-3) transcriptional factors (4). These two SGK substrates are also involved in survival signaling by the closely related kinase Akt.
Interleukin-6 (IL-6) is a multifunctional cytokine that mediates diverse tissue responses in response to environmental stimuli (21). The expression of IL-6 is increased during infection, trauma, or cellular stresses. In addition to a major role in host cell defense and the regulation of inflammatory responses, IL-6 signaling pathways have been shown to contribute to tumor progression in epithelial (e.g., cholangiocarcinoma) as well as hematopoeitic (e.g., multiple myeloma) human tumors (41). These may involve IL-6 effects on either cell proliferation or cell survival. Cell survival resulting from inhibition of apoptosis by IL-6 has also been described in several settings (6, 17, 29). Although signaling pathways mediating proliferative responses by IL-6 are well characterized, signaling for cell survival remains poorly understood (20, 21). The activation of cell survival signaling can promote tumor growth by providing neoplastic cells with the ability to survive under adverse environmental conditions such as limited growth factor availability or during treatment with cytotoxic drugs.
The p38 mitogen-activated protein kinase (MAPK), like SGK, acts as a critical intracellular regulator of environmental changes. The p38 MAPK pathway can be activated by heat shock, bacterial lipopolysaccharide, inflammatory cytokines, UV radiation, or hormones and is an important signal transduction mechanism mediating cellular responses to environmental changes. Activation of p38 MAPK has diverse downstream signaling effects, including cell cycle progression, apoptosis, differentiation, and cellular inflammatory responses (33). Several lines of evidence support a critical role of p38 MAPK signaling in IL-6-mediated signaling during growth of human tumors. IL-6 is an autocrine factor involved in human cholangiocarcinoma growth (34). We have shown that IL-6 stimulation activates the p38 MAPK in malignant but not in nonmalignant biliary tract epithelia (35, 37). Furthermore, we have also demonstrated that inhibition of p38 MAPK signaling reduces anchorage-independent growth in vitro and decreases xenograft growth in immunodeficient mice (42, 49). In addition, p38 MAPK signaling activates a dominant cell survival pathway in response to double-stranded RNA, a potent inducer of IL-6 expression (43). These observations suggest that IL-6 may promote cell survival by p38 MAPK-dependent pathways. However, the downstream mediators of survival signaling by p38 MAPK are poorly characterized. Our results show that IL-6 can regulate SGK by multiple p38 MAPK-dependent mechanisms, including regulation of transcriptional expression as well as phosphorylation of SGK at the Ser78 residue. Furthermore, regulation of SGK involves constitutive p38 MAPK activity. Several distinct isoforms of p38 MAPK, p38-α, -β, -γ, and -δ have been identified in mammalian cells. Although these isoforms have 60–70% identity, there is considerable overlap in their substrate specificities in vitro. Our studies show that SGK regulation by IL-6 specifically involves the p38α isoforms. In addition, chemotherapeutic drug toxicity in malignant biliary epithelial cells is increased in cells in which SGK expression is reduced. Taken together, these results suggest that survival signaling by IL-6 may involve transcriptional as well as posttranslational regulation of the survival kinase SGK via a p38 MAPK-dependent pathway.
MATERIALS AND METHODS
Reagents and plasmids
Recombinant human IL-6 was purchased from Biosource International (Camarillo, CA). Wortmannin, LY-294002, SB-203580, and SB-202190 were obtained from Calbiochem (La Jolla, CA). The FLAG-tagged wild-type and dominant negative forms (AF or KM) of human p38α, p38β, p38γ, and p38δ were generated as previously described (15, 16, 22, 23, 38). The AFs are p38 mutants that cannot be phosphorylated because the TGY dual phosphorylation site has been changed to AGF, whereas the kinase-dead KM mutants were generated by a mutation of the ATP-binding site Lys to Met (K to M). Rabbit polyclonal antibodies against p38α, p38β, p38γ, p38δ, and peroxidase-conjugated anti-rabbit and anti-mouse secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal antibodies against total SGK, phospho-SGK (Ser78), total p38 MAPK, and mouse monoclonal phospho-p38 (Thr180/Tyr182) antibodies were from Cell Signaling (Beverly, MA). Phospho-SGK (Thr256) antibody was purchased from Upstate Biotechnology (Lake Placid, NY). Rabbit polyclonal antibodies against SGK-2 and SGK-3 were obtained from Abgent (San Diego, CA). Monoclonal α-tubulin, β-actin, and anti-FLAG antibodies were obtained from Sigma (St. Louis, MO).
Cell lines and cultures
Malignant human cholangiocarcinoma cell lines KMCH-1 and Mz-ChA-1 were obtained as previously described (42, 43). KMCH-1 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, and Mz-ChA-1 cells were cultured in CMRL 1066 medium with 10% fetal bovine serum, 1% l-glutamine, and 1% antimycotic antibiotic mix. Cell culture media and supplements were obtained from Invitrogen (St. Louis, MO) unless otherwise noted. Cells were cultured in 100-mm plates and used when 75–80% confluent. Cell lines overexpressing IL-6 for xenograft studies were obtained from Mz-ChA-1 cells stably transfected with the pTarget (Promega, Madison, WI) expression plasmid containing full-length IL-6 under the control of a cytomegalovirus (CMV) promoter and selected by growth in medium containing G418. These cell lines were designated as Mz-IL-6, and empty vector controls cells were designated as Mz-1, respectively, and comprised a mixed population of stable transfectants without isolation of specific clones. For studies involving IL-6, cells were serum starved for 12 h before being stimulated with IL-6. For inhibitor studies, cells were preincubated with inhibitors 1 h before the addition of IL-6, except for studies with phosphatidylinositol 3-kinase (PI3-kinase) inhibitors, which were preincubated for 2 h.
Transient transfections
Cells (2 × 106) were plated in 100-mm dishes in culture media for 24 h. The media were then replaced, and plasmids were transfected in serum-free media using 15–18 μg of plasmid DNA per dish and TransIT-LT1 transfection reagent (Mirus, Madison, WI). After 4–6 h, the media were replaced with regular media containing 10% serum and the cells were incubated for 48 h before study.
Construction of recombinant adenoviruses encoding wild-type and mutant p38α
The recombinant adenoviruses encoding the wild-type and mutant p38α were produced according to the method of He et al. (19) with some modifications. Briefly, the FLAG-tagged wild-type and mutant human p38α cDNAs were cloned into the shuttle vector pAdTrack-CMV, linearized, and cotransformed into Escherichia coli BJ5183 cells along with the adenoviral backbone vector pAdEasy-1. Recombinants were selected for kanamycin resistance and confirmed with the use of restriction endonuclease analyses. Finally, the linearized recombinant plasmid was transfected into an adenovirus packaging cell line: human embryonic kidney-293 cells. Recombinant adenoviruses were collected 10–14 days after infection and were concentrated using a CsCl gradient. The shuttle vector pAdTrack-CMV also encodes green fluorescent protein (GFP) driven by a separate CMV promoter, and thus the titers of the viral stocks were estimated by counting GFP-expressing cells. An adenovirus-expressing GFP tag (AdGFP) under the control of a separate CMV promoter, which was a gift from Dr. Kim Heidenreich (Dept. of Pharmacology, University of Colorado HSC, Denver, CO), was used as a control.
Preparation of nuclear and cytoplasmic extracts
Nuclear and cytoplasmic fractions were obtained using the NE-PER extraction kit (Pierce, Rockford, IL) according to the manufacturer’s instructions. Protein concentrations in nuclear and cytoplasmic fractions were determined using the Bradford method, and reagents were obtained from Bio-Rad (Hercules, CA).
Immunoprecipitation and in vitro kinase assay
KMCH cells were stimulated with IL-6 in the presence or absence of p38 MAPK inhibitors. Cells were placed on ice and extracted with lysis buffer containing 50 mM β-glycerophosphate, pH 7.3, 1.5 mM EDTA, 1 mM EGTA, 1 mM DTT, and phosphatase inhibitor cocktails I and II (Sigma). Lysates were centrifuged for 15 min at 12,000 g, and SGK was immunoprecipitated from 150 μg of cell extract using anti-SGK monoclonal antibody (Cell Signaling, Beverly, MA) and the Seize immunoprecipitation kit (Pierce, Rockford, IL). In vitro kinase assays were then performed using the SGK kinase assay kit (StressGen, Vancouver, BC, Canada).
Western blot analysis
After treatment, confluent cell monolayers in 100-mm dishes were washed twice with ice-cold phosphate-buffered saline and lysed by incubation for 20 min in 1 ml of ice-cold cell lysis buffer (1% Nonidet P-40, 50 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM phenylmethylsulfonyl fluoride, 1 mM sodium vanadate, 1 mM sodium fluoride, and 1× protease mixture) and stored at −70°C. Protein concentrations were measured using a Bradford protein assay kit (Bio-Rad). Equivalent amounts of protein were resolved and mixed with 6 × SDS-PAGE sample buffer and then subjected to SDS-PAGE in a 4–20% linear gradient Tris · HCl-ready gel (Bio-Rad). The resolved proteins were transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk (except for phospho-SGK and p38 Western blots, in which 5% bovine serum albumin was used) in Tris-buffered saline, pH 7.4, containing 0.05% Tween 20, and were incubated with primary antibodies and horseradish peroxidase-conjugated secondary antibodies according to the manufacturer’s instructions. The protein of interest was visualized with enhanced chemiluminescence (Amersham Biosciences) and a charge-coupled device camera-based imager (ChemiImager 4000, Alpha Innotech, San Leandro, CA) and quantitated using NIH Image software.
Isolation of RNA and real-time PCR quantification
RNA was isolated using an RNA isolation kit (Bio-Rad), and reverse transcription was performed using 1 μg of total RNA and the reverse transcription kit (Invitrogen) as described by the manufacturer. To measure the mRNA levels of SGK, quantitative real-time PCR was performed on a MX 3000P real-time PCR Instrument (Stratagene, San Diego, CA) using the resulting total cDNA. The mRNA level of β-actin was used as a control. For the quantitative SYBR Green real-time PCR, 100 ng of cDNA was used per reaction. Each 20-μl SYBR Green reaction consisted of 2 μl of cDNA (50 ng/μl), 10 μl of 2× Universal SYBR Green PCR Master Mix (Sigma), and 2 μl of 20 nM forward and reverse primers. Optimization was performed for each gene-specific primer prior to the experiment to confirm that primer concentrations and reaction conditions did not produce artificial amplification signals in the no-template control tubes. Primer sequences were designed using Primer Express software (PerkinElmer Life Sciences) and were as follows: SGK forward, CCT TGT GGA TAT GCT GTG TGA ACC G; SGK reverse, 5′-TGG GGC ATT GGT CCA TAA AAA CC-3′; IL-6 forward, 5′-GCA GAA TGA GAT GAG TTG TC-3′; IL-6 reverse, 5′-GCC TTC GGT CCA GTT GCC TT-3′; β-actin forward, 5′-CCA AGG CCA ACC GCG AGA AGA TGA C-3′; and β-actin reverse, 5′-AGG GTA CAT GGT GGT GCC GCC AGA C-3′. PCR parameters were as follows: 10 min at 95°C and then 40 cycles of 30 s at 95°C, 1 min at 68°C, and 1 min at 72°C. Specificity of the produced amplification product was confirmed by melting curve analysis of the reaction products using SYBR Green as well as by visualization on ethidium bromide-stained 1.8% agarose gels to confirm a single band of the expected size. Each sample was tested in triplicate with quantitative real-time PCR, and samples obtained from at least three independent experiments were used for calculation. Threshold values were determined for each sample-primer pair, and means ± SD values were calculated. SGK or IL-6 mRNA expression was normalized against β-actin expression.
Nude mouse xenograft model
Eight-week-old male athymic nu/nu mice were obtained from Charles River Laboratories (Wilmington, MA) and fed food and water ad libitum. The mice were housed 4 per cage, and fluorescent light was controlled to provide alternate light and dark cycles of 12 h each. The animals received a subcutaneous injection of either Mz-1 or Mz-IL-6 cells (3 × 106 viable cells suspended on 0.5 ml of extracellular matrix gel) on their right flanks. Tumor volume was estimated by serial measurements obtained twice per week. The xenografts were excised. Tissue was divided and homogenized to obtain cell lysates or used for extraction of nuclear proteins or mRNA isolation. Animal protocols were approved by the Institutional Animal Care and Use Committee.
RNA interference
RNA interference for gene silencing was performed using small interfering 21-nucleotide double-stranded RNA (siRNA) molecules. SiRNA specific for SGK and control siRNA were obtained from Ambion (Austin, TX). KMCH cells were transfected as previously described (49). Briefly, 0.1 μg of siRNA was mixed with 6 μl of transfection agent (TransIt TKO, Mirus, Madison, WI), and the mixture was incubated in 1 ml of medium at room temperature for 15–20 min before being added to cultured cells grown to 50–60% confluence for 48 h. The efficacy of gene silencing was assessed by immunoblot analysis.
Cytotoxicity assay
Transfected cells were seeded into 96-well plates (10,000 viable cells/well) and incubated with gemcitabine, 5-fluorouracil, or appropriate diluent controls in a final volume of 200-μl medium. After 24 h, cell viability was assessed using a commercially available tetrazolium bioreduction assay for viable cells (CellTiter 96 AQ; Promega, Madison, WI), and cytotoxicity was assessed as previously described (43).
Statistical analysis
Data are expressed as the means ± SE from at least three separate experiments performed in triplicate, unless otherwise noted. The differences between groups were analyzed using a double-sided Student’s t-test when only two groups were present. Statistical significance was considered to be P < 0.05. Statistical analyses were performed with the GB-STAT statistical software program (Dynamic Microsystems, Silver Spring, MD).
RESULTS
Involvement of p38 MAPK in IL-6 activation of SGK
Activation of intracellular kinase signaling pathways after IL-6 receptor ligation can mediate survival signaling in human cholangiocarcinoma cells. We have previously shown that IL-6 activates the p38 MAPK signaling pathway. Because of the established role of SGK as a survival kinase, we evaluated the role of SGK as a downstream substrate of p38 MAPK signaling by IL-6. We began by assessing the temporal relationship between p38 MAPK and SGK phosphorylation by IL-6 stimulation. After incubation with 10 ng/ml IL-6, p38 MAPK was rapidly activated. Phosphorylation at the Thr180/Tyr182 sites of p38 MAPK was observed as early as 5 min after stimulation of KMCH cells with IL-6 (Fig. 1, A and B). In contrast, phosphorylation of SGK became detectable after 20–30 min. Similar results were also observed in Mz-ChA-1 cells, derived from metastatic gallbladder cancer. IL-6 stimulated SGK phosphorylation at Ser78 after 30 min, whereas the activation of p38 MAP kinase was also detectable at 5 min (Fig. 1, C and D). Thus phosphorylation of p38 MAPK precedes that of SGK after stimulation by IL-6. In KMCH malignant cholangiocytes, p38 MAPK is predominantly located in the cytoplasm and translocates to the nucleus in response to stimuli that activate p38 MAPK. Phosphorylation of p38 MAPK was rapidly followed by translocation of p38 MAPK to the nucleus, which occurred within 10 min and preceded nuclear translocation of SGK (Fig. 2A).
Fig. 1.
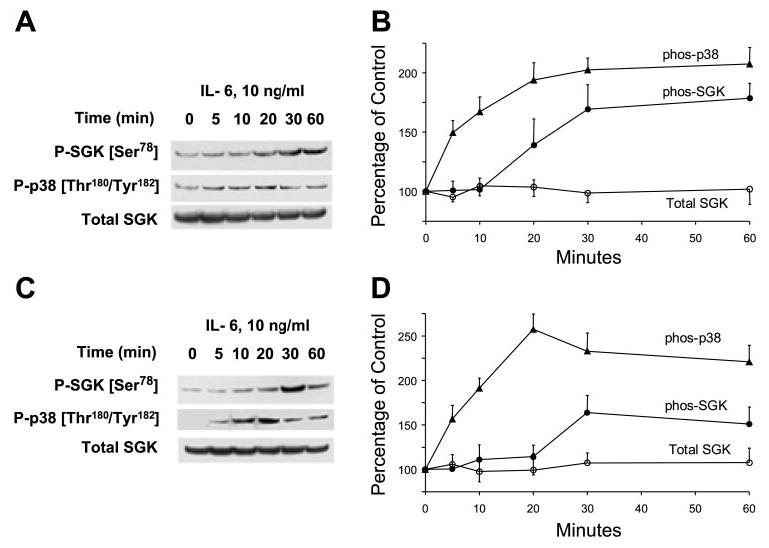
Interleukin-6 (IL-6) stimulates p38 MAP kinase activation and glucocorticoid-stimulated kinase (SGK) phosphorylation at Ser78 site. A and C: representative immunoblots are shown with quantitative data after densitometric analysis from 3 separate experiments (B and D). Malignant human cholangiocarcinoma cell lines KMCH (A and B) and Mz-ChA-1 (C and D) were serum starved for 12 h before incubation with 10 ng/ml IL-6. At the indicated times, cell lysates were prepared and analyzed by immunoblotting with the use of a Thr180/Tyr182 phospho-specific p38 MAPK antibody (P-p38). The blot was stripped and reprobed with antibodies specific for phospho-Ser78 and phosphorylation site-independent SGK (Total SGK). In both KMCH and Mz-ChA-1 cells, p38 MAPK phosphorylation occurred within 5 min of stimulation with IL-6 and preceded increases in SGK phosphorylation, which were first detected 20–30 min after stimulation.
Fig. 2.
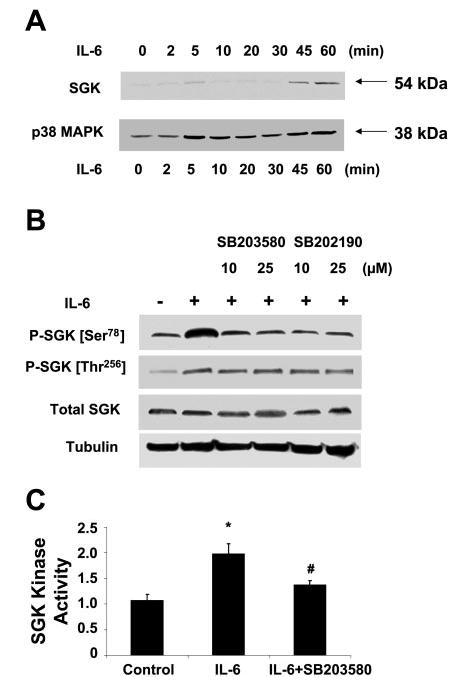
IL-6-induced SGK activation is p38 MAPK dependent. A: KMCH cells were incubated with 10 ng/ml IL-6 for various time periods (time 0–60 min). At the indicated times, the cells were lysed, and nuclear extracts obtained as described in materials and methods. Immunoblot analysis was performed using antibodies specific for p38 MAPK or SGK. An increase in nuclear expression of p38 MAPK occurs rapidly and within a few minutes after stimulation with IL-6, whereas the increase in nuclear SGK expression occurs later. A representative immunoblot from 3 separate experiments is shown. B: IL-6-induced SGK phosphorylation at Ser78 is blocked by p38 MAPK inhibitors. KMCH cells were grown to near-confluence and serum starved for 12 h. Cells were preincubated with the p38 MAPK inhibitors SB-203580 and SB-202190 at the indicated concentrations for 1 h before stimulation with IL-6 (10 ng/ml) for 40 min. Immunoblot analysis was performed using SGK Ser78 and Thr256 phosphorylation site-specific antibodies and then stripped and reprobed with phosphorylation state-independent antibodies to SGK (total SGK) or α-tubulin, used as a loading control. IL-6-dependent phosphorylation at SGK Ser78 was reduced to basal levels by preincubation with either p38 MAPK inhibitor. A representative immunoblot from 3 separate experiments is illustrated. C: IL-6-induced SGK activation is blocked by p38 MAPK inhibitor. KMCH cells were grown to near-confluence and serum starved for 12 h. Cells were preincubated with the p38 MAPK inhibitor SB-203580 (10 μM) for 1 h before stimulation with IL-6 (10 ng/ml) for 40 min. SGK activity was assessed after immunoprecipitating SGK from cell lysates prepared from the cells treated as described above. Bars show means ± SE of at least 3 experiments. *P < 0.01 when compared with control group. #P < 0.01 when compared with IL-6-treated group.
To further examine the relationship between activation of SGK by IL-6 and p38 MAPK activation, we used SB-203580, a pyridinyl imidazole compound that inhibits the catalytic activity of p38 MAPK. Treatment with SB-203580 reduced SGK phosphorylation by IL-6 (Fig. 2B). A similar reduction in SGK phosphorylation at Ser78 by IL-6 was observed in the presence of SB-202190, which is pharmacologically distinct from SB-203580. Previous studies (10) have demonstrated that both SB-203580 and SB-202190 are highly selective against p38 MAPK and have minimal effects on SGK activity or many other kinase activities. Indeed, incubation with SB-203580 decreased SGK activity after IL-6 stimulation (Fig. 2C). In combination, these findings suggest that activation of p38 MAPK may result in SGK activation and that SGK may be a downstream target substrate of p38 MAPK activation by IL-6.
IL-6-dependent SGK Ser78 phosphorylation is PI3-kinase independent
IL-6 receptor ligation results in the activation of several intracellular signaling pathways, such as JAK-STAT, ERK, p38 MAPK, and PI3-kinase pathways (20). Our studies with pharmacological inhibitors indicated that PI3-kinase or p38 MAPK pathways can mediate the survival effect of IL-6 (data not shown). Thus SGK activation by IL-6 may involve signaling by activation of PI3-kinase signaling. The mechanism of SGK activation by PI3-kinase signaling involves reversible phosphorylation at the Thr256 site in the activation loop of SGK (36). Thr256 resides in the action loop of the catalytic domain and is a major site for activation of SGK by phosphorylation by the 3-phosphoinositide-dependent kinases. Although considered to be the major site for the activation of SGK, the presence of additional active phosphorylation sites is suggested by studies showing that mutagenesis of Thr256 does not abrogate total cellular phosphorylation of SGK and is supported by the identification of Ser78 as a phosphorylation-dependent site for SGK activation by the BMK-1 (18). Thus we evaluated phosphorylation of SGK at both Thr256 and Ser78 using phosphorylation site-specific antibodies. As expected, phosphorylation at both sites occurred following stimulation by IL-6. However, incubation with the PI3-kinase inhibitor wortmannin inhibited IL-6-induced SGK phosphorylation at Thr256 but not Ser78 (Fig. 3). Furthermore, the increased translocation of SGK to the nucleus after IL-6 treatment was not substantially reduced by either wortmannin or by LY-294002, two specific inhibitors of PI3-kinase, even when used at relatively high concentrations (Fig. 3B). Similarly, translocation of p38 MAPK to the nucleus and reduction of SGK levels in the cytoplasm was not affected by wortmannin (data not shown). Taken together, these observations place SGK phosphorylation downstream of p38 MAPK following IL-6 stimulation in a pathway distinct from PI3-kinase signaling.
Fig. 3.
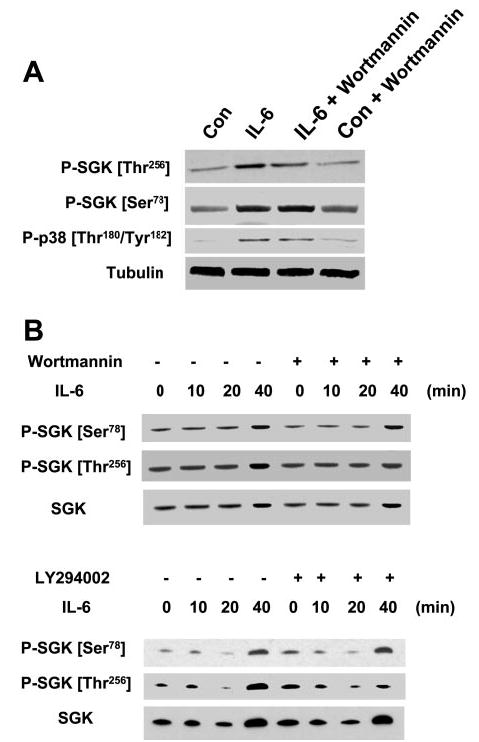
Inhibition of phosphatidylinositol 3-kinase (PI3-kinase) blocks IL-6-induced phosphorylation of SGK at the Thr256 site but not at the Ser78 site. A: KMCH cells were incubated with or without 10 ng/ml IL-6 in the presence or absence of the PI3-kinase inhibitor wortmannin (1 μM). After 40 min, cells were lysed and immunoblot analysis performed using phosphorylation site-specific antibodies to SGK Thr256, SGK Ser78, p38 MAPK Thr180/Tyr182, and α-tubulin. Wortmannin blocked SGK phosphorylation at Thr256, but not at Ser78. Identical results were obtained with LY-294002 (100 μM) and lower concentrations of wortmannin (100–500 nM) (not shown). B: KMCH cells were incubated with IL-6 (10 ng/ml) in the presence or absence of 500 nM wortmannin or 50 μM LY-294002. At the indicated times, nuclear extracts were obtained as described in materials and methods. Immunoblot analysis was performed using SGK Ser78 and Thr256 phosphorylation site-specific and total SGK antibodies. Nuclear expression of total SGK and SGK Ser78, indicating phosphorylation site-specific activation and translocation of SGK, occurred despite inhibition of PI3-kinase.
p38 MAPK isoform-specific activation of SGK by IL-6
To further examine p38 MAPK signaling in SGK activation by IL-6, we began by assessing the involvement and role of the specific p38 MAPK isoforms involved. First, we assessed the expression of p38 isoforms in KMCH and Mz-ChA-1 cells by performing immunoblot analysis using isoform-specific antibodies. The cell lines differed in the specific p38 MAPK isoforms expressed (Fig. 4A). p38α (38 kDa) and p38δ (39 kDa) were expressed in both cell lines, whereas p38β (40 kDa) was expressed in Mz-ChA-1 cells only. However, p38γ was not expressed in either cell line. The isoforms migrated differently on SDS-PAGE due to their different molecular weights. The higher levels of p38α expression relative to that of the other isoforms in both cell lines indicated that p38α is the predominant p38 MAPK isoform expressed in these cells. Next, we evaluated the individual roles of each p38 MAPK isoform in the signal transduction pathways leading to SGK activation. Cells were transfected with dominant negative (kinase inactive) p38 isoforms, and their effects on SGK phosphorylation after stimulation with IL-6 was assessed. The the dual phosphorylation residue TGY (AF mutants) or have an inactive ATP-binding site (KM mutants). A p38 isoform-specific effect on IL-6-induced SGK activity was observed (Fig. 4B). Transfection of dominant negative p38α, but not p38β, p38γ, or p38δ, decreased the IL-6-dependent SGK phosphorylation at the Ser78 site (Fig. 4B) as well as nuclear translocation (Fig. 4C). Although phosphorylation at the Thr256 site was also observed in response to IL-6 stimulation, expression of the dominant negative p38 isoforms did not have a significant effect on phosphorylation at this site. These studies support the presence of both a p38α-dependent pathway involving phosphorylation at Ser78, as well as a p38 MAPK independent pathway involving phosphorylation at Thr256 during activation of SGK by IL-6.
Fig. 4.
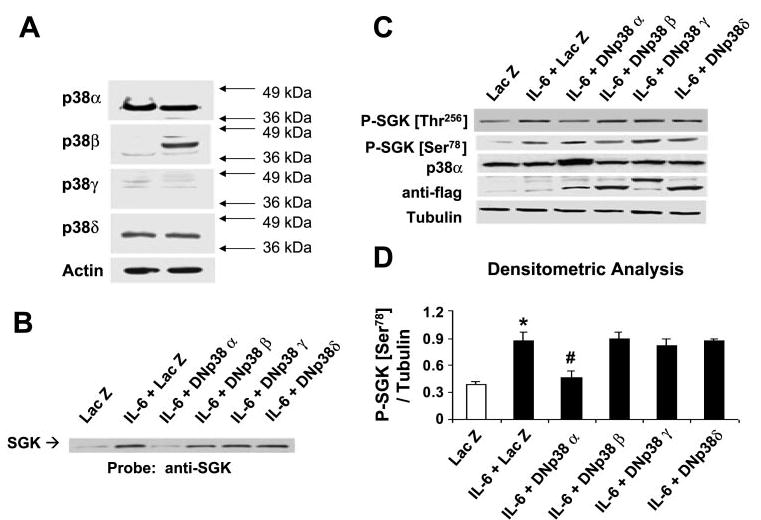
p38 MAPK isoforms are differentially expressed and selectively activate SGK in cholangiocarcinoma cells. A: cell lysates were obtained from unstimulated KMCH (left) or Mz-ChA-1 (right) cholangiocarcinoma cells. Protein (50 μg) was separated by SDS-PAGE, and the membrane was stripped and reprobed with antibodies specific for p38α-, -β, -γ, or -δ MAPK isoforms, or for β-actin used as a loading control. The results are representative of 3 separate experiments. KMCH cells were transfected with FLAG-tagged, isoform-specific, dominant negative p38 MAPK or LacZ-expressing control plasmids as described in materials and methods. After 48 h, cells were stimulated with 10 ng/ml IL-6 for 40 min. Cells were lysed, and an aliquot of the whole cell lysate was used for Western blot analysis, whereas nuclear extracts were obtained from the remainder. B: SGK expression in nuclear extracts was identified using an antibody against SGK. A representative immunoblot of three separate studies is shown. Dominant negative p38α decreases nuclear translocation of SGK. C: immunoblot analysis of whole cell lysate using an antibody against phospho-Ser78 SGK. The same blot was reprobed with specific antibodies against phospho-Thr256 SGK, p38α MAPK, FLAG, and α-tubulin. Expression of dominant negative (DN) constructs was confirmed using anti-FLAG antibodies. D: expression of phospho-Ser78 SGK was normalized to α-tubulin and quantitated with the use of NIH Image software. The means ± SD of densitrometric analysis from 3 separate determinations are shown. Dominant negative p38α selectively decreases phosphorylation at Ser78. *P < 0.05, compared with LacZ control group. #P < 0.05, compared with IL-6-treated LacZ group.
Overexpression of p38α MAPK increases SGK expression and phosphorylation at Ser78
To more directly assess the role of the p38α isoform in activation of SGK, we overexpressed p38α using an adenoviral construct and examined the effect on SGK phosphorylation. Adenoviral delivery of p38α resulted in a robust increase in total p38 MAPK levels (Fig. 5A). An increase in p38α MAPK correlated with increased phosphorylation at SGK Ser78, suggesting that SGK phosphorylation at Ser78 involves constitutive expression of the p38α MAPK. Next, we examined the time course of SGK expression and phosphorylation after infection of KMCH with adenoviral constructs expressing p38α. Increased phosphorylation of SGK at Ser78 but not at the Thr256 site was detectable after 48 h. Interestingly, in addition to inducing SGK phosphorylation, overexpression of p38α caused an increase in the level of total SGK expression. An increase in p38 MAPK expression was seen by 24 h and preceded the increase in SGK expression observed at 36 h (Fig. 5, B and C). The increased p38 levels resulted in a significant increase in p38 MAPK and SGK nuclear translocation (Fig. 6A), suggesting an increase in constitutively active p38 MAPK and SGK. As shown in Fig. 6, B and C, and consistent with the idea that SGK is a downstream target of p38 MAPK after IL-6 stimulation, overexpression of p38α caused an increase in SGK phosphorylation at Ser78. Adenoviral delivery of DN-p38α, on the other hand, which would be expected to inhibit p38 MAP kinase activity, showed reduced SGK Ser78 phosphorylation compared with control cells that overexpressed GFP (Fig. 6B). Similar effects of overexpression of p38α on SGK Ser78 phosphorylation were also observed in Mz-ChA-1 cells (Fig. 6C). Furthermore, total SGK expression correlated with p38α expression and was reduced to a level slightly below that seen in GFP-expressing cells by dominant negative (kinase dead) p38α (Fig. 6, B and C).
Fig. 5.
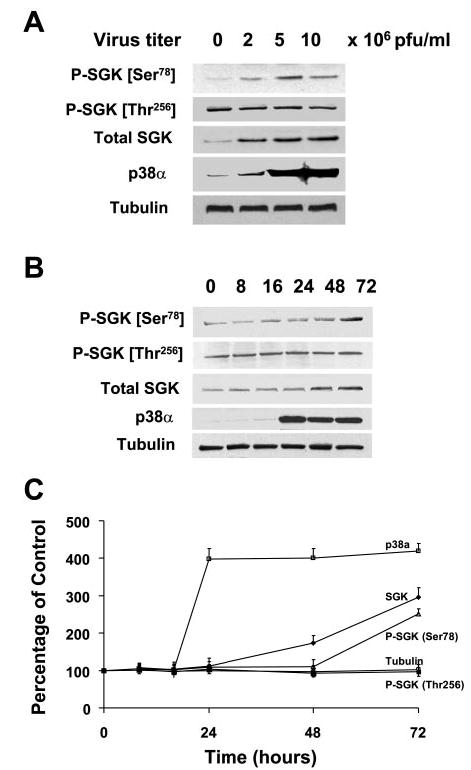
Overexpression of p38α stimulates SGK phosphorylation at Ser78 in the absence of IL-6. A: KMCH cells were infected with 0, 2, 5, and 10 × 106 plaque-forming units (PFU)/ml of adenovirus encoding p38α MAPK cDNA. Whole cell lysates were prepared 48 h after infection and assessed by immunoblot analysis with a Ser78 phospho-specific SGK antibody. The blots were stripped and reprobed with phospho-specific antibodies against Thr256 SGK or phosphorylation state independent antibodies to SGK (Total SGK), p38 MAPK, and α-tubulin (loading control). SGK phosphorylation at Ser78 but not at Thr256 increased proportionally to p38 MAPK expression. B: representative blot. C: means ± SD from 3 separate experiments. In B and C, cell lysates were obtained at various time points after infection as indicated above the lanes. Immunoblot analysis was performed using Ser78 or Thr256 phospho-specific SGK antibodies. The blot was stripped and reprobed with an antibody against total SGK, p38α MAPK, and α-tubulin. Increased p38α expression was seen within 24 h, whereas SGK expression was evident only 36 h after infection. An increase in Ser78 but not Thr256 phosphorylation was observed after 48 h.
Fig. 6.
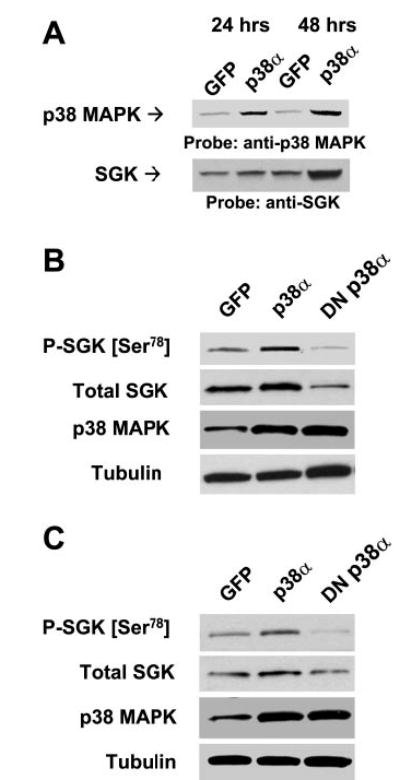
Correlation between p38α expression and SGK. A: KMCH cells were infected with 5 × 106 PFU/ml adenoviral (Adv)-p38α or Adv-green fluorescent protein (GFP) (controls). Nuclear extracts were prepared after 24 or 48 h, and immunoblot analysis was performed using specific antibodies to p38 MAPK or SGK. KMCH cells (B) or Mz-ChA-1 cells (C) were infected with Adv constructs encoding GFP, p38α, or DNp38α. Whole cell lysates were prepared 48 h after infection and assessed by performing immunoblot analysis using a Ser78 phospho-specific SGK antibody. The blots were stripped and reprobed with phosphorylation state-independent antibodies to SGK (total SGK), p38 MAPK, and α-tubulin (loading control).
SGK generally refers to SGK-1, one of three closely related members of the SGK gene family (24). The transcriptional regulation of the other two closely related isoforms, SGK-2 and SGK-3 remains poorly understood. We assessed the expression of the SGK-1, SGK-2, and SGK-3 isoforms by immunoblot analysis using isoform-specific antibodies. In contrast to SGK-1, neither IL-6 stimulation nor p38α overexpression altered SGK-2 or SGK-3 expression in cell lysates obtained after incubation with 0 or 10 ng/ml IL-6 for 6 h or in cells infected with adenoviral constructs expressing p38α or GFP (controls) for 48 h.
Upregulation of SGK mRNA expression by IL-6 and/or p38α MAPK stimulation
To further examine the effects of p38α overexpression on total SGK protein expression, we performed real-time PCR to quantitate alterations in SGK transcript levels. Overexpression of p38α caused a concentration-dependent increase in SGK mRNA, supporting a direct transcriptional rather than posttranslational mechanism for increased SGK protein expression after overexpression of p38α (Fig. 7, A and B). On the basis of these observations, we postulated that transcriptional upregulation of SGK may represent an additional mechanism involved in survival signaling by IL-6. To further examine the mechanism, we quantitated alterations in transcript levels with IL-6 stimulation. Basal levels of SGK mRNA were detectable by quantitative real-time PCR and increased in a concentration-dependent manner after treatment with IL-6 (Fig. 7C). A profound induction of mRNA was observed, with a remarkable >4-fold induction with IL-6 (10 ng/ml) after 6 h. The mRNA levels of β-actin were nearly identical in all of these samples, indicating the specificity of the response. These quantitative real-time PCR data demonstrate, for the first time, concentration-dependent transcriptional regulation of SGK by IL-6 stimulation as well as by p38α overexpression. Furthermore, the increased SGK mRNA expression correlated well with total SGK protein expression assessed by immunoblot analysis (Fig. 7D). The rapidity of the transcriptional response suggests that SGK expression may mediate the survival effects of IL-6. To establish whether IL-6 induces SGK mRNA by altering transcription or by altering mRNA stability, the expression of SGK was assessed in the presence of the transcriptional inhibitor actinomycin D. First, we assessed the effect of preincubation with actinomycin D on SGK mRNA expression induced by IL-6 or by p38α overexpression. The increase in SGK mRNA in response to either IL-6 stimulation or enforced expression of p38α was blocked in the presence of actinomycin D. Next, we assessed the time course of SGK mRNA expression in the presence of actino-mycin D and did not observe any significant effect on the rate of loss of SGK mRNA levels. Thus IL-6 increases SGK mRNA by a transcriptional mechanism and does not alter SGK mRNA stability.
Fig. 7.
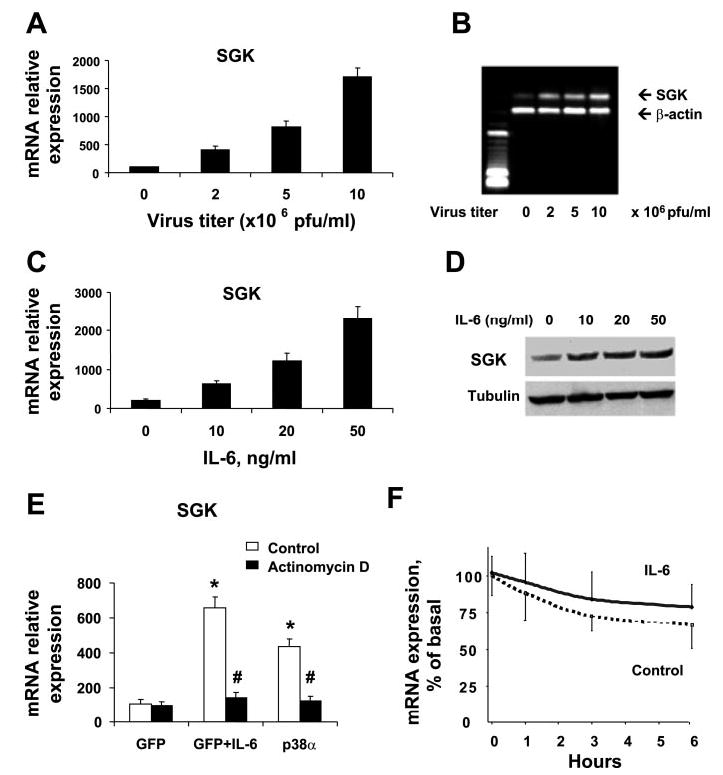
Overexpression of p38α increases SGK mRNA expression. A and B: KMCH cells were infected with Adv-p38α constructs at the indicated viral titers. After 48 h, total RNA was isolated and reverse transcribed to obtain cDNA. SGK mRNA expression was quantitated by real-time PCR and expressed relative to β-actin mRNA expression concurrently assessed in the same samples. Overexpression of p38α increased SGK mRNA expression in a concentration-dependent manner. The PCR products were verified by agarose gel electrophoresis of the PCR product, as well as by melting curve analysis (not shown). The graph in A depicts the means ± SD from 3 separate experiments, whereas a single representative gel is shown in B. C: KMCH cells grown to near-confluence were serum starved for 12 h. Cells were then stimulated for 6 h with IL-6 at the concentrations indicated. Total RNA was isolated and cDNA was generated after reverse transcription. The expression of SGK and β-actin mRNA was quantitated by real-time RT-PCR. SGK mRNA expression is depicted relative to β-actin. Results represent means ± SD of 4 experiments. D: cell lysates were obtained from KMCH cells stimulated with IL-6 as above. Total protein (50 μg) was subjected to gel electrophoresis and immunoblotting using polyclonal anti-SGK antibody; the blots were then stripped and reprobed with antitubulin as a loading control. A representative immunoblot of 4 experiments is shown at right. IL-6 increased SGK mRNA and protein expression in a concentration-dependent manner. E: IL-6- and p38α-dependent increase of SGK expression is transcriptionally mediated. KMCH cells were infected with adenoviral-GFP or p38α constructs. After 24 h, by which time p38α had been expressed (Fig. 5B), 5 μM actinomycin D was added for another 24 h. The adenoviral-GFP-infected cells were then treated with or without IL-6 (10 ng/ml) for 6 h, after which total RNA was isolated, cDNA was generated, and the expression of SGK and β-actin mRNA was quantitated by real-time RT-PCR. *P < 0.05 compared with GFP group. #P < 0.05 compared with control group in the absence of actinomycin D. F: IL-6 does not alter the stability of SGK mRNA. KMCH cells were incubated with 5 μM actinomycin D in the presence or absence of 10 ng/ml IL-6. At each time point, total cellular RNA was isolated, and real-time PCR for SGK mRNA was performed. The means ± SD of the percentage of basal SGK mRNA expression from three individual experiments are shown. Differences between the groups were not significant (P > 0.05).
SGK expression is increased after enforced expression of IL-6 in vivo
To determine whether IL-6 can modulate SGK in vivo, we performed studies in which Mz-ChA-1 human cholangiocarcinoma cells were stably transfected to produce cells overexpressing IL-6 (Mz-IL-6) compared with controls (Mz-1). The effect of enforced IL-6 expression was then assessed in xenograft studies in immunodeficient mice. Consistent with its role as a potential autocrine promoter of tumor growth, xenograft growth and chemoresistance were increased in xenografts of cells overexpressing IL-6 compared with controls (data not shown). Expression of phospho-SGK or p38 MAPK was significantly increased in homogenates from IL-6-overexpressing tumor cell xenografts compared with controls (Fig. 8). Similarly, there was an increase in constitutive expression in nuclear extracts, consistent with an increased nuclear translocation, and a 2.8-fold increase in SGK expression following enforced expression of IL-6 in vivo. These observations indicate that overexpression of IL-6 increases SGK expression and basal phosphorylation in vivo.
Fig. 8.
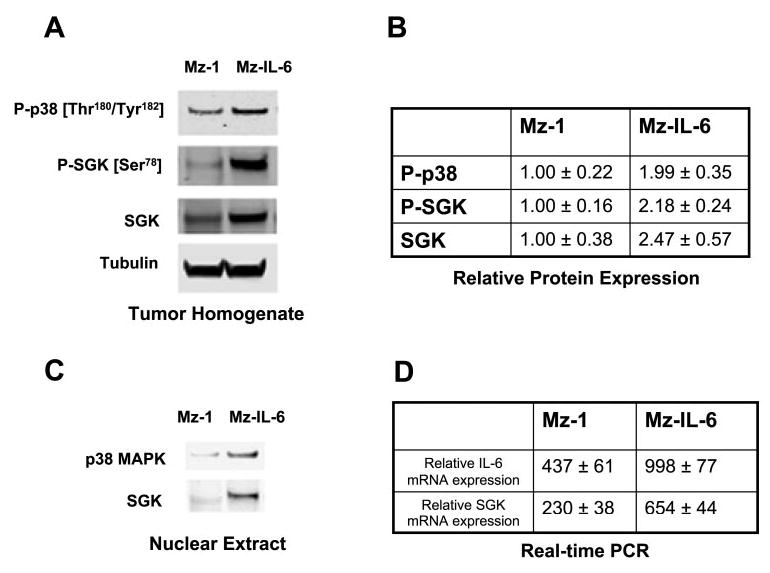
SGK expression is increased after enforced expression of IL-6 in vivo. Mz-ChA-1 cells were stably transfected with full-length IL-6 to generate Mz-IL-6 cells, which overexpressed IL-6 compared with their Mz-1 controls. Cells (3 × 106) were injected subcutaneously into the flank of athymic BALB/c mice. A: Western blot analysis was performed in cell lysates from tumor cell xenografts using antibodies to phos-pho-p38 MAP kinase, phospho-SGK, total SGK, and tubulin. A representative panel is shown. B: the expression of phospho-p38 and SGK expression was normalized to α-tubulin and quantitated using NIH Image software (n = 4). Overexpression of IL-6 is associated with increased expression of SGK in vivo. C: nuclear extracts were obtained from xenograft lysates, and nuclear expression of p38 MAPK and SGK expression was assessed. Nuclear expression of both p38 MAP kinase and SGK was increased in Mz-IL-6 xenografts. D: RNA was extracted from xenograft tissues and IL-6 and SGK mRNA expression was assessed by quantitative real-time PCR. IL-6 and SGK mRNA expression was normalized to β-actin mRNA expression. The data represents results from 3 experiments performed in triplicate.
SGK mediates resistance to chemotherapy
Activation of survival signaling can result in increased resistance to cytotoxic drugs. To assess the functional role of modulation of SGK expression, siRNA to SGK was used to reduce cellular SGK expression. Compared with a control siRNA, siRNA to SGK increased cell viability during incubation with the chemotherapeutic agents gemcitabine or 5-fluorouracil (Fig. 9). Because IL-6 decreases the sensitivity of malignant cholangiocarcinoma cells to chemotherapy, these results support a role for the modulation of SGK expression in mediating the cytoprotective effects of IL-6.
Fig. 9.
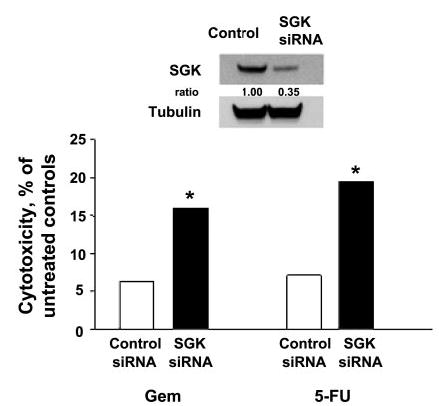
siRNA to SGK increases cytotoxicity. KMCH cells were transfected with siRNA to SGK or to a control siRNA for 48 h. SGK expression after transfection was assessed by immunoblot analysis. An immunoblot of SGK expression in cells transfected with the siRNA to SGK or control siRNA is shown in the inset. The media were changed, and cells were plated (10,000/well) in 96-well plates and incubated with 10 μM gemcitabine (Gem) or 10 μM 5-fluorouracil (5-FU) or their respective diluent controls. Cytotoxicity was assessed after 24 h using a viable cell assay. Cytotoxicity in cells treated with each drug is expressed relative to that observed in untreated (diluent only) controls. An increased drug-induced cytotoxicity was observed in cells transfected with siRNA to SGK compared with control siRNA. Results are means ± SE of 6 studies (SE in each case was <1%). *P < 0.01 compared with the siRNA control group.
DISCUSSION
In addition to a well-characterized role as a mediator of the cellular response to inflammation and regulator of hepatic growth and regeneration, IL-6 plays a central role as an autocrine regulator of tumor growth in some human cancers. Survival signaling by IL-6 may contribute to these effects. In the present study, we have investigated the mechanisms by which IL-6 activated intracellular signaling regulates the survival kinase SGK. These studies show that 1) IL-6 activates SGK in cholangiocarcinoma cells, 2) p38α MAPK is necessary for SGK activation by IL-6, and 3) overexpression of p38α MAPK is sufficient for regulation of SGK transcription and phosphorylation at the Ser78 residue. p38 MAPK regulation of SGK has been reported in response to environmental changes such as in human hepatoma cells during cell shrinkage (1, 45). However, the present studies are the first to demonstrate the specific role of the p38α isoform in SGK activation by IL-6 and the first to show that p38α MAPK expression is sufficient for SGK activation. These studies define SGK as both a downstream kinase substrate and a transcriptionally regulated gene product of p38 MAPK signaling in response to IL-6.
Although it was initially identified as being under acute transcriptional control by serum and glucocorticoids, SGK has subsequently been shown to be stimulated by diverse stimuli such as osmotic and oxidative stress, growth factors, or hormones (1, 5, 28, 30, 32, 46). In contrast to that of most protein kinases, the regulation of SGK is complex and can occur at multiple levels involving transcriptional control, subcellular localization, and posttranslational modifications involving kinase phosphorylation/dephosphorylation (see Ref. 13 for review). These mechanisms are differentially regulated in a cell-type and stimulus-specific manner. The presence of multiple regulatory mechanisms for SGK activation suggests that this kinase acts as a common downstream integrator of the response to diverse stimuli and signaling cascades. The activation of SGK by IL-6 and p38 MAPK activation involves phosphorylation at the Ser78 residue in preference to classical SGK phosphorylation sites at Thr256 or Ser422 (36). Although Ser78 is not located within the catalytic domain, SGK activation has been shown to occur following phosphorylation at this site by BMK1, a member of the MAP kinase family (18). Unlike Thr256, the Ser78 site is not phosphorylated by members of the 3-phosphoinositide-dependent kinase family. The presence of PI3-kinase-independent mechanisms has been described previously for SGK activation under specific conditions such as by cell detachment, but the precise nature was not defined (40). Although p38α MAPK dependent phosphorylation at Ser78 residue occurs independently of the PI3-kinase pathway, our studies do not exclude an additional contribution of this or other p38 MAPK-independent signaling pathways during SGK activation by IL-6. Indeed, our studies indicate that IL-6 can phosphorylate the Thr256 site independent of p38 MAPK activation. Taken together, these observations indicate that SGK activation by IL-6 can occur via temporally separated mechanisms involving activation by different signaling cascades, phosphorylation at different sites, and transcriptional regulation. Concomitant activation of stimulus-dependent signaling cascades can activate different intracellular pathways that act in concert and converge onto SGK. Upstream regulators of SGK can thereby provide the mechanism for stimulus specificity by which SGK coordinates specific cellular responses. Thus SGK is well positioned to act as a central integration point of the cellular survival response to diverse physiological or pathophysiological stimuli mediated by either the p38 MAPK or the PI3-kinase pathways.
These studies identify SGK as a downstream target kinase for p38α. Many substrates of p38 MAPK are themselves serine/threonine kinases. These include the MAPK-activated protein kinases (MK) such as MK2 and MK3, the p38-regulated and -activated protein kinases or MK5, the MAPK kinase-interacting kinases 1 and 2 (MNK-1 and MNK-2), and the mitogen- and stress-activated kinases 1 and 2. Downstream kinases can mediate physiologically relevant effects of p38 MAPK signaling as demonstrated in MK2-knockout mice, which are able to resist stress and survive endotoxin-induced septic shock (26). Transcriptional regulation of SGK expression by p38 MAPK provides an additional level of regulation of SGK by p38 MAPK. Several transcription factors have been identified as direct targets of the p38 MAPK signaling. These include activating transcription factor 2, CHOP/GADD153, and myocyte enhancer factor 2. Additional studies will be required to identify specific transcriptional factors involved in regulation of SGK expression by p38 MAPK. Because of the common responses to environmental stresses described for both p38 MAPK and SGK as well as the multiple mechanisms by which p38 MAPK can regulate SGK expression, it seems probable that the p38 MAPK/SGK axis is a critical mediator of the cellular response to perturbations in the extracellular environment.
Unlike most other kinases involved in p38 MAPK signaling, SGK is regulated transcriptionally as well as posttranslationally, raising the intriguing possibility that SGK may serve to amplify specific stress responses after p38 MAPK activation. Recently, the mitogen-activated protein kinase kinase kinase 3 (MEKK3) has been shown to be a target substrate for SGK (7). MEKK3 lies upstream of mitogen-activated protein kinase kinase (MKK) 3 and 6, which are activators of p38 MAPK signaling. This raises the possibility of feedback loops involving the p38 MAPK-SGK axis with the potential for self-limiting the activation cascade independent of phosphatase activity by inhibition of an upstream activator.
Considerable similarity exists in the mechanisms of activation and downstream effects of SGK and Akt. Both Akt and SGK can be activated by growth factors in a PI3-kinase-dependent manner. The preponderance of available evidence shows that Akt and SGK constitute major cell survival pathways. Furthermore, several identical downstream targets of Akt, such as Bad and FOXO-3, are also downstream substrates for SGK. These observations suggest that SGK and Akt may act in an analogous fashion to promote cellular survival. While unique functions for SGK have been reported, these may not directly mediate cell survival (39). However, the cell and stimulus specificity of activation of survival signaling by Akt and SGK are undefined. We have observed a lack of Akt activation by IL-6 and the inability of dominant negative Akt to ameliorate the survival effects of IL-6 in cholangiocarcinoma cells (data not shown). These observations support the primary involvement of SGK during survival signaling by IL-6.
IL-6 increases resistance of cholangiocarcinoma cells to undergo apoptosis in response to chemotherapeutic agents, such as gemcitabine or 5-fluorouracil (48). Indeed, production of IL-6 has been correlated with induction of drug resistance in several tumors such as myeloma, lung, breast, ovarian, prostate, and colorectal cancer (3, 9, 11, 14, 25, 41). High levels of IL-6 are produced by multidrug-resistant cancer cells, whereas some cancer cells that are sensitive to drug treatment do not express IL-6 (9). In human hepatoma cells, endogenous production of IL-6 can confer resistance to chemotherapy through induction of multidrug-resistant protein (27). Furthermore, anti-IL-6 has been shown to restore the ability of hematopoietic K562 mutant cells to undergo chemotherapy-induced apoptosis (12). It is therefore noteworthy that manipulation of SGK expression by siRNA can also modulate cellular responses to chemotherapeutic agents. Thus our observations identify SGK as a functionally relevant intermediate in survival signaling by IL-6 and implicate the p38 MAPK-SGK signaling axis in IL-6-induced chemoresistance.
IL-6 is an autocrine factor important in the growth of several tumors such as cholangiocarcinoma and selectively activates p38 MAPK signaling in malignant cholangiocarcinoma cells. We propose that dysregulation of p38 MAPK/SGK-mediated signaling in tumor cells permits the survival of transformed cells under otherwise adverse environmental conditions, such as exposure to chemotherapeutic drugs or to hypoxia or under growth-factor limited conditions. Additional studies to assess the contribution of SGK in biliary tract tumorigenesis and transformed cell behavior are warranted.
Footnotes
GRANTS
This work was supported by the Scott and White Hospital Foundation and by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-069370.
References
- 1.Bell LM, Leong ML, Kim B, Wang E, Park J, Hemmings BA, Firestone GL. Hyperosmotic stress stimulates promoter activity and regulates cellular utilization of the serum- and glucocorticoid-inducible protein kinase (Sgk) by a p38 MAPK-dependent pathway. J Biol Chem. 2000;275:25262–25272. doi: 10.1074/jbc.M002076200. [DOI] [PubMed] [Google Scholar]
- 2.Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001;20:4380–4390. doi: 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borsellino N, Bonavida B, Ciliberto G, Toniatti C, Travali S, D’Alessandro N. Blocking signaling through the Gp130 receptor chain by interleukin-6 and oncostatin M inhibits PC-3 cell growth and sensitizes the tumor cells to etoposide and cisplatin-mediated cytotoxicity. Cancer. 1999;85:134–144. [PubMed] [Google Scholar]
- 4.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buse P, Tran SH, Luther E, Phu PT, Aponte GW, Firestone GL. Cell cycle and hormonal control of nuclear-cytoplasmic localization of the serum- and glucocorticoid-inducible protein kinase, Sgk, in mammary tumor cells. A novel convergence point of anti-proliferative and proliferative cell signaling pathways. J Biol Chem. 1999;274:7253–7263. doi: 10.1074/jbc.274.11.7253. [DOI] [PubMed] [Google Scholar]
- 6.Chauhan D, Kharbanda S, Ogata A, Urashima M, Teoh G, Robertson M, Kufe DW, Anderson KC. Interleukin-6 inhibits Fas-induced apoptosis and stress-activated protein kinase activation in multiple myeloma cells. Blood. 1997;89:227–234. [PubMed] [Google Scholar]
- 7.Chun J, Kwon T, Kim DJ, Park I, Chung G, Lee EJ, Hong SK, Chang SI, Kim HY, Kang SS. Inhibition of mitogen-activated kinase kinase kinase 3 activity through phosphorylation by the serum- and glucocorticoid-induced kinase 1. J Biochem (Tokyo) 2003;133:103–108. doi: 10.1093/jb/mvg010. [DOI] [PubMed] [Google Scholar]
- 8.Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J. 2003;22:4202–4211. doi: 10.1093/emboj/cdg407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conze D, Weiss L, Regen PS, Bhushan A, Weaver D, Johnson P, Rincon M. Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Res. 2001;61:8851–8858. [PubMed] [Google Scholar]
- 10.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Vita F, Orditura M, Auriemma A, Infusino S, Roscigno A, Catalano G. Serum levels of interleukin-6 as a prognostic factor in advanced non-small cell lung cancer. Oncol Rep. 1998;5:649–652. [PubMed] [Google Scholar]
- 12.Dedoussis GV, Mouzaki A, Theodoropoulou M, Menounos P, Kyrtsonis MC, Karameris A, Maniatis A. Endogenous interleukin 6 conveys resistance to cis-diamminedichloroplatinum-mediated apoptosis of the K562 human leukemic cell line. Exp Cell Res. 1999;249:269–278. doi: 10.1006/excr.1999.4442. [DOI] [PubMed] [Google Scholar]
- 13.Firestone GL, Giampaolo JR, O’Keeffe BA. Stimulus-dependent regulation of serum and glucocorticoid inducible protein kinase (SGK) transcription, subcellular localization and enzymatic activity. Cell Physiol Biochem. 2003;13:1–12. doi: 10.1159/000070244. [DOI] [PubMed] [Google Scholar]
- 14.Frassanito MA, Cusmai A, Iodice G, Dammacco F. Autocrine interleukin-6 production and highly malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood. 2001;97:483–489. doi: 10.1182/blood.v97.2.483. [DOI] [PubMed] [Google Scholar]
- 15.Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 16.Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem. 1996;271:2886–2891. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- 17.Hardin J, MacLeod S, Grigorieva I, Chang R, Barlogie B, Xiao H, Epstein J. Interleukin-6 prevents dexamethasone-induced myeloma cell death. Blood. 1994;84:3063–3070. [PubMed] [Google Scholar]
- 18.Hayashi M, Tapping RI, Chao TH, Lo JF, King CC, Yang Y, Lee JD. BMK1 mediates growth factor-induced cell proliferation through direct cellular activation of serum and glucocorticoid-inducible kinase. J Biol Chem. 2001;276:8631–8634. doi: 10.1074/jbc.C000838200. [DOI] [PubMed] [Google Scholar]
- 19.He XS, Rivkina M, Robinson WS. Construction of adenoviral and retroviral vectors coexpressing the genes encoding the hepatitis B surface antigen and B7-1 protein. Gene. 1996;175:121–125. doi: 10.1016/0378-1119(96)00136-9. [DOI] [PubMed] [Google Scholar]
- 20.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirano T. Interleukin 6 and its receptor: ten years later. Int Rev Immunol. 1998;16:249–284. doi: 10.3109/08830189809042997. [DOI] [PubMed] [Google Scholar]
- 22.Jiang Y, Chen C, Li Z, Guo W, Gegner JA, Lin S, Han J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38β) J Biol Chem. 1996;271:17920–17926. doi: 10.1074/jbc.271.30.17920. [DOI] [PubMed] [Google Scholar]
- 23.Jiang Y, Gram H, Zhao M, New L, Gu J, Feng L, Di Padova F, Ulevitch RJ, Han J. Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem. 1997;272:30122–30128. doi: 10.1074/jbc.272.48.30122. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi T, Deak M, Morrice N, Cohen P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J. 1999;344:189–197. [PMC free article] [PubMed] [Google Scholar]
- 25.Komoda H, Tanaka Y, Honda M, Matsuo Y, Hazama K, Takao T. Interleukin-6 levels in colorectal cancer tissues. World J Surg. 1998;22:895–898. doi: 10.1007/s002689900489. [DOI] [PubMed] [Google Scholar]
- 26.Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999;1:94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- 27.Lee G, Piquette-Miller M. Influence of IL-6 on MDR and MRP-mediated multidrug resistance in human hepatoma cells. Can J Physiol Pharmacol. 2001;79:876–884. [PubMed] [Google Scholar]
- 28.Leong ML, Maiyar AC, Kim B, O’Keeffe BA, Firestone GL. Expression of the serum- and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem. 2003;278:5871–5882. doi: 10.1074/jbc.M211649200. [DOI] [PubMed] [Google Scholar]
- 29.Lichtenstein A, Tu Y, Fady C, Vescio R, Berenson J. Interleukin-6 inhibits apoptosis of malignant plasma cells. Cell Immunol. 1995;162:248–255. doi: 10.1006/cimm.1995.1076. [DOI] [PubMed] [Google Scholar]
- 30.Mizuno H, Nishida E. The ERK MAP kinase pathway mediates induction of SGK (serum- and glucocorticoid-inducible kinase) by growth factors. Genes Cells. 2001;6:261–268. doi: 10.1046/j.1365-2443.2001.00418.x. [DOI] [PubMed] [Google Scholar]
- 31.Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol. 2004;15:161–170. doi: 10.1016/j.semcdb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 32.Naray-Fejes-Toth A, Fejes-Toth G, Volk KA, Stokes JB. SGK is a primary glucocorticoid-induced gene in the human. J Steroid Biochem Mol Biol. 2000;75:51–56. doi: 10.1016/s0960-0760(00)00136-9. [DOI] [PubMed] [Google Scholar]
- 33.Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 34.Okada K, Shimizu Y, Nambu S, Higuchi K, Watanabe A. Interleukin-6 functions as an autocrine growth factor in a cholangiocarcinoma cell line. J Gastroenterol Hepatol. 1994;9:462–467. doi: 10.1111/j.1440-1746.1994.tb01275.x. [DOI] [PubMed] [Google Scholar]
- 35.Park J, Gores GJ, Patel T. Lipopolysaccharide induces cholangiocyte proliferation via an interleukin-6-mediated activation of p44/p42 mitogen-activated protein kinase. Hepatology. 1999;29:1037–1043. doi: 10.1002/hep.510290423. [DOI] [PubMed] [Google Scholar]
- 36.Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999;18:3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park J, Tadlock L, Gores GJ, Patel T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology. 1999;30:1128–1133. doi: 10.1002/hep.510300522. [DOI] [PubMed] [Google Scholar]
- 38.Pramanik R, Qi X, Borowicz S, Choubey D, Schultz RM, Han J, Chen G. p38 isoforms have opposite effects on AP-1-dependent transcription through regulation of c-Jun. The determinant roles of the isoforms in the p38 MAPK signal specificity. J Biol Chem. 2003;278:4831–4839. doi: 10.1074/jbc.M207732200. [DOI] [PubMed] [Google Scholar]
- 39.Sakoda H, Gotoh Y, Katagiri H, Kurokawa M, Ono H, Onishi Y, Anai M, Ogihara T, Fujishiro M, Fukushima Y, Abe M, Shojima N, Kikuchi M, Oka Y, Hirai H, Asano T. Differing roles of Akt and serum- and glucocorticoid-regulated kinase in glucose metabolism, DNA synthesis, and oncogenic activity. J Biol Chem. 2003;278:25802–25807. doi: 10.1074/jbc.M301127200. [DOI] [PubMed] [Google Scholar]
- 40.Shelly C, Herrera R. Activation of SGK1 by HGF, Rac1 and integrin-mediated cell adhesion in MDCK cells: PI-3K-dependent and -independent pathways. J Cell Sci. 2002;115:1985–1993. doi: 10.1242/jcs.115.9.1985. [DOI] [PubMed] [Google Scholar]
- 41.Szczepek AJ, Belch AR, Pilarski LM. Expression of IL-6 and IL-6 receptors by circulating clonotypic B cells in multiple myeloma: potential for autocrine and paracrine networks. Exp Hematol. 2001;29:1076–1081. doi: 10.1016/s0301-472x(01)00682-8. [DOI] [PubMed] [Google Scholar]
- 42.Tadlock L, Patel T. Involvement of p38 mitogen-activated protein kinase signaling in transformed growth of a cholangiocarcinoma cell line. Hepatology. 2001;33:43–51. doi: 10.1053/jhep.2001.20676. [DOI] [PubMed] [Google Scholar]
- 43.Tadlock L, Yamagiwa Y, Marienfeld C, Patel T. Double-stranded RNA activates a p38 MAPK-dependent cell survival program in biliary epithelia. Am J Physiol Gastrointest Liver Physiol. 2003;284:G924–G932. doi: 10.1152/ajpgi.00355.2002. [DOI] [PubMed] [Google Scholar]
- 44.Waldegger S, Barth P, Raber G, Lang F. Cloning and characterization of a putative human serine/threonine protein kinase transcriptionally modified during anisotonic and isotonic alterations of cell volume. Proc Natl Acad Sci USA. 1997;94:4440–4445. doi: 10.1073/pnas.94.9.4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waldegger S, Gabrysch S, Barth P, Fillon S, Lang F. h-sgk serine-threonine protein kinase as transcriptional target of p38/MAP kinase pathway in HepG2 human hepatoma cells. Cell Physiol Biochem. 2000;10:203–208. doi: 10.1159/000016351. [DOI] [PubMed] [Google Scholar]
- 46.Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL. Characterization of sgk, a novel member of the serine/threonine protein kinase gene family, which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol. 1993;13:2031–2040. doi: 10.1128/mcb.13.4.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64:1757–1764. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- 48.Yamagiwa Y, Marienfeld C, Meng F, Holcik M, Patel T. Translational regulation of XIAP by interleukin-6: a novel mechanism of tumor cell survival. Cancer Res. 2004;64:1293–1298. doi: 10.1158/0008-5472.can-03-2517. [DOI] [PubMed] [Google Scholar]
- 49.Yamagiwa Y, Marienfeld C, Tadlock L, Patel T. Translational regulation by p38 mitogen-activated protein kinase signaling during human cholangiocarcinoma growth. Hepatology. 2003;38:158–166. doi: 10.1053/jhep.2003.50257. [DOI] [PubMed] [Google Scholar]