


Abstract
Dpb11 is required for the loading of DNA polymerases α and ɛ on to DNA in chromosomal DNA replication and interacts with the DNA damage checkpoint protein Ddc1 in Saccharomyces cerevisiae. The interaction between the homologs of Dpb11 and Ddc1 in human cells and fission yeast is thought to reflect their involvement in the checkpoint response. Here we show that dpb11-1 cells, carrying a mutated Dpb11 that cannot interact with Ddc1, are defective in the repair of methyl methanesulfonate (MMS)-induced DNA damage but not in the DNA damage checkpoint at the permissive temperature. Epistatic analyses suggested that Dpb11 is involved in the Rad51/Rad52-dependent recombination pathway. Ddc1 as well as Dpb11 were required for homologous recombination induced by MMS. Moreover, we found the in vivo association of Dpb11 and Ddc1 with not only the HO-induced double-strand break (DSB) site at MAT locus but also the donor sequence HML during homologous recombination between MAT and HML. Rad51 was required for their association with the HML donor locus, but not with DSB site at the MAT locus. In addition, the association of Dpb11 with the MAT and HML locus after induction of HO-induced DSB was dependent on Ddc1. These results indicate that, besides the involvement in the replication and checkpoint, Dpb11 functions with Ddc1 in the recombination repair process itself.
INTRODUCTION
DNA replication in eukaryotic cells is carried out by three essential DNA polymerases, Polα, Polδ and Polɛ, which are loaded onto chromatin at replication origins by a number of loading factors, including Dpb11 (1). DPB11 was first cloned as a multi-copy suppressor of a temperature-sensitive mutant of the DNA polymerase ɛ subunit, Dpb2, and acts as a suppressor of certain mutants of the catalytic subunit (Pol2) of Polɛ, which is essential for DNA replication in eukaryotic cells (2). One of the initial steps in eukaryotic chromosomal DNA replication is the assembly of minichromosome maintenance (MCM) proteins into pre-replicative complexes (pre-RCs) at the end of mitosis (3). The association of Dpb11 with origins of replication is dependent on the presence of certain components of the MCM complex, RPA, and Dpb2, but not Polα-primase. Dpb11 co-immunoprecipitates with Polɛ from S-phase cell extracts, but only weakly from G1 cell extracts (4). After replication origin firing, the MCM complex dissociates from origins and translocates to neighboring DNA sites with kinetics that resemble those of replication fork progression (4). In one temperature sensitive dpb11 mutant, dpb11-1, the MCM complex binds to origin DNA but is unable to dissociate at the restrictive temperature. RPA binds to origin DNA independently of Dpb11 function, but neither Polɛ nor Polα is loaded on to origin DNA in dpb11-1 mutants at the non-permissive temperature (4). Thus, although pre-RCs containing MCM and RPA are formed in dpb11-1 mutants, DNA synthesis is not initiated in these cells.
Dpb11 is associated with Sld2, which is a substrate for the cyclin-dependent kinase (CDK1) in Saccharomyces cerevisiae (5,6). The phosphorylation of multiple S/T residues in Sld2 at the onset of replication initiation increases Sld2 affinity for Dpb11 and is necessary for the initiation of DNA replication (6). Thus, Dpb11 performs its DNA polymerase loading function at origins by forming a complex with Sld2.
Besides its function in the initiation of DNA replication, Dpb11 also plays a role in the DNA replication checkpoint (2,4). In addition, Dpb11 interacts with Ddc1 of budding yeast (7), which is one of the components of the Ddc1–Rad17–Mec3 complex involved in the DNA damage checkpoint (8). The Ddc1–Rad17–Mec3 complex is structurally related to proliferating cell nuclear antigen (PCNA), ‘sliding clamp’, whose homotrimeric structure allows it to encircle DNA (9,10). Rad24–Rfc2-5, which is also required for the DNA damage checkpoint (11), loads the Ddc1–Mec3–Rad17 complex onto damaged sites on DNA in a manner analogous to that of RFC-mediated loading of PCNA onto sites of DNA replication (12–14). Interestingly, fission yeast Rad9 and human RAD9, which are the homologs of budding yeast Ddc1, interact with the Dpb11 homologs, fission yeast Cut5 and human TopBP1, respectively (15,16). Thus, this interaction has been conserved throughout evolution. The interaction is dependent on the phosphorylation of Rad9 in fission yeast, and is required for both checkpoint signaling and DNA metabolism (15). Similar to its yeast counterpart, human TopBP1 is required for cell survival, DNA replication, DNA damage checkpoint and transcriptional regulation (17). However, the meaning of the interaction between Dpb11 and Ddc1 has remained obscure in budding yeast.
The dpb11-1 mutant is sensitive to methyl methanesulfonate (MMS) and ultraviolet (UV) light even at the permissive temperature (2), although the mutant is proficient in the G2/M DNA damage checkpoint following UV irradiation at the non-permissive temperature (7). This suggests that Dpb11 is involved in the DNA repair process itself, although little is known about what this function might be. To elucidate the repair function of Dpb11, we performed a number of experiments with the dpb11-1 mutant of S.cerevisiae, whose gene product fails to interact with Ddc1 (7).
MATERIALS AND METHODS
Yeast strains
Yeast strains used in this study were listed in Supplementary Table 1. The dpb11-1 mutant was constructed by two-step gene replacement using the PstI fragment of the plasmid YIplac121dpb11-1ΔN, which was provided by Dr H. Araki. Complementation of the thermosensitivity of the dpb11-1 mutant was confirmed by transforming a single-copy plasmid carrying the wild-type DPB11 gene, which was also provided by Dr H. Araki. The apn1::hisG-URA3-hisG was constructed by one-step gene replacement using the BamHI–EcoRI fragments of plasmids pSCP108 (these plasmids were from Dr B. Demple). Complete null mutants, Myc-tagged and HA-tagged alleles were made by standard PCR-based gene disruption and insertion methods (18–20). Gene disruption was confirmed by genomic PCR. Some double-mutant strains were isolated by tetrad dissection. Complete sequences of the primers used for the construction of DNA used to disrupt an appropriate gene or to check a disruption will be provided upon request, along with the details of the strains. All analyses were performed at 23°C (i.e. at the permissive temperature of the dpb11-1 mutant) unless otherwise indicated.
Analysis of MMS and UV sensitivity
Strains with the MR93-28c background were constructed. Logarithmically growing cells were serially diluted by 10-fold with distilled water and spotted onto YPAD plates or YPAD plates containing the indicated concentrations of MMS, or irradiated with the indicated doses of UV light. The plates were incubated for 4 days and photographed. Alternatively, logarithmically growing cells were diluted to 107 cells/ml and cultured in the presence of 15 μg/ml nocodazole for 200 min, which synchronized them in the G2/M phase. The G2/M-arrested cells were exposed to 0.1% MMS for 1 h, washed to remove MMS, and then cultured in YPAD medium with or without nocodazole. At specific intervals, the cells were washed to remove nocodazole, and then diluted and inoculated onto YPAD plates.
Assay of the frequency of interchromosomal homologous recombination between heteroalleles
Strains with the MR101 background were constructed such that recombination between the heteroalleles his1-1 and his1-7 in a diploid could be detected by the restoration of histidine prototrophy. As described previously (21), logarithmically growing cells were inoculated onto SC-His plates containing various concentrations of MMS and on to YPAD plates to evaluate survival. When UV-induced recombination was assayed, cells were inoculated onto SC-His plates and onto YPAD plates, and irradiated with given doses of UV. After 4 days incubation, colonies were enumerated. The numbers of His+ colonies per 106 survivors are indicated. The data show a typical result of at least three independent experiments, and line bars indicate the standard deviation.
Pulsed-field gel electrophoresis
Logarithmically growing cells were diluted to 107 cells/ml and cultured in the presence of 15 μg/ml nocodazole for 200 min, which synchronized them in the G2/M phase. Cells arrested in the G2/M phase were exposed to 0.1% MMS for 1 h and were then washed to remove MMS and cultured in YPAD medium containing nocodazole for specific periods. Agarose plugs of chromosomal DNA were prepared following the protocol described previously (22). After electrophoresis, the gel was stained with 0.5 μg/ml ethidium bromide for 30 min, destained in deionized water for 20 min, and photographed.
G2/M phase DNA damage checkpoint
Logarithmically growing cells were arrested with nocodazole and treated with 0.3% MMS for 30 min. At the indicated times after release from nocodazole treatment, the percentage of uninucleate large budded cells was scored by DAPI staining. To detect eventual modification of Rad53-13Myc and Chk1-13Myc in response to DNA damage, cells were arrested in the G2/M phase in YPAD medium containing nocodazole for 3 h, after which 0.2% MMS was added. After 2.5 h, the cells were harvested for immunoblotting analysis. The cells were incubated at 30°C for 5 min in 100 mM K-EDTA and 10 mM DTT, followed by incubation at 30°C for 5 min in 0.6 M sorbitol, 50 mM KH2PO4/K2HPO4, 10 mM DTT and 2.5 mg/ml zymolyase 100T. The cells were washed twice with wash buffer (0.4 M sorbitol, 100 mM KCl, 2.5 mM MgCl, 50 mM HEPES–KOH, 1 mM phenylmethlysulfonyl fluoride and protease inhibitors). A total of 50 μl of the cell pellet was resuspended in 900 μl of wash buffer and the cells were lysed by the addition of 1 vol of lysis buffer (wash buffer with 2% Triton X-100). Lysis was performed on ice for 10 min, after which SDS–polyacrylamide sample buffer was added. For western blot analysis, equivalent volumes of whole cell extract (WCE) were loaded. The proteins were detected by immunoblotting with anti-Myc.
Chromatin immunoprecipitation (ChIP)
ChIP was carried out as described previously with minor modifications (23,24). Asynchronous cultures were grown overnight at 30°C in YP medium containing 2% raffinose. When cultures reached logarithmic phase, expression of HO endonuclease was induced by adding 2% galactose. The cultures were washed to remove galactose 1 h after the addition of galactose and treated with 2% glucose to repress the expression of HO endonuclease. Cells were harvested, and proteins were cross-linked to DNA with 1% formaldehyde for 20 min, followed by quenching with glycine at a final concentration of 125 mM for 5 min. The cells were lysed with glass beads, and the extracts were sonicated to shear DNA to an average size of 1 kb. Extracts were then divided into immunoprecipitate (IP) and input samples (20:1 ratio). Immunoprecipitation of cross-linked DNA was carried out with a monoclonal anti-Myc antibody (9E10) (Santa Cruz Biotechnology, Inc.) and Dynabeads Protein G (Dynal Biotech) for 4 h at 4°C. After a series of wash of the protein-bound beads, proteins were released from beads by reversal of cross-linking (6 h at 65°C). The samples were treated with proteinase K, and DNA for PCR was prepared by phenol extraction and ethanol precipitation.
PCR amplification
To amplify immunoprecipitated DNA, primers, P1 (5′-TCCCCATCGTCTTGCTCT-3′) and P2 (5′-GCATGGGCAGTTTACCTTTAC-3′), which span the HO recognition site, or P1 (5′-TCCCCATCGTCTTGCTCT-3′) and P3 (5′-CCCAAGGCTTAGTATACACATCC-3′), which span the HMLα locus were used in combination with primers, SMC2-FW1 (5′-GACGACCTTGTAACAGTCCAGACAG-3′) and SMC2-RV1 (5′-GGCGAATTCCATCACATTATACTAACTACGG-3′), which anneal to the SMC2 locus used as the control. PCR products were separated by agarose gel electrophoresis, and the amount of the product was determined using NIH Image. Since the IP efficiencies of different loci were often very different, the signal from the interested locus was first normalized with the signal from an independent locus (SMC2) on chromosome VI, namely, by dividing each MAT or HML IP signal by the corresponding SMC2 IP signal. Then, MAT or HML IP signals at later time points were normalized to the IP signal at 0 h as 1. ChIP data are representative of at least three independent experiments.
Detection of DSB induction and strand invasion
Double-strand break (DSB) induction (MATa locus) and strand invasion were detected by PCR on input DNA. The primers used for detection of DSB and strand invasion were 5′-CTTTTAGTTTCAGCTTTCCG-3′ (pI)/5′-ACTCTATAAGGCCAAATGTACAAAC-3′ (pJ) and 5′-GCAGCACGGAATATGGGACT-3′ (pA)/5′-ATGTGAACCGCATGGGCAGT-3′ (pB), respectively. PCR products were separated by agarose gel electrophoresis. For quantification, the signal from the interested locus was normalized with the signal from an independent locus (SMC2) on chromosome VI.
RESULTS
Dpb11 is required for DNA repair
The dpb11-1 mutant is sensitive to MMS and UV light even at the permissive temperature (2), although the mutant is proficient in the G2/M DNA damage checkpoint following UV irradiation at the non-permissive temperature (7). This suggests that Dpb11 is involved in the DNA repair process itself, although little is known about what this function might be.
To elucidate the repair function of Dpb11, we performed a number of experiments with the dpb11-1 mutant. DPB11 is essential for cell growth and a mutant allele, dpb11-1, causes cell death at non-permissive temperatures (2). All assays in this study were performed at the permissive temperature since dpb11-1 shows an S-phase defect at non-permissive temperatures (2). We first examined whether Dpb11 is required for the repair of MMS-induced DNA damage in G2/M-arrested cells to avoid any complications due to the S-phase defect of the dpb11-1 mutant. When cells arrested in the G2/M phase by nocodazole were treated with MMS and then cultured in MMS-free medium containing nocodazole to maintain their G2/M-arrested state, the distinctive bands corresponding to each chromosome disappeared upon exposure to MMS in both wild-type and dpb11-1 cells and a low-molecular weight smear due to damaged DNA appeared instead (Figure 1A). The restoration of chromosome-sized DNA bands occurred in both wild-type and dpb11-1 cells after culture in MMS-free medium; however, the restoration occurred less efficiently in dpb11-1 cells than in wild-type cells (Figure 1A). Indeed, dpb11-1 cells arrested in the G2/M phase showed higher sensitivity to MMS than wild-type cells (Figure 1B). We clarified whether the G2/M DNA damage checkpoint is intact in dpb11-1 cells by monitoring their mitotic division following exposure to MMS. As shown in Figure 1C, wild-type and dpb11-1 cells showed delayed nuclear division while the rad24 mutant, which has a defective DNA damage checkpoint, proceeded through mitosis faster than wild-type cells. Thus, the checkpoint function of dpb11-1 cells remains intact upon exposure to MMS (Figure 1C), as it does upon exposure to UV (7). To confirm this, we examined the phosphorylation of Rad53 and Chk1, which are involved in the damage checkpoint and are phosphorylated during its activation (25). Phosphorylated forms of Rad53 and Chk1 appeared in G2/M-arrested dpb11-1 cells as well as in wild-type cells upon exposure to MMS (Figure 1D). These results indicate that Dpb11 is itself required for DNA repair.
Figure 1.
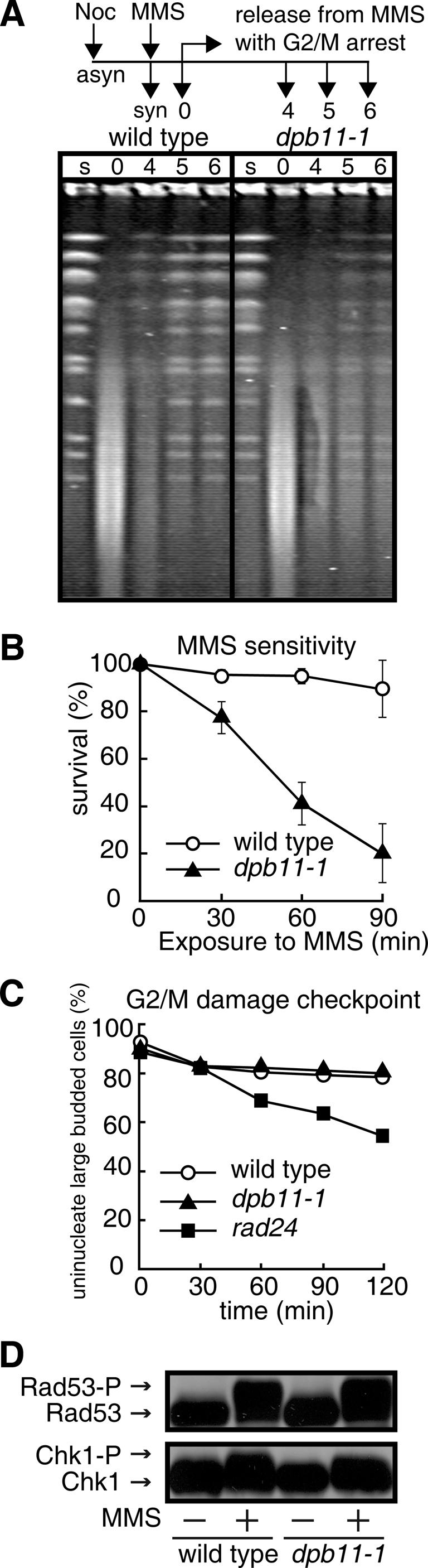
Involvement of Dpb11 in DNA repair. (A) Pulsed-field gel electrophoresis of chromosomal DNA. Cells (wild type, MR101; dpb11-1, YHO201) arrested in G2/M phase by nocodazole were exposed to 0.1% MMS at 23°C for 1 h, washed to remove MMS, and cultured in MMS-free medium containing nocodazole for the indicated periods to allow them to repair the damage to their DNA. The cells were then harvested and their DNA was analyzed by PFGE as described in the Materials and Methods. Lane s, G2/M-arrested cells; lane 0, cells treated with MMS for 1 h; lanes 4–6, cells treated with MMS for 1 h and cultured in MMS-free medium for 4, 5 and 6 h, respectively. (B) Sensitivity to MMS of cells arrested in G2/M. Cells (wild type, MR101; dpb11-1, YHO201) arrested in G2/M were exposed to 0.1% MMS for the indicated periods, washed to remove MMS, and inoculated onto YPAD plates containing neither MMS nor nocodazole. (C) Induction of the G2/M DNA damage checkpoint in response to MMS exposure. Cells (wild type, MR101; dpb11-1, YHO201; rad24, YHO210) arrested in the G2/M phase were exposed to 0.3% MMS for 30 min and washed to remove MMS and nocodazole. The cells were incubated in medium containing neither MMS nor nocozadole for the periods indicated. (D) Modification of Rad53 and Chk1 in response to DNA damage. Wild type (YHO302) and dpb11-1 (YHO304) cells expressing Rad53-13Myc (upper panel) or wild type (YHO303) and dpb11-1 (YHO305) cells expressing Chk1-13Myc (lower panel) were arrested in G2/M phase in YPAD medium containing nocodazole at 23°C and then exposed to 0.2% MMS for 2.5 h. The cells were then harvested and proteins prepared from these cells were subjected to SDS–PAGE. Rad53 and Chk1 were detected by western blotting using anti-Myc antibody.
Dpb11 is involved in MMS-induced recombination repair
We next performed an epistatic analysis of dpb11-1 and mutants of various repair pathways involved in base excision repair (BER: apn1), nucleotide excision repair (NER: rad1), post-replication-repair (PRR: rad18) and homologous recombination repair (HR: rad52). The double mutants apn1 dpb11-1, rad1 dpb11-1 and rad18 dpb11-1 showed an additive or synergistic sensitivity to MMS and UV compared with the corresponding single mutants (Figure 2A), suggesting that Dpb11 functions in a pathway other than BER, NER and PRR. However, the sensitivity to MMS of rad52 dpb11-1 cells was not additive to that of rad52 cells, while their sensitivity to UV was greater than that of either single mutant. Although the above results seem to indicate the involvement of Dpb11 in the Rad52 repair pathway, this notion is not firmly supported by the low sensitivity of dpb11-1 cells to the concentration of MMS employed. To confirm that Dpb11 actually functions in the Rad52-dependent recombination repair, we examined whether Dpb11 is involved in MMS-induced interchromosomal recombination between the heteroallelic loci his1-1 and his1-7 in diploid dpb11-1 cells. In wild-type cells, the recombination frequency was significantly increased by exposure to MMS or UV. The induction of UV-induced recombination in dpb11-1 cells is at the same level as that of wild-type cells (Figure 2C). However, the induction level of recombination in dpb11-1 cells upon exposure to MMS was low compared with that of wild-type cells (Figure 2B). Taken together, it seems likely that Dpb11 is required for MMS-induced interchromosomal recombination.
Figure 2.

Involvement of Dpb11 in the homologous recombination repair pathway. (A) Epistatic analyses of the effect of dpb11-1 and mutations in various repair pathways on the sensitivity to MMS and UV. apn1, base excision repair; rad1, nucleotide excision repair; rad18, post-replication repair; rad52, homologous recombination repair. (B) MMS-induced interchromosomal recombination. [The viability of the cells cultured with 0.008% MMS was 98 and 92% for wild-type (MR101) and dpb11-1 (YHO201) cells, respectively.] (C) UV-induced interchromosomal recombination. [The viability of cells at a UV dose of 30 J/m2 was 97 and 70% for wild-type (MR101) and dpb11-1 (YHO201) cells, respectively.]
Homologous recombination mediated by Rad52, as well as by Rad51, Rad54, Rad55 and Rad57, is the dominant pathway for the repair of DSBs in S.cerevisiae. On the other hand, DSBs can be also repaired by Rad52-dependent recombination pathway requiring Rad59 function (26–29). We, therefore, carried out another epistatic analysis with dpb11-1 and rad51 or rad59 in cells exposed to MMS. The dpb11-1 mutation increased the MMS sensitivity of the rad59 mutant (Figure 3A) but not of the rad51 mutant (Figure 3B). We further examined the relationship between Dpb11 and Rad51, Rad52, or Rad59 in MMS-induced interchromosomal recombination. The frequency of MMS-induced recombination in the dpb11-1 rad59 mutant was lower than those of the single mutants, unlike dpb11-1 rad51 and dpb11-1 rad52 mutants (Figure 3C). These results suggest that Dpb11 plays a role in recombination repair and is involved in the Rad52/Rad51-dependent homologous recombination pathway upon exposure to MMS.
Figure 3.

Involvement of Dpb11 in damage-induced interchromosomal recombination. (A) Epstatic analysis of the effect of dpb11-1 and rad59 on the sensitivity to MMS. (B) Epstatic analysis of the effect of dpb11-1 and rad51 on the sensitivity to MMS. (C) MMS-induced recombination frequency in various mutants. Wild type (MR101), dpb11-1 (YHO201), rad59 (YHO206) and rad59 dpb11-1 (YHO207), rad51 (YHO204), rad51 dpb11-1 (YHO205), rad52 (YHO202), rad52 dpb11-1 (YHO203) cells. [The viability of the cells cultured with 0.001% MMS was 98, 95, 89, 71, 89, 91, 71 and 71% for wild type, dpb11-1, rad59, rad59 dpb11-1, rad51, rad51 dpb11-1, rad52 and rad52 dpb11-1 cells, respectively.] Cells were inoculated onto YPAD plates or SC plates lacking His that contained the indicated concentrations of MMS and then incubated at 23°C for 4 days. The numbers of His+ colonies per 106 survivors are indicated and the data shown are the typical results of at least three independent experiments. The line bars indicate standard deviations.
Ddc1, which interacts with Dpb11, is also required for the recombination repair
Dpb11 physically interacts with Ddc1 (7), which is involved in the damage checkpoint (8). However, dpb11-1 encodes a protein that is unable to interact with Ddc1 (7). The biological significance of the interaction between Dpb11 and Dcd1 is not clear at present but it is worth noting that the fission yeast and human homologs of Dpb11 (Cut5 and TopBP1, respectively) also interact with their Ddc1 counterpart (Rad9) (15,16). To examine whether Ddc1 mediates the repair function of Dpb11, we monitored DNA repair in G2/M phase-arrested ddc1 cells (to avoid the deficient of checkpoint function) as well as the frequency of MMS-induced interchromosomal recombination. As in dpb11-1 cells, the repair of DNA damages induced by MMS was considerably impaired in ddc1 cells compared with wild-type cells (Figure 4A), suggesting that Ddc1 is also required for the repair of MMS-induced DNA damages.
Figure 4.

Involvement of the checkpoint clamp Ddc1 and the checkpoint clamp loader Rad24 in the same recombination repair pathway as Dpb11. (A) Pulsed-field gel electrophoresis of chromosomal DNA. Wild type (MR101), ddc1 (YHO208), rad24 (YHO210) cells arrested in G2/M were treated with 0.1% MMS at 23°C for 1 h, washed to remove MMS, and cultured in MMS-free medium containing nocodazole for the indicated periods to be allowed to repair their damaged DNA. The cells were then harvested and their DNA was analyzed by PFGE as described in Materials and Methods. Lane s, G2/M-arrested cells; 0, cells treated with MMS for 1 h; lanes 4–6, cells treated with MMS for 1 h and cultured in MMS-free medium for 4, 5 and 6 h, respectively. (B) MMS-induced interchromosomal recombination. [The viability of the cells cultured with 0.004% MMS was 98, 61 and 66% for wild type (MR101), ddc1 (YHO208), rad24 (YHO210), respectively.] (C) MMS-induced interchromosomal recombination in wild-type, ddc1, rad24, ddc1 rad24 cells. [The viability of the cells cultured with 0.001% MMS were 98, 94, 95 and 94% for wild-type (MR101), ddc1 (YHO208), rad24 (YHO210) and ddc1 rad24 (YHO211) cells, respectively.] (D) MMS-induced interchromosomal recombination in wild-type, dpb11-1, ddc1 and ddc1 dpb11-1 cells. [The viability of cells cultured with 0.001% MMS was 98, 95, 94 and 88% for wild-type (MR101), dpb11-1 (YHO201), ddc1 (YHO208) and ddc1 dpb11-1 (YHO209) cells, respectively.] (B–D) The numbers of His+ colonies per 106 survivors are indicated and the data shown are the typical results of at least three independent experiments. The line bars indicate standard deviations.
This reveals that the Ddc1–Mec3–Rad17 checkpoint clamp is directly involved in DSB repair. We also examined whether Ddc1 is required for recombination repair as well as Dpb11. As shown in Figure 4B, MMS-induced recombination was severely affected in ddc1 cells compared with wild-type cells. It is known that the Ddc1–Mec3–Rad17 and Rad24–RFC2-5 (RAD24–RFC) complexes function in a DNA damage checkpoint pathway (9,10). Moreover, it is known that Ddc1–Mec3–Rad17 shares sequence and structural similarities with the DNA polymerase sliding clamp, PCNA while Rad24–RFC shares sequence and structural similarities with the PCNA clamp loader (replication factor C complex RFC1-5) (30). Therefore, Ddc1–Mec3–Rad17 and Rad24–RFC are referred to as the checkpoint clamp and checkpoint clamp loader, respectively. Similar to the ddc1 mutant, the rad24 mutant showed less efficient DNA repair (Figure 4A) and a defect in the induction of homologous recombination upon exposure to MMS (Figure 4B). Similar results were obtained with the mec3 and rad17 mutants (data not shown). To elucidate the relationship between Ddc1 and Rad24 in MMS-induced interchromosomal recombination, we measured the recombination frequencies in various mutants. The recombination frequency in ddc1 rad24 double mutants was almost the same as that in ddc1 or rad24 single mutants (Figure 4C). These results indicated that the Ddc1–Mec3–Rad17 complex functions in the same pathway as Rad24–RFC2-5 (RAD24–RFC) in the recombination repair as well as the checkpoint response.
Ddc1 functions in the same recombination pathway as Dpb11
Dpb11 and Ddc1 physically interact with each other (7). We showed each of Dpb11 and Ddc1 is involved in recombination repair (Figures 2B and 4B). We further confirmed the relationship of Dpb11 and Ddc1 in recombination repair. dpb11-1 and ddc1 cells showed a reduced frequency of recombination compared with wild-type cells, but the frequency of MMS-induced recombination in ddc1 dpb11-1 cells was reduced to the same level as that in ddc1 cells (Figure 4D), indicating that DPB11 is epistatic with DDC1 in the MMS-induced recombination repair pathway. These results indicated that the Dpb11-interacting protein Ddc1 plays a role in facilitating recombination repair in addition to its DNA damage checkpoint function.
Dpb11 and Ddc1 are recruited onto not only the recipient MAT locus but also the donor HML locus upon induction of DSB by HO endonuclease
One of the best studied homologous recombination events is the HO endonuclease-induced MAT switching, using HML as the donor template (31). Introduction of a galactose-inducible GAL::HO gene into cells provides the means to induce DSB synchronously in all cells of the population. It has been shown that the DSB end of MAT DNA produced by HO endonuclease is resected by 5′−3′ exonucleases (32) to make a 3′ ended ssDNA, which recruits the Rad51-strand exchange protein, and the DNA–protein complex invades into donor DNA. Thus, Rad51 associates not only with the HO-cut MAT locus but also the HML donor strand (23,24,33,34). To confirm the involvement of Dpb11 and Ddc1 in recombination repair, we used this system. DSB formation at the HO recognition site and invasion of the MAT recipient strand into the donor strand (HML) were monitored by PCR using the primers shown in Figure 5A. The amount of PCR product of MATa locus was considerably decreased 1 h after induction of endonuclease, i.e. cleavage at the MAT locus occurred within 1 h with high efficiency, and strand invasion was detected 2 and 4 h after induction (Figure 5B). Association of Ddc1 and Dpb11 with the MAT locus and the HML donor strand was monitored by ChIP assay using primers shown in Figure 5C. As reported previously (35,36), the association of Ddc1 to the MAT locus was observed 1 h after induction of HO endonucelease when DSB was formed (Figure 5B and D). Importantly, Ddc1 also associated with the HML donor strand (Figure 5D). Similar results were obtained with Dpb11, showing the association of Dpb11 with the MAT locus and the HML donor strand (Figure 5E).
Figure 5.

Recruitment of Dpb11 and Ddc1 at HML locus as well as the DSB site during DSB-induced gene conversion in vivo. JKM161 background strains carrying the HMLα donor sequence were treated with 2% galactose to induce expression of HO endonuclease, and then with 2% glucose after 1 h to repress HO endonuclease. Chromatin prepared from Ddc1-13Myc (wild-type; YHO401, rad51 null mutants; YHO402) and Dpb11-13Myc (wild-type, YHO403; rad51 null mutants, YHO404; ddc1 null mutants, YHO405) cells at the indicated time points was immunoprecipitated with anti-Myc antibody. (A) Schematic representation of the postsynaptic complex during HO endonuclease-induced recombination and illustration for the design of PCR primers used in the experiment B. (B) Detection of DSB formation and strand invasion intermediates upon HO endonuclease induction. DNA was purified from the cells (wild type, YHO403; rad51 cells, YHO404; ddc1 cells, YHO405) transiently expressing HO endonuclease, and PCR was performed using primers that flank the MATa locus to detect DSB at the site (pI and pJ). Strand invasion step was monitored by PCR using a primer distal to MAT and a primer within the Yα sequence in HML (pA and pB). Primers specific to the SMC2 locus were included in the PCR as a control. (C) Diagram of MAT and HML showing the location of primers for ChIP experiments used in the experiments D and E. The primers 189 and 483 bp distal to the DSB site at MAT-Z (P1 and P2) and 189 and 467 bp from the uncleaved HO recognition site at HML (P1 and P3) were used to amplify immunoprecipitated DNA by PCR to detect association of Ddc1 and Dpb11 at the DSB site and the HML locus. (D and E) ChIP analysis. DNA fragments were amplified using the primers shown in (D) as MAT (left panel) and HML (right panel) on the DNA template prepared by ChIP with (D) Ddc1-13Myc (wild type, open circle; rad51 mutants, closed triangle) or (E) Dpb11-13Myc (wild type, open circle; rad51 mutants, closed triangle; ddc1 mutants, open square). PCR-amplified SMC2 signals from the same DNA in the same test tubes were used as the loading control. Fold of increase was calculated as described in Materials and Methods. Average values of three independent experiments are shown and error bars indicate the standard deviation.
Rad51 is required for the association of Dpb11 and Ddc1 with the donor HML locus but not the DSB site at MAT locus
Dpb11 and Ddc1 were required for recombination repair (Figures 3 and 4) and associated with the DSB site and the donor strand for recombination repair after induction of DSB (Figure 5). Next, we examined whether the association of Dpb11 and Ddc1 onto the DSB site and the donor strand is dependent on the recombination protein Rad51. In rad51 null mutant cells, the cleavage at the MAT locus occurred within 1 h with high efficiency, but strand invasion was not detected even 4 h after induction of HO endonuclease (Figure 5B). The association of Ddc1 and Dpb11 with the DSB site at MAT locus was not affected in rad51 mutant cells (Figure 5D and E). However, Dpb11 and Ddc1 did not associate with the HML donor strand in rad51 mutant cells (Figure 5D and E). These results suggest that Dpb11 and Ddc1 are recruited to the HML donor strand following Rad51-mediated strand invasion.
Ddc1 is required for the association of Dpb11 with not only the DSB site but also the donor strand
We next examined whether the association of Dpb11 with the damaged DNA is dependent on Ddc1. In ddc1 null mutant cells, the cleavage at the MAT locus occurred within 1 h with high efficiency and strand invasion occurred with similar kinetics to those of wild-type cells (Figure 5B). However, in ddc1 mutant cells, the increase of association of Dpb11 with the MAT and HML locus was not observed after induction of HO endonuclease (Figure 5E). These results indicate that Dpb11 is recruited on to the DSB site and the donor site by Ddc1.
DISCUSSION
In budding yeast, Dpb11 is involved in the initiation of DNA replication and replication checkpoint (2,4). The Ddc1–Rad17–Mec3 complex is involved in the activation of DNA damage checkpoint pathway as a damage sensor (8). The budding yeast Dpb11 interacts with Ddc1, while the meaning of the interaction has remained obscure. Here we show that both Dpb11 and Ddc1 function in the same pathway of MMS-induced recombination repair.
Using the dpb11-1 mutant that expresses the protein truncated the C-terminal region to be incapable of interaction with Ddc1 (2,7), we showed that dpb11-1 cells are not defective in the function of DNA damage checkpoint unlike ddc1 cells (7,8) but both dpb11-1 and ddc1 cells are defective in MMS-induced recombination (Figures 2B and 4B). After the induction of HO endonuclease, Dpb11 and Ddc1 associated with not only the MAT/DSB site but also the HML donor region (Figure 5D and E). Importantly, the association of Dpb11 and Ddc1 with the HML donor region did not occur in rad51 mutants (Figure 5B). These results suggest that Dpb11 and Ddc1 function in some step of recombination repair after strand invasion mediated by Rad51 in the DSB-induced recombination repair process. Thus, we propose that besides the involvement in checkpoint responses, Dpb11 and Ddc1 are also involved in the recombination repair process itself.
How do the checkpoint clamp and the checkpoint clamp loader participate in DSB-induced recombination? In the models of DSB-induced recombination repair, the two ends of DSB are resected by 5′–3′ exonucleases, which allows the 3′ ends to invade an intact homologous template. This process involves DNA-strand exchange proteins such as Rad52 and Rad51, before new DNA synthesis initiates from 3′ ends at 3′-primer/template junctions (37). In vitro study showed that the checkpoint clamp loader has been shown to load the checkpoint clamp onto DNA substrates, including 3′-recessed primer templates in an ATP-dependent manner (14). The checkpoint clamp loader Rad24–RFC loads the checkpoint clamp Ddc1–Mec3–Rad17 at DSB site created in the MAT locus by HO endonuclease (35,36), that is thought to be resulted in the activation of checkpoint response. Interestingly, Lisby et al. (38) reported that Ddc1 foci appear almost simultaneously with Mre11 foci after gamma irradiation. However, in contrast to Mre11 foci they last much longer and the turnover kinetics of Ddc1 foci are similar to the turnover of Rad52 foci. Importantly, we observed that Ddc1 bound to the homologous donor strand depending on Rad51 after induction of DSB (Figure 5D). These results seem to support our notion that Ddc1 as well as Dpb11 are involved in the later step of homologous recombination.
How does Dpb11 participate in recombination? It has been reported that DNA polymerase ɛ and δ are redundantly involved in recombination repair (39). Dpb11 loads DNA polymerase ɛ at origins of DNA replication (4). The CDK1 is important for a latter step in homologous recombination that occurs after strand invasion but before the initiation of DNA synthesis (40). In this context, it is worth noting that Dpb11 forms a complex with the CDK1 phosphorylation substrate, Sld2, and that the Dpb11–Sld2 complex is essential for chromosomal DNA replication (5,6). We show here that Dpb11 associates with the homologous donor strand depending on Rad51. In addition, the association of Dpb11 with the homologous donor strand is also dependent on Ddc1 (Figure 5E). These results suggest that Dpb11 functions in the later step of recombination repair by the aid of Ddc1.
Considering these observations together with our results, in the later step of recombination repair, i.e. after the strand invasion by Rad51, Ddc1–Mec3–Rad17 checkpoint clamp associates with 3′-primer/template junctions of the homologous donor strand, and recruits Dpb11. And then, these proteins may recruit DNA polymerases to the recombination intermediates for DNA polymerization at damaged site. Interestingly, the interaction between Dpb11 and Ddc1 has been conserved throughout evolution. Indeed, fission yeast Rad9 and human RAD9, which are homologs of budding yeast Ddc1, interact with the Dpb11 homologs, fission yeast Cut5 and human TopBP1, respectively (15,16). The interaction with Cut5 is dependent on the phosphorylation of Rad9 in fission yeast, and is required for both checkpoint signaling and DNA metabolism (15). Thus, to date, the interaction between the homologs of Dpb11 and Ddc1 has been thought to reflect their involvement in the checkpoint response (41–43). The results presented here suggest that the interaction between Dpb11 and Ddc1 is required for recombination repair process itself. It is known that efficient homologous recombination contributes to maintenance of genome stability. Recent analysis of physiological function of human TopBP1, which is the homolog of yeast Dpb11, in S-phase by using small interfering RNA, revealed that TopBP1 seems to monitor ongoing DNA replication and play a critical role in the maintenance of genomic stability during normal S-phase as well as following genotoxic stress (44). Taking into account our current data concerning the function of yeast Dpb11, it is interesting to examine whether the human counterpart, TopBP1, is also involved in damage-induced homologous recombination in the future.
Thus, on a more general level, our observations provide a novel insight about genome stability into how proteins involved in replication, repair and checkpoint engage in more intimate crosstalk than previously held up until now.
Acknowledgments
We thank H. Araki, A. Sugino, Y. Kawasaki, M. Arisawa, B. Demple, J. E. Haber and T. Weinert for plasmids or strains used in the study. We thank all members of Enomoto's laboratory. This work was supported by Grants-in-Aid for Scientific Research on Priority Areas from The Ministry of Education, Science, Sports and Culture of Japan, and by Health Sciences Research Grants from the Ministry of Health and Welfare of Japan. Funding to pay the Open Access publication charges for this article was provided by Grants-in-Aid for Scientific Research on Priority Areas from The Ministry of Education, Science, Sports and Culture of Japan.
Conflict of interest statement: None declared.
REFERENCES
- 1.Kawasaki Y., Sugino A. Yeast replicative DNA polymerases and their role at the replication fork. Mol. Cells. 2001;12:277–285. [PubMed] [Google Scholar]
- 2.Araki H., Leem S.H., Phongdara A., Sugino A. Dpb11, which interacts with DNA polymerase II (epsilon) in Saccharomyces cerevisiae, has a dual role in S-phase progression and at a cell cycle checkpoint. Proc. Natl Acad. Sci. USA. 1995;92:11791–11795. doi: 10.1073/pnas.92.25.11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aparicio O.M., Weinstein D.M., Bell S.P. Components and dynamics of DNA replication complexes in S.cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- 4.Masumoto H., Sugino A., Araki H. Dpb11 controls the association between DNA polymerases alpha and epsilon and the autonomously replicating sequence region of budding yeast. Mol. Cell. Biol. 2000;20:2809–2817. doi: 10.1128/mcb.20.8.2809-2817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamimura Y., Masumoto H., Sugino A., Araki H. Sld2, which interacts with Dpb11 in Saccharomyces cerevisiae, is required for chromosomal DNA replication. Mol. Cell. Biol. 1998;18:6102–6109. doi: 10.1128/mcb.18.10.6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masumoto H., Muramatsu S., Kamimura Y., Araki H. S-Cdk-dependent phosphorylation of Sld2 essential for chromosomal DNA replication in budding yeast. Nature. 2002;415:651–655. doi: 10.1038/nature713. [DOI] [PubMed] [Google Scholar]
- 7.Wang H., Elledge S.J. Genetic and physical interactions between DPB11 and DDC1 in the yeast DNA damage response pathway. Genetics. 2002;160:1295–304. doi: 10.1093/genetics/160.4.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Longhese M.P., Paciotti V., Fraschini R., Zaccarini R., Plevani P., Lucchini G. The novel DNA damage-checkpoint protein Ddc1p is phosphorylated periodically during the cell cycle and in response to DNA damage in budding yeast. EMBO J. 1997;16:5216–5226. doi: 10.1093/emboj/16.17.5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green C.M., Erdjument-Bromage H., Tempst P., Lowndes N.F. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr. Biol. 2000;10:39–42. doi: 10.1016/s0960-9822(99)00263-8. [DOI] [PubMed] [Google Scholar]
- 10.Kondo T., Matsumoto K., Sugimoto K. Role of a complex containing Rad17, Mec3, and Ddc1 in the yeast DNA damage-checkpoint pathway. Mol. Cell. Biol. 1999;19:1136–1143. doi: 10.1128/mcb.19.2.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinert T.A., Kiser G.L., Hartwell L.H. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev. 1994;6:652–665. doi: 10.1101/gad.8.6.652. [DOI] [PubMed] [Google Scholar]
- 12.Bermudez V.P., Lindsey-Boltz L.A., Cesare A.J., Maniwa Y., Griffith J.D., Hurwitz J., Sancar A. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl Acad. Sci. USA. 2003;100:1633–1638. doi: 10.1073/pnas.0437927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellison V., Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol. 2003;1:231–243. doi: 10.1371/journal.pbio.0000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Majka J., Burgers P.M. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc. Natl Acad. Sci. USA. 2003;100:2249–2254. doi: 10.1073/pnas.0437148100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furuya K., Poitelea M., Guo L., Caspari T., Carr A.M. Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 2004;18:1154–1164. doi: 10.1101/gad.291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makiniemi M., Hillukkala T., Tuusa J., Reini K., Vaara M., Huang D., Pospiech H., Majuri I., Westerling T., Makela T.P., et al. BRCT domain-containing protein TopBP1 functions in DNA replication and damage response. J. Biol. Chem. 2001;276:30399–30406. doi: 10.1074/jbc.M102245200. [DOI] [PubMed] [Google Scholar]
- 17.Garcia V., Furuya K., Carr A.M. Identification and functional analysis of TopBP1 and its homologs. DNA Repair. 2005;4:1227–12239. doi: 10.1016/j.dnarep.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Goldstein A.L., McCusker J.H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 19.Kitada K., Yamaguchi E., Arisawa M. Cloning of the Candida glabrata TRP1 and HIS3 genes, and construction of their disruptant strains by sequential integrative transformation. Gene. 1995;165:203–206. doi: 10.1016/0378-1119(95)00552-h. [DOI] [PubMed] [Google Scholar]
- 20.Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S.cerevisiae. Yeast. 1996;12:259–265. doi: 10.1002/(SICI)1097-0061(19960315)12:3%3C259::AID-YEA901%3E3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 21.Ui A., Satoh Y., Onoda F., Miyajima A., Seki M., Enomoto T. The N-terminal region of Sgs1, which interacts with Top3, is required for complementation of MMS sensitivity and suppression of hyper-recombination in sgs1 disruptants. Mol. Genet. Genomics. 2001;265:837–850. doi: 10.1007/s004380100479. [DOI] [PubMed] [Google Scholar]
- 22.Ui A., Seki M., Ogiwara H., Onodera R., Fukushige S., Onoda F., Enomoto T. The ability of Sgs1 to interact with DNA topoisomerase III is essential for damage-induced recombination. DNA Repair. 2005;4:191–201. doi: 10.1016/j.dnarep.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Sugawara N., Wang X., Haber J.E. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 24.Wolner B., Van Komen S., Sung P., Peterson C. Recruitment of the recombinational repair machinery to a DNA double strand break in yeast. Mol. Cell. 2003;12:221–232. doi: 10.1016/s1097-2765(03)00242-9. [DOI] [PubMed] [Google Scholar]
- 25.Sanchez Y., Bachant J., Wang H., Hu F., Liu D., Tetzlaff M., Elledge S.J. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166–1171. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- 26.Bai Y., Symington L.S. A Rad52 homolog is required for RAD51-independent mitotic recombination in Saccharomyces cerevisiae. Genes Dev. 1996;10:2025–2037. doi: 10.1101/gad.10.16.2025. [DOI] [PubMed] [Google Scholar]
- 27.Malkova A., Ivanov E.L., Haber J.E. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc. Natl Acad. Sci. USA. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malkova A., Signon L., Schaefer C.B., Naylor M.L., Theis J.F., Newlon C.S., Haber J.E. RAD51-independent break-induced replication to repair a broken chromosome depends on a distant enhancer site. Genes Dev. 2001;15:1055–1060. doi: 10.1101/gad.875901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Signon L., Malkova A., Naylor M.L., Klein H., Haber J.E. Genetic requirements for RAD51- and RAD54-independent break-induced replication repair of a chromosomal double-strand break. Mol. Cell. Biol. 2001;21:2048–2056. doi: 10.1128/MCB.21.6.2048-2056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venclovas C., Thelen M.P. Structure-based predictions of Rad1, Rad9, Hus1 and Rad17 participation in sliding clamp and clamp-loading complexes. Nucleic Acids Res. 2000;28:2481–2493. doi: 10.1093/nar/28.13.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haber J.E. Switching of Saccharomyces cerevisiae mating-type genes. In: Craig N.L., Craigie R., Gellert M., Lambowitz A.M., editors. Mobile DNA II. Washington DC: ASM Press; 2002. pp. 927–952. [Google Scholar]
- 32.White C.I., Haber J.E. Intermediates of recombination during mating type switching in Saccharomyces cerevisiae. EMBO J. 1990;9:663–673. doi: 10.1002/j.1460-2075.1990.tb08158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyazaki T., Bressan D.A., Shinohara M., Haber J.E., Shinohara A. In vivo assembly and disassembly of Rad51 and Rad52 complexes during double-strand break repair. EMBO J. 2004;23:939–949. doi: 10.1038/sj.emboj.7600091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X., Haber J.E. Role of saccharomyces single-stranded DNA-binding protein RPA in the strand invasion step of double-strand break repair. PLoS Biol. 2004;2:104–112. doi: 10.1371/journal.pbio.0020021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kondo T., Wakayama T., Naiki T., Matsumoto K., Sugimoto K. Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science. 2001;294:867–870. doi: 10.1126/science.1063827. [DOI] [PubMed] [Google Scholar]
- 36.Melo J.A., Cohen J., Toczyski D.P. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev. 2001;15:2809–2821. doi: 10.1101/gad.903501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haber J.E. DNA recombination: the replication connection. Trends Biochem. Sci. 1999;24:271–275. doi: 10.1016/s0968-0004(99)01413-9. [DOI] [PubMed] [Google Scholar]
- 38.Lisby M., Barlow J.H., Burgess R.C., Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 39.Wang X., Ira G., Tercero J.A., Holmes A.M., Diffley J.F., Haber J.E. Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 2004;24:6891–6899. doi: 10.1128/MCB.24.16.6891-6899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ira G., Pellicioli A., Balijja A., Wang X., Fiorani S., Carotenuto W., Liberi G., Bressan D., Wan L., Hollingsworth N.M., et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;43:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Greer D.A., Besley B.D., Kennedy K.B., Davey S. hRad9 rapidly binds DNA containing double-strand breaks and is required for damage-dependent topoisomerase II beta binding protein 1 focus formation. Cancer Res. 2003;63:4829–4835. [PubMed] [Google Scholar]
- 42.Harris S., Kemplen C., Caspari T., Chan C., Lindsay H.D., Poitelea M., Carr A.M., Price C. Delineating the position of rad4+/cut5+ within the DNA-structure checkpoint pathways in Schizosaccharomyces pombe. J. Cell Sci. 2003;116:3519–3529. doi: 10.1242/jcs.00677. [DOI] [PubMed] [Google Scholar]
- 43.Saka Y., Yanagida M. Fission yeast cut5+, required for S phase onset and M phase restraint, is identical to the radiation-damage repair gene rad4+ Cell. 1993;74:383–393. doi: 10.1016/0092-8674(93)90428-s. [DOI] [PubMed] [Google Scholar]
- 44.Kim J.E., McAvoy S.A., Smith D.I., Chen J. Human TopBP1 ensures genome integrity during normal S phase. Mol. Cell. Biol. 2005;24:10907–10915. doi: 10.1128/MCB.25.24.10907-10915.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]