

Abstract
Using small-angle x-ray scattering, we have observed the cGMP-induced elongation of an active, cGMP-dependent, monomeric deletion mutant of cGMP-dependent protein kinase (Δ1–52PKG-Iβ). On saturation with cGMP, the radius of gyration of Δ1–52PKG-Iβ increases from 29.4 ± 0.1 Å to 40.1 ± 0.7 Å, and the maximum linear dimension increases from 90 Å ± 10% to 130 Å ± 10%. The elongation is due to a change in the interaction between structured regulatory (R) and catalytic (C) domains. A model of cGMP binding to Δ1–52PKG-Iβ indicates that elongation of Δ1–52PKG-Iβ requires binding of cGMP to the low-affinity binding site of the R domain. A comparison with cAMP-dependent protein kinase suggests that both elongation and activation require cGMP binding to both sites; cGMP binding to the low-affinity site therefore seems to be a necessary, but not sufficient, condition for both elongation and activation of Δ1–52PKG-Iβ. We also predict that there is little or no cooperativity in cGMP binding to the two sites of Δ1–52PKG-Iβ under the conditions used here. Results obtained by using the Δ1–52PKG-Iβ monomer indicate that a previously observed elongation of PKG-Iα is consistent with a pure change in the interaction between the R domain and the C domain, without alteration of the dimerization interaction. This study has revealed important features of molecular mechanisms in the biochemical network describing PKG-Iβ activation by cGMP, yielding new insight into ligand activation of cyclic nucleotide-dependent protein kinases, a class of regulatory proteins that is key to many cellular processes.
Cyclic guanosine monophosphate (cGMP) is a second messenger signaling molecule that is central to the regulation of many physiological processes, including smooth muscle tone, visual transduction, platelet aggregation, bone growth, and electrolyte and fluid homeostasis (see reviews in refs. 1–3). Its known targets are ion channels, which can alter cellular cation or anion transport; phosphodiesterases, which degrade cyclic nucleotides; and the cGMP-dependent protein kinases (PKGs), which are believed to be responsible for most intracellular cGMP actions. The PKG [first discovered by Kuo and Greengard (4)] is a serine/threonine protein kinase whose substrates include ion channels and pumps, receptors, and enzymes that control intracellular Ca2+ concentrations. In its active form, PKG can transfer the γ-phosphate from ATP to target proteins, preferentially targeting serines in substrates having consensus sequence RRXSX, with some exceptions (1, 5). Phosphorylation of target proteins by PKG effects smooth muscle relaxation by reduction of cellular Ca2+. The PKGs and cAMP-dependent protein kinases (PKA) are homologous enzymes, and the two enzyme families share many similarities in biochemical function and domain organization.
PKG is a dimer comprised of two identical monomers (1–3). Each monomer of PKG contains a regulatory domain (R) and a catalytic domain (C) on a single polypeptide chain. The monomers dimerize via interactions between the extreme N-terminal sequences of the R domain. Each R domain in PKG contains two homologous cyclic nucleotide-binding sites arranged in tandem; cyclic nucleotide binding to these sites causes activation. There are two homologous forms of PKG: type I, with an acetylated N terminus, is usually associated with the cytoplasm; type II, with a myristylated N terminus, is generally associated with the membrane. Types I and II have not been shown to coexpress in any tissue. Two closely related isoforms of type I are known, differing only in the first ≈100 aa: PKG-Iα and PKG-Iβ.
Beginning at the N terminus, type I PKG consists of the following functional domains in order of sequence: (1–116), the amino-terminal dimerization/autoinhibitory domain, with two autophosphorylation sites; (117–234), the high-affinity cGMP binding domain, containing cGMP-binding site A; (235–355), the low-affinity cGMP binding domain, containing cGMP-binding site B; (356–491), the Mg2+/ATP binding site; (492–616), a target protein interaction domain; and (617–686), C-terminal residues of unknown function. The domains in the N-terminal region of the enzyme (1–355) comprise the R domain, and the remaining residues (356–686) comprise the C domain. Residue numbers are given in parentheses for bovine isoform β; the α isoform is identical to β except in the N-terminal domain, which has 16 fewer residues. The overall organization of the homologous PKA is similar to that of the PKGs, except that in PKA the R and C domains reside on different polypeptide chains, and are thus referred to as “subunits.”
Binding of two cGMP molecules fully activates one molecule of PKG monomer. Activation can also be achieved by autophosphorylation in the autoinhibitory domain. Because cGMP binding stimulates autophosphorylation, it is likely that cellular activation of PKG involves the concerted actions of cGMP binding and autophosphorylation. In both the PKAs and the PKGs, activation occurs only after cyclic nucleotide binding releases the autoinhibitory domain of the R subunit/domain from its interaction with the catalytic cleft of the C subunit/domain.
Although no atomic structure data on PKG are currently available, crystal structures have been determined for both the truncated R subunit (6, 7) and C subunit (8, 9) of PKA. Sequence identity between PKG-Iβ and PKA is >30% for the R subunit, and >40% for the C subunit. An atomic structure of the PKA heterodimer (i.e., the R subunit in complex with the C subunit) is not available, but a recent model based on high-resolution R and C subunit structures, small-angle scattering data, and a known inter-subunit ion pair (10) predicts the arrangement of the R and C subunits within the heterodimer, and also predicts specific interactions that are important for formation and stabilization of the complex between the R and C subunits.
In a previous study of mechanisms associated with cGMP binding and activation of PKG, we observed a dramatic elongation of PKG-Iα on binding cGMP (11). Fourier transform infrared spectroscopy showed that this conformational change did not involve significant secondary structure changes, indicating that the elongation could be well-explained by a rearrangement of structured domains. A study of PKG-Iβ using ion exchange chromatography, gel filtration chromatography, and native gel electrophoresis also showed that either cGMP-binding or autophosphorylation of the more C-terminal phosphorylation site (Ser-79) activated the protein and was correlated with an apparent elongation of the protein (12).
To better understand the conformational changes related to cGMP activation of PKG, we engineered a monomeric PKG-Iβ mutant with the 52 N-terminal residues deleted (Δ1–52PKG-Iβ), thus eliminating the dimerization domain before the autophosphorylation sites. This monomeric PKG-Iβ is highly dependent on cGMP and retains function comparable to that of dimeric PKG-Iβ. We studied this deletion mutant of PKG-Iβ using small-angle scattering in the presence of varying levels of cGMP, with results that complement and expand on our previous studies of cGMP activation of the dimer: (i) the general shape of cGMP-free Δ1–52PKG-Iβ has been characterized, and that shape is consistent with the recently published model of the PKA R/C interaction (10); (ii) as occurred in the PKG dimer, a dramatic elongation of the Δ1–52PKG-Iβ molecule is observed on adding saturating levels of cGMP; (iii) the scattering at intermediate levels of cGMP implies that the monomer elongation is well-explained by a simple two-state model (compact and extended structures); (iv) a model of cGMP binding to Δ1–52PKG-Iβ reveals that binding to the low-affinity cGMP-binding site is required for elongation, and comparison with PKA suggests that binding to both cGMP-binding sites is required for both elongation and activation; (v) the success of the model suggests that there is little or no cooperativity in cGMP binding under the conditions used here; and (vi) the elongation of monomeric PKG subunits can completely account for the previously observed cGMP-induced elongation of dimeric PKG (11).
Methods
Mutagenesis, Expression, Purification, and Characterization.
Human PKG-Iβ cDNA was ligated into the EcoRI and SmaI sites of the baculovirus expression vector pVL 1392 (13, 14). The EcoRI/SacI fragment for Δ1–52PKG-Iβ was subcloned into the SacI/SmaI fragment of pVL 1392-PKG-Iβ. The protein was expressed and purified to apparent homogeneity by using cAMP-affinity chromatography essentially as described for WT PKG-Iβ (14). Protein was characterized by using specific kinase catalytic activity (−/+ cGMP), cGMP-binding activity, and purity on 10% SDS/PAGE gels (15). Cyclic nucleotide content of purified Δ1–52PKG-Iβ was <1% of total cyclic nucleotide-binding sites (16). By matrix-assisted laser desorption ionization mass spectrometry, protein had a molecular mass of 71.5 kDa, in agreement with the amino acid sequence. The amino-terminal sequence of the protein was verified by sequential Edman degradation (13).
Small-Angle Scattering.
Small-angle scattering was used as a probe of large-scale shape changes (see ref. 17 and references therein). Experiments were performed by using a stock solution of 0.0783 mM Δ1–52PKG-Iβ. The concentration of PKG was determined spectrophotometrically by measuring absorbance at 280 nm and assuming an extinction coefficient of 73,000 M−1⋅cm−1 [estimated from the amino acid sequence by using the expasy protparam tool at http://us.expasy.org/tools/protparam.html, based on an algorithm by Gill and Von Hippel (18)]. The buffer was 10 mM potassium phosphate, pH 6.8, 2 mM EDTA, 25 mM 2-mercaptoethanol, 150 mM NaCl. A stock solution of 6.676 mM cGMP sodium salt (spectrophotometric determination at 252 nm by using an extinction coefficient of 13.7 M−1⋅cm−1) in buffer was prepared for titration experiments. Scattering samples were prepared by weighing amounts of PKG and cGMP stock solutions to final volumes between 15 and 25 μl, and were mixed by using a vortexer. Samples were spun in a microcentrifuge, then loaded into a capillary sample chamber via pipette and centrifuge. The chamber was rinsed thoroughly with buffer before loading new sample; buffer was removed from the chamber by centrifugation. To conserve material, the chamber was not rinsed with sample solution before a final loading with sample solution. Lack of rinsing thus caused a small unknown decrease in the overall sample concentration (estimated 20% maximum effect, with no effect, however, on the cGMP:PKG ratio), preventing accurate analysis of I0/c. Scattering experiments were performed at a temperature of 13.3°C, as described in Heidorn and Trewhella (19), by using lysozyme as a standard for calibration. End point samples (cGMP-free and 9.4:1 cGMP:PKG) were exposed to monochromatic x-rays for >12 h. Intermediate points were exposed between 2 and 4 h. Buffer samples were exposed overnight. Scattering experiments yielded measurements of the intensity profile I(Q), where Q is the scattering vector length (see below description of P(R) analysis). We saw no evidence of aggregation for cGMP-free PKG samples. Some aggregation was apparent in the highest cGMP:PKG ratio sample (9.4:1). This level of aggregation can cause an apparent increase in the radius of gyration Rg, but mainly leads to overestimate and increased uncertainty in the Dmax parameter (see below description of Dmax). To avoid analysis complications due to aggregation, we used the precisely saturated 2:1 cGMP:PKG sample for analysis of the cGMP-saturated state because it did not show evidence of aggregation.
P(R) Analysis and Estimation of Structural Parameters.
We used the program gnom (20, 21) to estimate the radial Patterson distribution P(R) from I(Q), yielding estimates of the radius of gyration (Rg), the maximum distance (Dmax) and I0 with estimated errors [parameters defined as in Zhao et al. (11)]. A slit smearing correction was used in gnom to deconvolute the effect of the beam slit height. Use of gnom had two subjective aspects: (i) a number of intensity points at low Q that seemed to be measurements that were obstructed by the beamstop were removed; and (ii) Dmax is a user-supplied constraint, and values over a range of approximately ±10% yielded similar quality fits.
Two-State Modeling.
Data points (Qi,Ii) in PKG scattering curves were modeled as a linear combination of the data points (Qi, Ii(1)) obtained from cGMP-free PKG and data points (Qi, Ii(2)) obtained from cGMP-saturated PKG as Ii(calc) = a(1)Ii(1) + a(2)Ii(2). Analytic expressions for a(1) and a(2) were calculated by minimizing the sum of the squared residuals Σi(Ii(calc) − Ii)2, and approximate analytic expressions for errors in a(1) and a(2) were obtained by differential analysis of the expressions for a(1) and a(2). Custom software for carrying out the calculations was written in the c programming language. The occupancy of the extended state was calculated as a(2)(a(1) + a(2))−1.
Singular Value Decomposition (SVD) Analysis.
In addition to two-state modeling, we used SVD analysis (see, e.g., ref. 22) to assess whether titrations could be explained by a linear combination of just two signals. Previous application of SVD analysis to small-angle scattering data includes detection of structural intermediates in lysozyme unfolding (23). A matrix was constructed with rows indexing the Q values and columns indexing the titration points. The singular value spectrum was analyzed to determine what fraction of the signal could be explained by the first two components, and the left singular vectors were inspected to determine whether individual scattering components were dominated by noise.
Model of cGMP Binding to Monomeric PKG.
Our model of cGMP binding to Δ1–52PKG-Iβ has concentrations of five distinct molecular species: (i) free PKG; (ii) free cGMP; (iii) PKG:A (cGMP bound to the high-affinity site A); (iv) PKG:B (cGMP bound to the low-affinity site B); and (v) PKG:AB (cGMP bound to both sites). We make the following simplifying assumptions: (a) there is negligible free cGMP below a cGMP:PKG ratio of 2:1; and (b) there is no cooperativity in cGMP binding, meaning that dissociation constants for cGMP binding to the A and B binding sites are independent. Assumption a is reasonable because PKG concentrations were in the neighborhood of 75,000 nM, which is 2–3 orders of magnitude larger than the measured cGMP-binding site dissociation constants (55 and 750 nM). Assumption b has not been experimentally confirmed but is a hypothesis that is tested by our model. We are interested in three functions: (i) the fraction of the population that has cGMP bound to site A, including binding to both sites, AR(x); (ii) the fraction of the population that has cGMP bound to site B, including binding to both sites, BR(x); and (iii) the fraction of the population that has cGMP bound to both sites ABR(x). Under our assumptions it can be shown that the following equations are valid for cGMP:PKG ≤ 2:
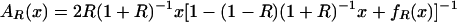 |
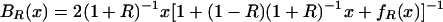 |
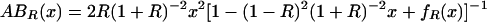 |
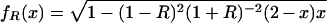 |
1 |
where x is the ratio cGMP:PKG, and R is the ratio of the dissociation constant for binding to site B over that for site A.
Results
Functional Characterization of Monomeric PKG.
Specific catalytic activity of Δ1–52PKG-Iβ was determined to be 2.72 ± 0.15 μmol⋅min−1⋅mg−1, which is similar to the value of ≈2.5 μmol⋅min−1⋅mg−1 for type I PKGs reported earlier (15). The kinase activity was highly dependent on cGMP for activation, and was stimulated >10-fold by addition of cGMP. The dissociation constant Kd for [3H]cGMP binding to Δ1–52PKG-Iβ at 30°C was determined to be 0.210 ± 0.008 μM (see Methods). The Δ1–52PKG-Iβ exhibits two kinetic components of cGMP binding (Kd values of 0.054 μM ± 0.007 and 0.750 μM ± 0.096) that correspond to the high-affinity and low-affinity cGMP-binding sites that also occur in native and WT PKG-Iβ (1). No cooperativity was observed in cGMP binding to Δ1–52PKG-Iβ.
Monomeric, cGMP-Free PKG.
Scattering experiments were performed on a solution of Δ1–52PKG-Iβ in the absence of cGMP. The experimental scattering curve I(Q) was analyzed to determine P(R) (Fig. 1; see Methods). The cGMP-free molecule is somewhat elongated, with a radius of gyration Rg = 29.4 ± 0.15 Å and a maximum linear dimension Dmax = 90 Å ± 10%. A perfectly spherical protein of the same molecular weight as the deletion mutant (71.5 kDa) would be predicted to have Rg = 22 Å and Dmax = 58 Å.
Figure 1.

P(R) distributions from all experiments. The apparent degree of elongation of the molecule increases with increasing amounts of cGMP, until saturation (see Table 1). The apparent isosbestic point in the P(R) distributions suggests a mixture of just two molecular species in solution.
A simulated scattering curve was calculated from the model of the R/C interaction in PKA. This curve is consistent with the experimental I(Q) for Δ1–52PKG-Iβ (Fig. 2); the fit has χ2/n = 1.07. The Rg of the raw PKA R/C model is calculated to be 28.1 Å, which is 4.4% less than that of the cGMP-free monomeric PKG. Allowing for the existence of a hydration layer (24), the model with the best-fit hydration shell has a calculated Rg = 29.0 Å, which just 1.4% less than the experimental value.
Figure 2.

I(Q) from Δ1–52PKG-Iβ without cGMP. Scattering data from Δ1–52PKG-Iβ (points) are similar to simulated scattering calculated from a model of the PKA R/C interaction (line); the fit has χ2/n = 1.07. crysol (39) was used to calculate the model scattering.
Monomeric PKG Elongates on Binding cGMP.
Scattering data were obtained from a solution of Δ1–52PKG-Iβ with a precisely saturating amount of cGMP (2:1 molar ratio). The experimental I(Q) curve yielded a P(R) characteristic of a highly elongated molecule (Fig. 1). The cGMP-bound molecule has Rg = 40.1 ± 0.7 Å, which is 11 Å greater than that of the cGMP-free molecule; and Dmax = 130 Å ± 10%, which is 40 Å longer than the cGMP-free molecule.
Two-State Model of Monomeric PKG Elongation.
To further characterize the dependence of the elongation of Δ1–52PKG-Iβ on binding cGMP, additional cGMP:Δ1–52PKG-Iβ solutions were prepared in molar ratios of 0.5:1, 1.5:1, and 9.4:1. Scattering data obtained from the 0.5:1 and 1.5:1 solutions have estimated P(R) distributions (Fig. 1) and structural parameters (Table 1) that seem to be intermediate in degree of elongation between the cGMP-free and cGMP-saturated samples. Scattering data from the 9.4:1 solution are similar to that from the 2:1 solution, with some evidence of aggregation that is also apparent in the “tailing” of the P(R) distribution out to R = 140 Å.
Table 1.
Structural parameters obtained from Δ1–52PKG-Iβ scattering experiments
| cGMP:PKG | Rg, Å | Dmax, Å | I0 |
|---|---|---|---|
| 0 | 29.4 ± 0.1 | 90 | 29,000 ± 200 |
| 0.56:1 | 31.1 ± 0.5 | 100 | 18,300 ± 300 |
| 1.47:1 | 36.3 ± 0.6 | 120 | 19,100 ± 400 |
| 1.98:1 | 40.1 ± 0.7 | 130 | 19,400 ± 400 |
| 9.4:1 | 41.4 ± 0.5 | 140 | 35,100 ± 400 |
The estimated molecular mass values determined from I0/c for the cGMP:PKG ratios between 0 and 2 gave an average of 68,000 Da ± 15%. The sample measured with excess cGMP (9.4:1) gave an estimate that was inflated by 30%, indicative of an amount of aggregation involving up to 26% of the Δ1–52PKG-Iβ monomers.
The apparent isosbestic point in the P(R) distributions (Fig. 1) suggests that all samples contain a cGMP-dependent mixture of molecules in one of two states, a hypothesis that was tested by fitting the titration data to a two-state model. Assuming that the cGMP-free PKG sample consists of molecules in a purely “compact” state, and that the molecules in the cGMP-saturated sample are in a purely “extended” state, the measured I(Q) at each titration point was modeled as a linear combination of the cGMP-free and cGMP-saturated (9.4:1) I(Q)s. A linear least-squares fit was performed to optimize the fit of each titration point by selection of relative weights of the compact and extended state. This simple two-state model accurately reproduces the data at each titration point (Fig. 3) and predicts the fraction of PKG in each sample that is in the extended state (Fig. 4). Singular value decomposition analysis (Fig. 5) also implies that the data can be explained by two states: the first two singular values account for 96% of the scattering signal; and I(Q) components beyond the first two are dominated by noise.
Figure 3.

Two-state model fit overlayed with 1.5:1 cGMP:PKG titration data. All titration point fits resulted in χ2/n ≤ 1.0.
Figure 4.

Modeling the cGMP-dependent elongation of PKG. (Left) Model of cGMP binding to monomeric PKG that is used to generate binding curves. States are annotated with respect to size and activity. (Right) Extended state occupancy vs. cGMP:PKG ratio for monomeric PKG. Predicted binding site occupancy curves for cGMP binding to site A, site B, and sites A and B combined are shown. In our experiments, the PKG:B state and the PKG:AB state cannot be distinguished.
Figure 5.

Singular value decomposition analysis of cGMP titration data. (Left) Normalized singular values are shown for SVD of the titration data. The first two components account for 96% of the signal. (Right) The first three SVD components of the scattering signal for the titration data are shown offset, sorted top-to-bottom. The third component and higher are dominated by noise. Combined, these plots provide evidence that the titration data are explained by two underlying signals, consistent with a two-state model of the elongation.
Model of the Effect of cGMP Binding on PKG Elongation.
To correlate the Δ1–52PKG-Iβ elongation with cGMP binding, the cGMP dependence of the modeled fraction of molecules in the extended state was compared with the predicted cGMP dependence of the occupancies of the cGMP binding sites. For this purpose, we developed a simple model of cGMP binding without cooperativity (Fig. 4; see Methods). The model has no free parameters. Dissociation constants for cGMP binding to the high-affinity and low-affinity sites were measured to be 55 nM and 750 nM, respectively, at 30°C (see Methods), yielding a ratio R = 13.6. Although the absolute value is quite sensitive to small temperature changes, the ratio of dissociation constants is not very sensitive to temperature changes of the magnitude of the difference between 30°C and the temperature of the scattering experiments (13.3°C).
The model shows that the occupancy of the high-affinity site alone is poorly correlated with occupancy of the extended state of the molecule (Fig. 4). The occupancy of the low-affinity site is much better correlated with the occupancy of the extended state. The occupancy of both states combined is also well correlated with the occupancy of the extended state. Although the data do not distinguish elongation on binding to the low-affinity site vs. elongation on binding to both sites, comparison of binding site organization in PKG and PKA suggests that the elongation occurs on binding of cGMP to both sites simultaneously (see Discussion).
Discussion
We have used small-angle x-ray scattering to characterize the cGMP-dependence of the shape of a monomeric deletion mutant of PKG, Δ1–52PKG-Iβ. This molecule retains the essential features of dimeric PKG, including high dependence on cGMP for activation, cGMP-binding affinity, and maximum catalytic activity. Without cGMP, the inactive Δ1–52PKG-Iβ is somewhat elongated, yet relatively compact, and the scattering data strongly agree with calculations from a recent model of the interaction between the R subunit and the C subunit of PKA (10). The PKG and PKA molecules have high enough sequence identity for homology modeling, and are expected to have the same overall shape. Our data are therefore consistent with the model of the interaction between the R subunit and C subunit of PKA, and can provide insight into mechanisms of cyclic nucleotide activation of both enzymes.
We have observed an elongation in Δ1–52PKG-Iβ on addition of cGMP. In addition, the scattering data obtained for any cGMP:PKG mixture is accurately modeled by a linear combination of scattering curves for a compact (smaller Rg, Dmax) and extended (larger Rg, Dmax) state, indicating that Δ1–52PKG-Iβ solutions in our experiments consist of a mixture of molecules in either of these two states. Insight into the elongation mechanism is gained by comparing this model with a model of cGMP binding to Δ1–52PKG-Iβ. Binding to the high-affinity site is not associated with appreciable elongation of Δ1–52PKG-Iβ, but binding to the low-affinity site or to both sites is associated with an overall elongation of the molecule. Thus, our modeling indicates that elongation of Δ1–52PKG-Iβ requires cGMP binding to the low-affinity site.
Although our modeling cannot indicate whether cGMP binding to the low-affinity site is sufficient to cause the elongation of Δ1–52PKG-Iβ, additional insights follow from comparison of PKG and PKA. Dissociation of PKA-I is greatly enhanced when exactly two cAMP molecules are bound to each R subunit (25), suggesting that two cAMP molecules are required to release the autoinhibitory pseudosubstrate sequence of the R subunit from the catalytic cleft of the C subunit and disrupt all other interactions between the PKA subunits. If we postulate that the cGMP-dependent elongation of Δ1–52PKG-Iβ is analogous to dissociation of PKA, it would therefore seem likely that the elongation of Δ1–52PKG-Iβ requires cGMP binding to both binding sites, and not just binding to site B. Moreover, the locations of the low-affinity and high-affinity sites are swapped between PKA and type I PKG, so that if (i) the structural mechanisms of dissociation/elongation of PKA and PKG-I are similar; and (ii) binding to the low-affinity site, or both sites, of either molecule is required for dissociation/elongation; then (iii) binding to both sites would be required for dissociation/elongation of either molecule. Indeed, for both the PKAs and the PKGs, cyclic nucleotide binding releases the autoinhibitory sequence from the catalytic cleft (see ref. 1), implying a shared mechanism in dissociation/elongation, at least in part. Support for these arguments comes from considering the model of the complex between the PKA R subunit and C subunit (10). In this model, the relatively small surface area of interaction between the R and C subunits (2,000 Å2), which presumably helps to mediate PKA dissociation, is dominated by interactions with the domain containing the low-affinity A site of the R subunit. In PKG-I, however, the A site is the high-affinity cGMP-binding site. Our scattering data suggest that the interactions between the R and C domains in PKG are similar to those between the subunits in PKA, and so we predict that the low-affinity domain of the PKG R domain has the smaller set of interactions with the C domain. These observations suggest that the low-affinity domain of R can have either large (for PKA) or small (for PKG) contact area with C; any likely mechanism for dissociation/elongation would therefore seem to involve cyclic nucleotide binding to both sites in either molecule. Thus, we are motivated to label the PKG:B state as “likely compact” in our annotated model of cGMP binding (Fig. 4).
In both PKG-I and PKG-II, disruption of cGMP-binding site A by point mutation has the greatest effect on activation of the enzymes by cGMP, despite the fact that in PKG-I this site is the high-affinity site, whereas in PKG-II it is the low-affinity site (14, 26). In addition, all biochemical studies of the PKGs and PKAs support involvement of both cyclic nucleotide-binding sites in activation of the enzymes, regardless of their order in the primary structure. We are therefore motivated to label the PKG:B state as “likely inactive” in our annotated model of cGMP binding (Fig. 4). Because activation requires release of pseudosubstrate from the catalytic cleft (see ref. 1), which in turn is necessary for dissociation of PKA, the existing data on cyclic nucleotide activation support our prediction that binding of cGMP to both sites is required for elongation of PKG. Conversely, the effect of elongation or dissociation on activity in cyclic nucleotide-dependent protein kinases is currently unknown. Although binding of cAMP was originally assumed to cause dissociation of the R and C subunits in PKA (27–29), fluorescent resonance energy transfer measurements suggest that dissociation may require interactions with the PKA inhibitor protein (30–33), and question whether dissociation is required for activation (34, 35). Under the conditions used here for Δ1–52PKG-Iβ, and in our previous study of PKG-Iα (11), we found that cGMP binding was sufficient to induce both elongation and activation. Our present data are consistent with a prediction that the PKG:B state of Δ1–52PKG-Iβ is compact and inactive, as suggested by previous studies of cyclic nucleotide activation and elongation/dissociation of the PKAs and PKGs. Future studies involving mutagenesis in the respective cGMP binding sites may be capable of clarifying uncertainties in the mechanism of cGMP-induced elongation of Δ1–52PKG-Iβ, and may yield clues concerning the effect of elongation on activation.
Our previous studies of the cGMP-dependent elongation of dimeric PKG-Iα demonstrated that the elongation of PKG-Iα was due to rearrangement of structured domains, but could not resolve whether it was solely due to conformational changes within the monomer (11). Remarkably, Dmax for the dimer was found to increase by 40 Å (11), whereas here we find that Dmax for the monomer increases by the same amount. The amount of elongation in the dimer can thus be easily accounted for by the elongation of two monomers. We therefore propose that binding of cGMP causes an elongation of the PKG dimer by inducing elongation of the monomers, without large-scale alterations of the dimer interaction.
Our model assumes no cooperativity in cGMP binding to Δ1–52PKG-Iβ. Although previous studies have clearly documented cooperativity in binding of cGMP to the dimeric PKG-Iα (36, 37), no cooperativity for PKG-Iβ has been reported in the literature. Because our model has successfully demonstrated a correlation between cGMP binding and elongation of Δ1–52PKG-Iβ, we predict no cooperativity in cGMP binding to PKG-Iβ. A difference in cooperativity between the isozymes would be interesting to observe experimentally: because the sequences of PKG-Iα and PKG-Iβ differ only in the dimerization/autoinhibitory domain, a difference could further support an allosteric influence of this region of the protein (15, 38).
In this study, we have probed a branched biochemical pathway with equilibrium intermediates by combining small-angle x-ray scattering with biochemical network modeling (Fig. 4). We have determined that free Δ1–52PKG-Iβ, a monomeric mutant of PKG-Iβ, is relatively compact and inactive; that Δ1–52PKG-Iβ with cGMP bound to only the high-affinity A site is also relatively compact and inactive; and that Δ1–52PKG-Iβ with cGMP bound into both the high-affinity A site and the low-affinity B site is elongated and active. We infer, based on comparison with PKA, that Δ1–52PKG-Iβ with cGMP bound to the low-affinity B site alone is relatively compact and inactive, and we infer that the elongation and activation of dimeric PKG-Iα is governed by the behavior of the monomer. Thus, we have provided structural and functional annotation of a model of cGMP binding to PKG, and have demonstrated the use of small-angle scattering not only to characterize the shapes of individual molecules, but also to elucidate the properties of biochemical networks.
Acknowledgments
We gratefully acknowledge Elaine Liong for matrix-assisted laser desorption ionization mass spectrometry analysis of samples. This work was supported by Department of Energy/Biological and Environmental Research Project KP110101 (J.T.) in support of the Oak Ridge Structural Molecular Biology Center; by the Department of Energy through contract W-7405-ENG-36 to the University of California via the Los Alamos National Laboratory Laboratory-Directed Research and Development Program; and by National Institutes of Health Project NIH DK 40029 (J.D.C. and S.H.F.).
Abbreviations
- PKG
cGMP-dependent protein kinase
- PKA
cAMP-dependent protein kinase
- R
regulatory
- C
catalytic
- SVD
singular value decomposition
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Francis S H, Corbin J D. Crit Rev Clin Lab Sci. 1999;36:275–328. doi: 10.1080/10408369991239213. [DOI] [PubMed] [Google Scholar]
- 2.Sausbier M, Schubert R, Voigt V, Hirneiss C, Pfeifer A, Korth M, Kleppisch T, Ruth P, Hofmann F. Circ Res. 2000;87:825–830. doi: 10.1161/01.res.87.9.825. [DOI] [PubMed] [Google Scholar]
- 3.Lohmann S M, Vaandrager A B, Smolenski A, Walter U, De Jonge H R. Trends Biochem Sci. 1997;22:307–312. doi: 10.1016/s0968-0004(97)01086-4. [DOI] [PubMed] [Google Scholar]
- 4.Kuo J F, Greengard P. J Biol Chem. 1970;245:2493–2498. [PubMed] [Google Scholar]
- 5.Yuasa K, Omori K, Yanaka N. J Biol Chem. 2000;275:4897–4905. doi: 10.1074/jbc.275.7.4897. [DOI] [PubMed] [Google Scholar]
- 6.Su Y, Dostmann W R, Herberg F W, Durick K, Xuong N H, Ten Eyck L, Taylor S S, Varughese K I. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- 7.Diller T C, Madhusudan, Xuong N H, Taylor S S. Structure (Cambridge) 2001;9:73–82. doi: 10.1016/s0969-2126(00)00556-6. [DOI] [PubMed] [Google Scholar]
- 8.Knighton D R, Zheng J H, Ten Eyck L F, Ashford V A, Xuong N H, Taylor S S, Sowadski J M. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- 9.Knighton D R, Zheng J H, Ten Eyck L F, Xuong N H, Taylor S S, Sowadski J M. Science. 1991;253:414–420. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 10.Tung C S, Walsh D A, Trewhella J. J Biol Chem. 2002;277:12423–12431. doi: 10.1074/jbc.M110298200. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J, Trewhella J, Corbin J, Francis S, Mitchell R, Brushia R, Walsh D. J Biol Chem. 1997;272:31929–31936. doi: 10.1074/jbc.272.50.31929. [DOI] [PubMed] [Google Scholar]
- 12.Chu D M, Francis S H, Thomas J W, Maksymovitch E A, Fosler M, Corbin J D. J Biol Chem. 1998;273:14649–14656. doi: 10.1074/jbc.273.23.14649. [DOI] [PubMed] [Google Scholar]
- 13.Sandberg M, Natarajan V, Ronander I, Kalderon D, Walter U, Lohmann S M, Jahnsen T. FEBS Lett. 1989;255:321–329. doi: 10.1016/0014-5793(89)81114-7. [DOI] [PubMed] [Google Scholar]
- 14.Reed R B, Sandberg M, Jahnsen T, Lohmann S M, Francis S H, Corbin J D. J Biol Chem. 1996;271:17570–17575. doi: 10.1074/jbc.271.29.17570. [DOI] [PubMed] [Google Scholar]
- 15.Wolfe L, Corbin J D, Francis S H. J Biol Chem. 1989;264:7734–7741. [PubMed] [Google Scholar]
- 16.Francis S H, Noblett B D, Todd B W, Wells J N, Corbin J D. Mol Pharmacol. 1988;34:506–517. [PubMed] [Google Scholar]
- 17.Wall M E, Gallagher S C, Trewhella J. Annu Rev Phys Chem. 2000;51:355–380. doi: 10.1146/annurev.physchem.51.1.355. [DOI] [PubMed] [Google Scholar]
- 18.Gill S C, von Hippel P H. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 19.Heidorn D B, Trewhella J. Biochemistry. 1988;27:909–915. doi: 10.1021/bi00403a011. [DOI] [PubMed] [Google Scholar]
- 20.Svergun D, Semenyuk A, Feigin L. Acta Crystallogr A. 1988;44:244–250. [Google Scholar]
- 21.Svergun D. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- 22.Press W H, Teukolsky S A, Vetterling W T, Flannery B P. Numerical Recipes in C. 2nd Ed. Cambridge, U.K.: Cambridge Univ. Press; 1992. pp. 59–70. [Google Scholar]
- 23.Chen L, Hodgson K O, Doniach S. J Mol Biol. 1996;261:658–671. doi: 10.1006/jmbi.1996.0491. [DOI] [PubMed] [Google Scholar]
- 24.Svergun D I, Richard S, Koch M H, Sayers Z, Kuprin S, Zaccai G. Proc Natl Acad Sci USA. 1998;95:2267–2272. doi: 10.1073/pnas.95.5.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houge G, Steinberg R, Ogreid D, Doskeland S. J Biol Chem. 1990;265:19507–19516. [PubMed] [Google Scholar]
- 26.Taylor M K, Uhler M D. J Biol Chem. 2000;275:28053–28062. doi: 10.1074/jbc.M004184200. [DOI] [PubMed] [Google Scholar]
- 27.Tsuzuki J, Kiger J A., Jr Biochemistry. 1978;17:2961–2970. doi: 10.1021/bi00608a004. [DOI] [PubMed] [Google Scholar]
- 28.Chau V, Huang L C, Romero G, Biltonen R L, Huang C. Biochemistry. 1980;19:924–928. doi: 10.1021/bi00546a016. [DOI] [PubMed] [Google Scholar]
- 29.Builder S E, Beavo J A, Krebs E G. J Biol Chem. 1980;255:3514–3519. [PubMed] [Google Scholar]
- 30.Sassone-Corsi P. Int J Biochem Cell Biol. 1998;30:27–38. doi: 10.1016/s1357-2725(97)00093-9. [DOI] [PubMed] [Google Scholar]
- 31.Lalli E, Sassone-Corsi P. J Biol Chem. 1994;269:17359–17362. [PubMed] [Google Scholar]
- 32.Fantozzi D A, Taylor S S, Howard P W, Maurer R A, Feramisco J R, Meinkoth J L. J Biol Chem. 1992;267:16824–16828. [PubMed] [Google Scholar]
- 33.Fantozzi D A, Harootunian A T, Wen W, Taylor S S, Feramisco J R, Tsien R Y, Meinkoth J L. J Biol Chem. 1994;269:2676–2686. [PubMed] [Google Scholar]
- 34.Johnson D A, Leathers V L, Martinez A M, Walsh D A, Fletcher W H. Biochemistry. 1993;32:6402–6410. doi: 10.1021/bi00076a013. [DOI] [PubMed] [Google Scholar]
- 35.Yang S, Fletcher W H, Johnson D A. Biochemistry. 1995;34:6267–6271. doi: 10.1021/bi00019a002. [DOI] [PubMed] [Google Scholar]
- 36.McCune R W, Gill G N. J Biol Chem. 1979;254:5083–5091. [PubMed] [Google Scholar]
- 37.Doskeland S O, Vintermyr O K, Corbin J D, Ogreid D. J Biol Chem. 1987;262:3534–3540. [PubMed] [Google Scholar]
- 38.Ruth P, Landgraf W, Keilbach A, May B, Egleme C, Hofmann F. Eur J Biochem. 1991;202:1339–1344. doi: 10.1111/j.1432-1033.1991.tb16509.x. [DOI] [PubMed] [Google Scholar]
- 39.Svergun D, Barberato C, Koch M. J Appl Crystallogr. 1995;28:768–773. [Google Scholar]