

Abstract
One of the principal goals of the structural genomics initiative is to identify the total repertoire of protein folds and obtain a global view of the “protein structure universe.” Here, we present a 3D map of the protein fold space in which structurally related folds are represented by spatially adjacent points. Such a representation reveals a high-level organization of the fold space that is intuitively interpretable. The shape of the fold space and the overall distribution of the folds are defined by three dominant trends: secondary structure class, chain topology, and protein domain size. Random coil-like structures of small proteins and peptides are mapped to a region where the three trends converge, offering an interesting perspective on both the demography of fold space and the evolution of protein structures.
The concept of protein folds originated from early observations that proteins of disparate evolutionary origins could adopt similar structures (1). As a result, the number of unique protein architectural types (or folds) was predicted to be much smaller than the number of protein families defined by sequence similarity (2, 3). One of the principal goals of the structural genomics initiative is to maximally populate the protein fold space, thereby providing structural templates for all existing protein families and laying a foundation for a global understanding of architecture, function, and fold evolution of protein sequences from genomics and proteomics (4–6).
Although the definition of protein folds is useful for counting purposes, it is well known that structural similarities can extend beyond the borders of fold types. For example, there are >20 scop folds with similar β-sandwich architectures, of which a vast majority contain a common substructural unit at one end of the double layer (7–9). An interesting question is whether the fold space is continuous with respects to topological arrangement of the secondary structure elements such that structures exist for all possible topologies allowed for a polypeptide chain. Surveys of known protein structures suggest that the protein fold space is highly nonuniformly populated. On one hand, there is a clear bias in the usage of the secondary structures and their connectivities by protein structures (1, 9, 10), which takes the form of well-segregated fold clusters at the level of structural domains (11). Such a bias is expected to impose a strong restriction on the shape of the fold space. On the other hand, even protein structures sharing common structural units such as the complete Greek key motif in β-sandwiches could show considerable structural diversity outside the common core (8, 9), and the degree of variations is probably limited only by the size of the protein (or protein domain) and, ultimately, the number of different protein sequences in nature.
The definitions of four broad structural classes, all-α, all-β, α/β, and α+β, based on secondary structure compositions and β-sheet topologies (12) represented the first step toward a global characterization of the protein fold space. These definitions have been generally accepted and are being used by many classification systems to organize the fold hierarchy (13, 14). However, there is a need for methods to represent the full range of structural relationships among folds for a better understanding of the organizing principles and features of the protein fold space. The fold family trees such as those built by Efimov (8), Zhang and Kim (9), and Taylor (15) are very informative, but the construction of such trees involves extensive manual operations and sometimes considerable human judgment. An alternative approach is to apply a uniform measure of the structural similarity across all fold types and map the structural relationships into a low dimensional space. Two such maps have been introduced, one by Orengo and colleages (16) and the other by Holm and Sander (17). Although the two maps were based on different structural alignment algorithms and multivariant analysis methods, they gave similar 2D projections that featured three large clusters corresponding to α, β, and α/β folds, respectively.
Here we revisit the mapping of protein fold space in light of the large number of new protein structures that have since been solved and the availability of high-quality protein fold catalogues such as those provided by scop (13). In particular, we focus our analysis on the full 3D representation of the fold space rather than 2D projections. We show that such a representation not only emphasizes the important role of secondary structure classes and chain topologies in the partitioning of the fold space, but also reveals the size of protein domain as an important factor in setting the overall distribution of folds. Our map thus provides a more complete view of the global trends in the protein fold space and has interesting implications for the evolution of protein structures. The representation can also be used to identify structural neighbors of novel folds revealed by structural genomics projects.
Methods
To assemble a structure data set that represents a majority of protein folds, we selected one domain structure from each of 498 common scop folds (taken from scop release 1.55). Only soluble protein domains that form a compact, globular structure are included in the present analysis. Pair-wise structural alignments of the 498 scop domains thus selected are carried out by using the program dali (17). Because our aim is to measure the extent of structural similarity rather than significance of a structure alignment, which is important for inferring evolutionary relationships, we used the original dali alignment score rather than the normalized Z score. The overall pair-wise comparisons of 498 folds lead to a 498 × 498 matrix of similarity scores Sijs, where Sij is the alignment score between the ith and jth folds. An appropriate method for handling such data matrices as a whole is metric matrix distance geometry (18). We first convert the similarity score matrix [Sij] to a distance matrix [Dij] by using Dij = Smax − Sij, where Smax is the maximum similarity score among all pairs of folds. We then transform the distance matrix to a metric (or Gram) matrix [Mij] by using
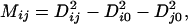 |
where Di0, the distance between the ith fold and the geometric centroid of all N = 498 folds, is determined by
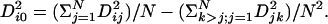 |
The eigen values of the metric matrix define an orthogonal system of axes, called factors. These axes pass through the geometric centroid of the points representing all observed folds and correspond to a decreasing order of the amount of information each factor represents.
The metric matrix that describes the relationships among 498 folds has three dominant eigen values (λ1, λ2, and λ3) (Fig. 1), suggesting that three dimensions are adequate for representing the essential features of the fold space. Each fold is thus represented by a point in the coordinate system defined by the first three principal factorial axes; the coordinate for the ith fold in the kth dimension (k = 1, 2, 3) is given by
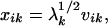 |
where vik is the ith element of the kth eigen vector that corresponds to eigen value λk. The distribution of these points provides a 3D map of the protein fold space.
Figure 1.

The first 20 eigen values of the metric matrix calculated from the 498 × 498 dali structural alignment scores.
Results
As described in Methods, we applied the metric matrix distance geometry method to all pair-wise “distances” (structural dissimilarities) to assign 3D coordinates to a set of 498 scop folds such that the relative distance between two folds is inversely correlated with the dali alignment score. The results of the mapping are shown in Fig. 2.
Figure 2.

A 3D representation of the protein fold space. Each sphere represents a protein fold family. Protein folds that belong to the α, β, and α/β scop classes are clustered mostly around three separate axes, α (red), β (yellow), and α/β (cyan). Most protein folds of the α+β class (blue) fall between the α and β axes and on or near the plane defined by the α and β axes. The α and β axes approximately intersect at one point indicated by the green ball.
The most salient feature of the map is the presence of four largely separated regions in the fold space. These regions correspond to the four-class definitions of protein structures by scop (α, β, α/β, and α+β), although the scop class assignment of the folds were not used in the mapping. However, Fig. 2 also reveals the existence of significant overlaps among different structural classes, with folds in the overlap regions containing features of both classes. In addition, there are discrepancies between the scop class assignments of a small number of folds and their physical locations in our map. In these cases, the scop classification appears to emphasize more localized structural features whereas the structure similarity-based mapping reflects the overall structural features of a fold. For example, the metallo-dependent phosphatase fold (exemplified by Protein Data Bank ID code 1ute) belongs to the α+β class according to scop because of an antiparallel β-sheet element at the C terminus. However, in Fig. 2 the same fold is mapped in the mainly α/β region, agreeing with its overall α/β-supersandwich architecture. Therefore, mapping the fold relationships by using a uniform measure of structural similarity seams to provide a more objective view of both the graded changes and the global partitioning of the protein fold space.
In Fig. 2, the α, β, and α/β folds are clustered around three separate axes (note that these axes are defined to represent the population distributions of α, β, and α/β fold for convenience, and they are different from the axes in the factorial space). The α and β axes are approximately coplanar and the protein folds of the α+β class are found on or near the plane and in between the α and β axes. The α/β axis is approximately perpendicular to and originates from the middle of the plane defined by the α and β axes. To exclude the possibility that this result is an artifact of the data set used, we repeated the mapping by using a smaller data set consisting of 125 scop domains randomly selected from the set of 498 domains and another data set including all 2,295 scop protein domains with <30% sequence identity to each other. Both calculations produced results strikingly similar to that presented in Fig. 2 in the overall shape of the fold distribution and the relative orientation of the three axes, suggesting that the observed features represent actual properties of the fold space.
Visual inspection of the representative structures in various regions of the map suggests that most of the protein folds on or near the plane defined by the α and β axes, designated here as the αβ plane, have simple up-and-down meander topologies, with α-α hairpin and β-β hairpin as the predominant connecting modes between secondary structure elements. In contrast, the protein folds along the α/β axis contain mainly β-α-β crossover connections (19). Furthermore, the deviation of the fold from the αβ plane is directly linked to the fractional abundance of the crossover connections in the structure, with folds farthest having virtually all crossover connections and folds closest to the plane incorporating the least amount of crossover and more meander connections. This trend also applies to protein folds assigned to other structural classes, although in these cases, the definition of crossover has to be extended. For example, the two β structures that are farthest away from the αβ plane, the β-roll fold and the single-stranded right-handed β-helix fold, both contain extensive β-β-β crossover connections.
The extensions of the α and β axes on the αβ plane approximately intersect at a point (Fig. 2). Mapped close to this point are small folds with irregular topologies. Moving away from the point, the size of the proteins generally increases, as do the length of the secondary structures and the complexity of the fold. In fact, the third factorial axis directly coincides with the change in protein size (Fig. 3). Because the evolution of proteins can only start with small peptides of random conformation, we hypothesize that structures of primordial peptides should also appear in the region near the intersection between the α and β axes. Because of the unique position of the α-β intersection in both the fold space and the evolution of protein structures, we designate this point as the “origin” of the fold space (Fig. 2).
Figure 3.

The three factorial axes defined by the eigen values of the metric matrix. (A) The first two axes discriminate among α, β, and α/β folds (color scheme the same as Fig. 2). (B) The third factorial axis correlates with the change in protein domain size, corresponding to a dominant trend in the distribution of folds in the fold space.
There is a large gap between the origin and the α/β-fold cluster. In fact, the α/β axis starts near the middle of the αβ plane, and not at the origin. This result has interesting implications for the evolution of this class of folds. It is conceivable that, of the primordial peptides, those containing fragments with high helix and/or strand propensity found their way to fold into small α, β, or α+β moieties, leading to the emergence of early α, β, and α+β folds. On the contrary, the α/β folds did not appear until proteins of sufficient size rose through evolution (moving away from the origin) and the formation of supersecondary structural units with the β-α−β crossover connections became possible. Therefore, the α/β folds may have arrived relatively late in the protein structure phylogeny compared with folds of other classes.
An indirect test of this hypothesis is provided by comparing the fold usage of organisms that occupy different levels of the tree of life (20). Two examples are shown in Fig. 4 where the fold usage of Chlamydia muridarum is compared with those of Aquifex aeolicus and Halobacterium sp., respectively. The Aquifex lineage is much more deeply rooted than Chlamydia in the (eu)bacterial tree (21), whereas Halobacterium belongs to the archael domain. In both cases, the most significant difference in fold usage involves the α/β folds (Fig. 4). The more evolved Chlamydia genome encodes a higher percentage of proteins in α/β folds than the less evolved Aquifex. Between domains, bacteria seem to have more α/β folds than archaea, and archaea have more α, β, and α+β folds. Although this analysis is limited by the fact that not all of the folds presented in an organism are known [sequence homology-based method allows for accurate structure annotation for only 30–40% of the protein domains (22, 23)], the consistent correlation seen between the α/β fold usage and the stage of evolutionary supports the notion that the topologically more elaborate α/β folds may be newer additions to the protein fold repertoire. Although evolving relatively late, the α/β fold is capable of accommodating a large number of different sequences. Of the 15 most populated folds in nature, more than half belong to the α/β class (24).
Figure 4.

Comparing the fold usages between two species in the eubacterial domain (Chlamydia versus Aquifex, A) and between two species representing two different domains (Chlamydia of bacteria versus Halobacterium of archaea, B). Structural annotations of the ORFs encoded by each genome are obtained by a blast search against the scop sequence database. Percentage of the annotated ORFs in a genome that adopts a particular fold defines the usage of the fold by the organism. The usages of the 498 folds by the second organism are subtracted from the fold usages by the first organism. A contour surface (mesh) is then constructed and set at the values of 0.4% for blue and −0.4% for red. Regions within the blue contour include folds that appear more frequently in the first organism, whereas regions within the red contour include folds that occur more frequently in the second organism.
Discussion
We present a 3D map of the protein fold space that incorporates most recent structural data and structure classification data. The map provides a more complete view of the major trends of the protein fold spaces that are intuitively interpretable in terms of factors that may underlie the design and evolution of protein structures.
Like the 2D projects of the fold space published earlier (16, 17), the 3D map shows a highly nonuniform distribution of the folds in the fold space. The 3D map thus encompasses and generalizes the global trends revealed by the 2D representation. As in the 2D map, the first two factorials in our representation discriminate between all-helical structures and structures with β-sheets and between mainly meander topologies and mainly crossover topologies, respectively. However, the addition of the third dimension and more structures representing distinct folds offers some important insights. For example, in the maps generated by Orengo and colleagues (16, 25), the α+β folds are tightly associated with folds of other classes, leading the authors to question the validity of separate α+β as a distinct architectural class. In fact, the maps generated by Holm and Sander (17, 26) treated the α/β and α+β folds as one class. In contrast, Fig. 2 shows a clear separation between α+β and other folds, supporting the original four class definition of Levitt and Chothia (12).
The most important insight provided by the 3D representation is the revelation of protein (domain) size as a major trend-setting factor in the fold space. Based on this result, we infer that the random coil-like structures of primordial peptides fall in a region near where the α- and β-fold clusters converge (the origin) and hypothesize that the α/β folds are not as deeply rooted as other structural classes in the structural phylogenic tree. The statistics of fold usages by organisms representing different stages of evolutionary seem to corroborate the latter prediction.
Existing structural alignment algorithms such as dali (17), ssp (27), vast (28), and ce (29) are powerful in identifying close structural homologues (i.e., structures of the same fold) for a newly solved structure. However, there are cases where new structures fall outside the detection radius defined by statistical significance levels assigned by these programs (30). More such structures are expected from the structural genomics projects where protein target selection is geared toward maximizing the chance of revealing novel folds or novel variations of known folds (31, 32). The 3D map presented here provides a framework for relating a newly determined fold to known folds in the structure databases based on weak structural similarities. The distance geometry procedure helps to position the new fold in the fold space in a way that is consistent with all of the similarity scores between the new structure and structures representing known folds. The mapping provides a structural context for a new fold such that the structural insights and structure–function relationships gleaned from other related folds can be used to provide a better interpretation and perspective on the new structural data (30, 33).
Acknowledgments
We thank Dr. Lisa Holm for providing the dali program and Drs. In-Geol Choi, Brian West, Steven Holbrook, Ken Frankel, and Se-Ran Jun for helpful discussions. This work was supported by National Science Foundation Grant DBI-0114707 and National Institutes of Health Grant GM 62412.
References
- 1.Richardson J S. Adv Protein Chem. 1981;34:167–339. doi: 10.1016/s0065-3233(08)60520-3. [DOI] [PubMed] [Google Scholar]
- 2.Chothia C. Nature. 1992;357:543–544. doi: 10.1038/357543a0. [DOI] [PubMed] [Google Scholar]
- 3.Zhang C, DeLisi C. J Mol Biol. 1998;284:1301–1305. doi: 10.1006/jmbi.1998.2282. [DOI] [PubMed] [Google Scholar]
- 4.Kim S H. Nat Struct Biol. 1998;5,Suppl.:643–645. doi: 10.1038/1334. [DOI] [PubMed] [Google Scholar]
- 5.Sanchez R, Pieper U, Melo F, Eswar N, Marti-Renom M A, Madhusudhan M S, Mirkovic N, Sali A. Nat Struct Biol. 2000;7,Suppl.:986–990. doi: 10.1038/80776. [DOI] [PubMed] [Google Scholar]
- 6.Stevens R C, Yokoyama S, Wilson I A. Science. 2001;294:89–92. doi: 10.1126/science.1066011. [DOI] [PubMed] [Google Scholar]
- 7.Ptitsyn O B, Finkelstein A V, Falk P. FEBS Lett. 1979;101:1–5. [PubMed] [Google Scholar]
- 8.Efimov A V. Proteins. 1997;28:241–260. doi: 10.1002/(sici)1097-0134(199706)28:2<241::aid-prot12>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 9.Zhang C, Kim S H. Proteins. 2000;40:409–419. doi: 10.1002/1097-0134(20000815)40:3<409::aid-prot60>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 10.Salem G M, Hutchinson E G, Orengo C A, Thornton J M. J Mol Biol. 1999;287:969–981. doi: 10.1006/jmbi.1999.2642. [DOI] [PubMed] [Google Scholar]
- 11.Holm L, Sander C. Science. 1996;273:595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- 12.Levitt M, Chothia C. Nature. 1976;261:552–558. doi: 10.1038/261552a0. [DOI] [PubMed] [Google Scholar]
- 13.Murzin A G, Brenner S E, Hubbard T, Chothia C. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 14.Orengo C A, Michie A D, Jones S, Jones D T, Swindells M B, Thornton J M. Structure (London) 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 15.Taylor W R. Nature. 2002;416:657–660. doi: 10.1038/416657a. [DOI] [PubMed] [Google Scholar]
- 16.Orengo C A, Flores T P, Taylor W R, Thornton J M. Protein Eng. 1993;6:485–500. doi: 10.1093/protein/6.5.485. [DOI] [PubMed] [Google Scholar]
- 17.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 18.Havel T F, Kuntz I D, Crippen G M. J Theor Biol. 1983;104:359–381. doi: 10.1016/0022-5193(83)90112-1. [DOI] [PubMed] [Google Scholar]
- 19.Richardson J S. Proc Natl Acad Sci USA. 1976;73:2619–2623. doi: 10.1073/pnas.73.8.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woese C R, Kandler O, Wheelis M L. Proc Natl Acad Sci USA. 1990;87:4576–4579. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burggraf S, Olsen G J, Stetter K O, Woese C R. Syst Appl Microbiol. 1992;15:352–356. doi: 10.1016/S0723-2020(11)80207-9. [DOI] [PubMed] [Google Scholar]
- 22.Teichmann S A, Chothia C, Gerstein M. Curr Opin Struct Biol. 1999;9:390–399. doi: 10.1016/S0959-440X(99)80053-0. [DOI] [PubMed] [Google Scholar]
- 23.Frishman D, Albermann K, Hani J, Heumann K, Metanomski A, Zollner A, Mewes H W. Bioinformatics. 2001;17:44–57. doi: 10.1093/bioinformatics/17.1.44. [DOI] [PubMed] [Google Scholar]
- 24.Zhang C, DeLisi C. Cell Mol Life Sci. 2001;58:72–79. doi: 10.1007/PL00000779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michie A D, Orengo C A, Thornton J M. J Mol Biol. 1996;262:168–185. doi: 10.1006/jmbi.1996.0506. [DOI] [PubMed] [Google Scholar]
- 26.Holm L, Sander C. Proteins. 1998;33:88–96. doi: 10.1002/(sici)1097-0134(19981001)33:1<88::aid-prot8>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 27.Taylor W R, Orengo C A. J Mol Biol. 1989;208:1–22. doi: 10.1016/0022-2836(89)90084-3. [DOI] [PubMed] [Google Scholar]
- 28.Gibrat J F, Madej T, Bryant S H. Curr Opin Struct Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 29.Shindyalov I N, Bourne P E. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 30.Dietmann S, Fernandez-Fuentes N, Holm L. Curr Opin Struct Biol. 2002;12:362–367. doi: 10.1016/s0959-440x(02)00332-9. [DOI] [PubMed] [Google Scholar]
- 31.Brenner S E. Nat Struct Biol. 2000;7,Suppl.:967–969. doi: 10.1038/80747. [DOI] [PubMed] [Google Scholar]
- 32.Frishman D. Protein Eng. 2002;15:169–183. doi: 10.1093/protein/15.3.169. [DOI] [PubMed] [Google Scholar]
- 33.Zhang C, Kim S-H. Curr Opin Chem Biol. 2003;7:28–32. doi: 10.1016/s1367-5931(02)00015-7. [DOI] [PubMed] [Google Scholar]