

Abstract
Neurodegenerative diseases such as Alzheimer's, Parkinson's and the transmissible spongiform encephalopathies (TSEs) are characterized by abnormal protein deposits, often with large amyloid fibrils. However, questions have arisen as to whether such fibrils or smaller subfibrillar oligomers are the prime causes of disease1,2. Abnormal deposits in TSEs are rich in PrPres, a protease-resistant form of the PrP protein with the ability to convert the normal, protease-sensitive form of the protein (PrPsen) into PrPres (ref. 3). TSEs can be transmitted between organisms by an enigmatic agent (prion) that contains PrPres (refs 4 and 5). To evaluate systematically the relationship between infectivity, converting activity and the size of various PrPres-containing aggregates, PrPres was partially disaggregated, fractionated by size and analysed by light scattering and non-denaturing gel electrophoresis. Our analyses revealed that with respect to PrP content, infectivity and converting activity peaked markedly in 17-27-nm (300-600 kDa) particles, whereas these activities were substantially lower in large fibrils and virtually absent in oligomers of ≤5 PrP molecules. These results suggest that non-fibrillar particles, with masses equivalent to 14-28 PrP molecules, are the most efficient initiators of TSE disease.
In designing strategies to limit TSE infections and their propagation within hosts, it remains important to identify the most infectious particles and their molecular composition. For several protein aggregation diseases, such as Alzheimer's disease and other amyloidoses, recent studies suggest that instead of being pathological, the formation of large amyloid fibrils might be a protective process that sequesters more dangerous subfibrillar oligomers of the amyloidogenic peptide or protein into relatively innocuous deposits1. Thus, efforts to disaggregate amyloid deposits might do more harm than good. Although TSE infectivity is associated with a wide range of PrPres aggregate states6, little is known about the relative levels of infectivity with respect to particle size, PrP content, or other potential constituents. Furthermore, the size of the smallest infectious unit remains under dispute. Reports that sodium dodecyl sulphate (SDS)-solubilized scrapie PrP monomers can be isolated as infectious units7-9 have not been confirmed10,11. Radiation inactivation12-14 and liposome solubilization studies15 have suggested that the minimal infectious unit is between 50-150 kDa, which would correspond to 2-6 PrP molecules, but no such small oligomers have been isolated and determined to be infectious. The present study compares the scrapie infectivity and in vitro PrP-converting activity associated with particles ranging in size between the ill-defined smallest infectious unit and large amyloid fibrils, and evaluates which are the most active with respect to PrP content. An infectivity bioassay in hamsters was used to measure infectivity directly, whereas much faster in vitro PrP conversion assays were used as surrogates and supplements for the bioassay. Previous studies have shown extensive correlation between infectivity and in vitro converting activity6,16.
To break down large PrPres aggregates and create a range of smaller particles for evaluation of these activities, preparations of largely purified PrPres were subjected to treatments with a variety of detergents and sonication. Initial results revealed that relatively small particles with high levels of converting activity were produced when SDS was used at a concentration of∼1% (Supplementary Fig. S1). Subsequent analyses of the alkyl sulphate family of detergents indicated that further optimization was obtained by switching to sodium n-undecyl sulphate (SUS) treatment (Supplementary Fig. S2). SUS-treated samples were diluted so that the final SUS concentration (0.1%) was well below the critical micelle concentration, and fractionated according to size using flow field-flow fractionation (FlFFF). Particle molar mass and radius measurements were obtained through in-line light scattering and refractive index detectors. PrP molecules eluted in two major peaks, one spanning fractions 5-8 and another spanning fractions 19-27 (Fig. 1a, filled circles). Dot-blot-based solid-phase conversion assays revealed that converting activity was first observed near fraction 10, rose to a plateau from fractions 12-18, and peaked around fraction 23 (Fig. 1a, open circles). Data obtained from suspension-based conversion assays were consistent with these results, and confirmed that the products observed in the solid-phase conversion assays represented proteinase K-resistant PrP fragments of the expected size3 (Supplementary Fig. S3a). To assess relative infectivity levels, fraction aliquots were also quickly diluted into normal hamster brain homogenate and inoculated intracerebrally into hamsters. A marked shortening of the incubation period of disease was observed near fraction 9, indicating a substantial increase in infectivity17, which peaked at fraction 12 and remained relatively steady throughout the rest of the elution (Fig. 1a, red squares). Because titration of both purified PrPres and scrapie brain homogenate stocks produced consistent incubation periods of disease with respect to levels of PrPres (Supplementary Fig. S4a), LD50 (dose lethal to 50% of animals tested) values derived from 263K strain scrapie brain homogenates were used to estimate the levels of infectivity in the fractionated material (Supplementary Fig. S4b). This analysis indicated that the 28-day shortening of incubation period between fractions 7 and 12 corresponded to a >600-fold increase in infectivity. Given limits to the resolution of the FlFFF technique, it is likely that the low levels of infectivity and converting activity detected in fractions 5-8 actually represent the leading edge of activities associated with larger particles that peak in fractions ≤9. These results revealed a major discordance between the levels of PrP, converting activity and infectivity with respect to the size of the particles found in preparations of PrPres.
Figure 1.
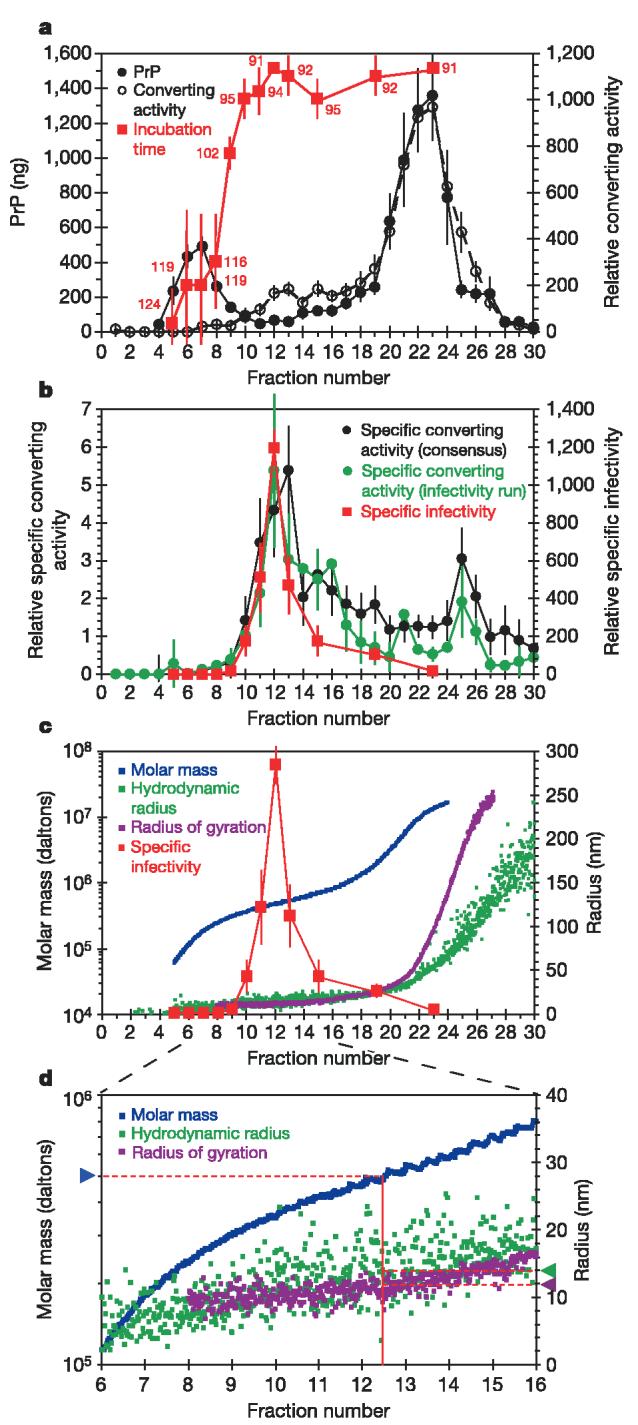
Analysis of fractionated PrPres. a, Quantification of PrP (per 1-ml fractions) by dot immunoblotting with anti-PrP monoclonal antibody 3F4 (n ranged from 3 to 16 from four FlFFF runs), analysis of converting activity by solid-phase conversion assay (n = 9 from four FlFFF runs) and scrapie infectivity analysis by incubation time assays in hamsters (n = 4 animals from one FlFFF run). Mean incubation periods (days) are indicated by red numbers in the plot. b, Calculated specific converting activity for the individual FlFFF run assessed for infectivity (n = 4 converting activity analyses), the consensus of all four FlFFF runs (n = 9 converting activity analyses) and specific infectivity (n = 4 animals). c, In-line light scattering analyses of FlFFF runs indicating MW (n = 3), rh (n = 4) and rg (n=4).The specific infectivity trace from b is superimposed. d, Expansion of a region of the plot shown in c. The solid red line indicates the centre of fraction 12, and the dashed red lines lead to arrowheads, which indicate the mass and radii of particles at this point in the elution. Mean values are shown; error bars represent ±standard error except for incubation time of disease, where they represent ±s.d.
Because PrP is often thought to be the major protein component of the infectious agent, the levels of converting activity and infectivity in the FlFFF fractions were divided by their total PrP content to give relative ‘specific converting activity’ (Fig. 1b, circles; see also Supplementary Fig. S3b) and ‘specific infectivity’ values (Fig. 1b, c, red squares). Both of these values peaked near fraction 12 and dropped off steeply to each side of the peak. Indeed, the early PrP-containing fraction 7 had∼3,000-fold lower specific infectivity than the peak fraction 12, and the large aggregates in fraction 23 had∼70-fold lower specific infectivity. This analysis revealed a tight correlation between specific infectivity and specific converting activity, and demonstrated that by far the most active particles per unit PrP peaked in fraction 12. Light scattering analyses of these particles indicated that they have apparent weight-average molar masses (MW) of several hundred kDa (Fig. 1c, d, blue squares) and apparent radii in the 12-14-nm range (Fig. 1c, d, green and violet squares).
Table 1 summarizes the size parameters determined from four independent FlFFF experiments assessed for converting activity, and a single fractionation analysed by infectivity bioassays. The ratio (ρ) of the radius of gyration (rg) to hydrodynamic radius (rh) was calculated to estimate the compactness of the particles with peak specific infectivity and converting activity (fraction 12). The values (∼0.90) are typical of fairly compact, spherical or ellipsoid shapes18. By comparison, much higher ρ values were obtained for fractions 21 and 26, indicating the predominance of highly extended structures. Mean apparent values of∼600 kDa (MW) and∼13.5 nm (rh) were determined for the material in fraction 12. These values presumably represent the size of the infectious particles including any bound SUS molecules. Analyses of a set of proteins (bovine serum albumin, ferritin, thyroglobulin) in the presence or absence of SUS showed that observed MW and rh values could be increased by as much as 73% and 60% respectively in the presence of SUS (data not shown), suggesting that the particles in fraction 12 may have detergent-subtracted MW values as low as∼300 kDa and rh values as low as∼8.5 nm. Thus, these data indicated that the particles with the highest specific infectivity and specific converting activity were approximately 300-600 kDa, roughly spherical-to-elliptical, and 17-27 nm in diameter.
Table 1.
Biophysical parameters of fractionated PrPres
| Fraction | MW (kDa) | (r g)z (nm) | (r h)z (nm) | p |
|---|---|---|---|---|
| 6 (monomer/small PrP oligomers) | 155 ± 62 | ND | 5.0 ± 1.7 | ND |
| 12 (peak specific infectivity) | 535 | 12.4 | 13.4 | 0.93 |
| 12 (peak specific converting activity) | 620 ± 331 | 12.1 ± 2.0 | 13.5 ± 0.9 | 0.90 |
| 21 (intermediate fibrils) | 7,770 ± 4,270 | 51.9 ± 11.5 | 37.4 ± 6.7 | 1.38 |
| 26 (largest fibrils) | 15,220 ±9,210 | 230.1 ±46.3 | 90.4 ±8.1 | 2.35 |
Weight-average molar mass (MW), z-average radius of gyration ((r g)z) and z-average hydrodynamic radius ((r h)z) values (mean ± s.d.) were determined from four individual fractionations of PrPres. Values for one fractionation of PrPres assayed for infectivity are also shown. p = r g/r h. ND, not determined.
To assess more accurately the oligomeric state of PrP in the early fractions (<9) of the FlFFF separation, samples were subjected to PAGE without further denaturation. Protein (silver) staining (Fig. 2a) and PrP immunoblot (Fig. 2b) analyses of these gels revealed that PrP monomers and a ladder of discrete oligomers up to apparent pentamers (most visible in the immunoblot) peaked in fractions 5-8, which were extremely low in specific infectivity and specific converting activity (Fig. 1b). However, in fractions 11-13, where the highest specific infectivity and specific converting activity were found, monomeric-to-pentameric structures were greatly reduced and larger oligomers above the 250 kDa marker predominated (Fig. 2a, b). The converting activity associated with these oligomers was directly assessed by subjecting detergent-treated PrPres to PAGE as described above, and then electro-blotting the material onto a polyvinylidene difluoride (PVDF) membrane for solid phase conversion analysis. Conversion products were only observed at the bands above the 150-250 kDa markers after SDS (Fig. 2d) or SUS (Supplementary Fig. S5c) treatments, even though the PrP content in the monomer and small oligomer bands was equivalent to, or greatly exceeded, the protein content in the higher bands (Fig. 2c; see also Supplementary Fig. S5a, b). Thus, results obtained from PAGE-based analyses were in excellent agreement with those obtained from dot-blot-based solid-phase conversion analyses (Fig. 1a), and revealed that infectivity and converting activity co-fractionated with structures larger than PrP pentamers, whereas PrP pentamers and smaller oligomers were devoid of converting activity.
Figure 2.
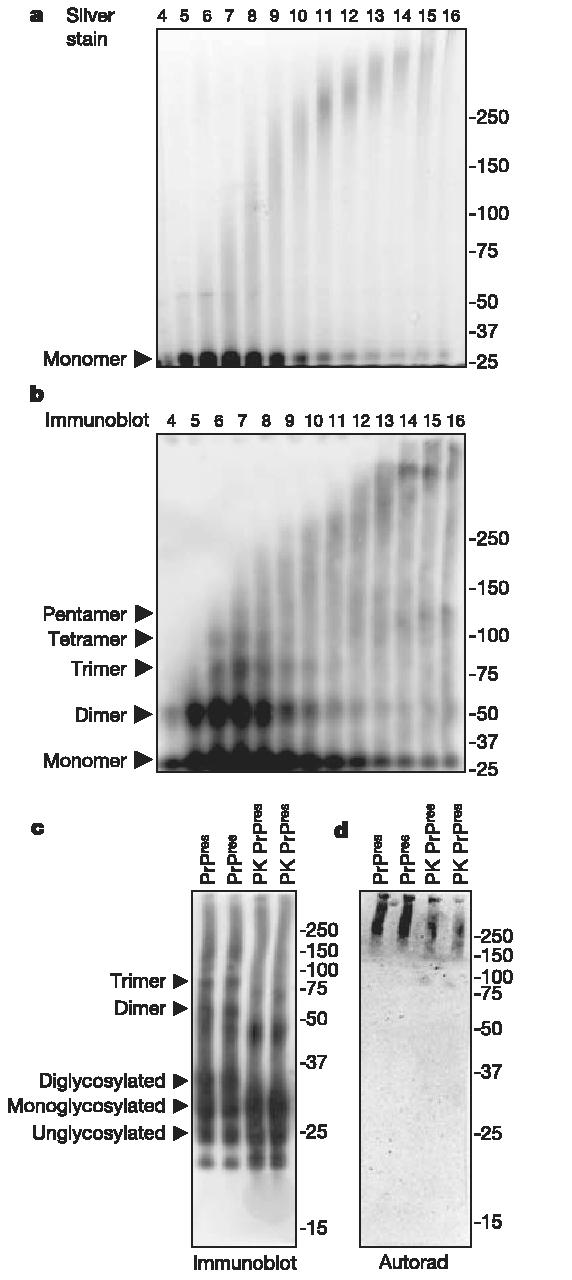
PAGE analyses of detergent-treated PrPres. a, b, Samples from FlFFF fractions were subjected to PAGE on 3-8% Tris-acetate gels and analysed by either silver stain (a) or immunoblotting with anti-PrP monoclonal antibody 3F4 after transfer to PVDF (b). c, d, Additional samples of purified PrPres were boiled in SDS-PAGE buffer without sonication, subjected to PAGE on 10% Bis-Tris gels, and analysed in duplicate by either immunoblotting (c) or solid-phase conversion (d). Fraction numbers and types of PrPres used (PK PrPres, proteinase K-digested PrPres) are shown at the top. PrP glycoforms and oligomers are indicated on the left, and molecular weight standards (kDa) are shown on the right.
To visualize the sizes and shapes of fractionated PrPres particles, samples from representative FlFFF fractions were analysed by transmission electron microscopy (Fig. 3). Consistent with the high r values noted in Table 1, fraction 26 contained a preponderance of long fibrils, whereas fraction 21 contained shorter fibrils in conjunction with more amorphous material. Analysis of material from fractions 10 and 15 revealed a collection of smaller amorphous and spherical particles. Although it was unclear whether all of the amorphous/spherical particles contained PrP, the results confirmed that there were no visible fibrils in these fractions. Thus, the size and shape of the particles detected by transmission electron microscopy agreed with the light scattering measurements, indicating that the most infectious particles of PrPres were roughly spherical or ellipsoidal in nature, and∼20-25 nm in diameter.
Figure 3.
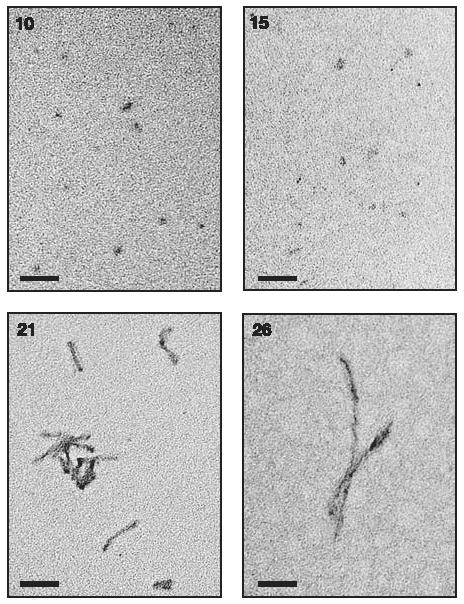
Transmission electron microscopy analyses of fractionated PrPres. Fraction numbers are indicated in the upper left of each panel. No particles were visible on control grids loaded with buffer alone. Scale bars, 100 nm.
TSE infectious units (prions) are likely to require both biochemical activity as initiators of PrP conversion and stability against degradation in the environment and the host. Whereas large PrPres aggregates might be expected to have greater stability, smaller oligomers (4-15-mers) have been predicted to have greater converting activity and infectivity per unit mass19,20. Our present data provide systematic evidence that although the infectivity per particle did not vary by more than approximately twofold between different sizes of infectious aggregates (see Supplementary Discussion), the most infectious units per mass of PrP, or the best apparent compromise between stability and activity, are∼17-27-nm particles of∼300-600 kDa. If these infectious particles are composed solely of PrP molecules averaging∼21.5 kDa each (which may not be the case, see Supplementary Discussion), this would correspond to an oligomer of 14-28 PrP molecules. Interestingly, this size range is consistent with the smallest disease-associated PrP aggregates (600 kDa) observed previously21. Our observations that the smallest stable unit with PrP converting activity is larger than a 5-mer concur with the results of a previous study that showed that SDS-generated oligomers comprising 4-6 PrP molecules with diameters of∼10 nm are not infectious22. Furthermore, we join others in failing to confirm previous reports of the generation of infectious PrP monomers in the presence of alkyl sulphate detergents7-11. However, although we screened deliberately for conditions that generate small filterable particles with converting activity, it remains possible that other conditions can stabilize infectious particles that are smaller than PrP hexamers.
The fact that the most infectious units are much smaller than the amyloid fibrils that are often observed in TSE-infected tissues and tissue extracts reinforces concerns that incomplete attempts to destabilize PrPres aggregates for the purposes of therapeutics or decontamination might result in unintended increases in infectivity. Consistent with this possibility, we observed that resonication (in 1% SUS) of large fibrillar PrPres fractions from the FlFFF separation decreased the average fibril length (according to electron microscopy) and increased converting activity by several-fold (data not shown), and others have reported that sonication of PrPres in the presence of phospholipids can increase scrapie infectivity levels15. This is not to say that the most infectious particles are necessarily derived from in vitro fragmentation of PrPres fibrils. Another strong possibility is that these particles are derived primarily from a distinct non-fibrillar PrP ultrastructure in TSE-infected brain tissue, such as the commonly observed amorphous membrane-associated deposits23. Collectively, our observations support the emerging view that with protein folding/aggregation diseases, smaller subfibrillar particles may be much more pathological than larger amyloid fibrils or plaques.
METHODS
Partial disaggregation of PrPres. PrPres was purified from scrapie-infected (263K strain) hamster brain and treated with proteinase K as described previously24 to produce a product of >90% purity. Unless designated otherwise, the samples were pelleted (20,800g, 20 min, 4 °C), re-suspended to 0.1 mg ml-1in 20 mM Tris pH 7.0 containing 1% SUS, sonicated for 1 min in a cup horn at maximum power, frozen in a dry ice/ethanol bath for 5 min, thawed, and incubated at 37 °C for 1 h. Additional FlFFF experiments in which the sonication step was omitted have shown that the intermediate-sized non-fibrillar oligomers with the highest specific converting activity are still generated, and the recovery of converting activity in fractions containing predominantly PrP monomers to pentamers is not enhanced (data not shown). In other designated experiments, samples of PrPres (0.25 mg ml-1) were simply boiled for 2 min in SDS-PAGE buffer (250 mM Tris pH 7.0, 1% SDS, 5% glycerol, 2.5% β -mercaptoethanol, 0.00025% bromophenol blue) without sonication.
Fractionation of PrPres. Samples of partially disaggregated PrPres (50μ g) were filtered through 0.2μ m Nanosep centrifugal devices (Pall Life Sciences) by centrifugation (500g, 20 min, 25 °C), and the filtrate was subjected to asymmetrical flow field-flow fractionation25 on an Eclipse F separation system (Wyatt Technology Europe). The channel was 26.5 cm in length and 350μ m in height, constructed with a trapezoidal spacer of maximal width 21 mm at the inlet, and lined with a 10-kDa cutoff polyethersulphone membrane at the accumulation wall. The sample was loaded in five 100-μ l injections, then eluted with 20 mM Tris pH 7.0 containing 0.1% SUS at a channel flow of 1 ml min-1 and a cross flow decreasing from 3 ml min-1 to 0 ml min-1 over 20 min while collecting 1-ml fractions.
Light scattering analyses. Static light scattering, refractive index and dynamic light scattering measurements were carried out on DAWN EOS, Optilab DSP and WyattQELS instruments, respectively (Wyatt Technologies), connected in-line to the FlFFF system. Weight-average molar mass (MW), z-average radius of gyration ((rg)z) and z-average hydrodynamic radius ((rh)z) values were calculated using ASTRA analysis software (version 4.90.07).
Solid-phase PrP conversion. Samples (50-250μ l) of fractionated PrPres were loaded onto PVDF membranes using a 96-well dot-blot apparatus as described previously26 or transferred by electro-blotting. Blocking and conversion on membrane sheets (∼55 cm2) were carried out as described27 under Gdn-free/ detergent-free conditions with the following exceptions: conversion reactions (5 ml) on each membrane sheet contained 500,000 counts per min 35S-labelled PrPsen, and 0.1% fetal bovine serum was replaced with 2% BSA. Proteinase K digestion (5 ml; 10μ gml-1) was terminated by drying the membrane, which was subjected to autoradiography and PhosphorImager analysis. Relative amounts of conversion in dot-blot analyses were obtained by comparison to a series of PrPres standards (1-1,000 ng) loaded onto each membrane sheet.
Incubation time bioassays for scrapie infectivity. Immediately after FlFFF separation, samples of PrPres were diluted fivefold into physiological phosphate buffer containing 1.25% normal hamster brain homogenate and 40 mM sucrose, and 50μ l aliquots were inoculated intracerebrally into Syrian golden hamsters. The incubation period of disease was defined as the number of days from inoculation to euthanization when confirmed to have clinical disease (recumbent animals).
Transmission electron microscopy. Samples of fractionated PrPres (∼5m l) were placed on 400-mesh parlodian-coated nickel grids, incubated for 1 h, washed with H2O (three times for 10 min), stained with 1% ammonium molybdate for 30 s, dried and visualized on an 80-kV Hitachi H7500 microscope fitted for image capture with a Hamamatsu side-mounted model C4742-95 CCD camera and Advantage HR/HR-B digital image software (AMT). All steps were performed at ambient temperature, and all reagents were passed through 0.02-m m filters within an hour of use.
Supplementary Material
Acknowledgments
Acknowledgements We thank C. Y. Huang and D. Follmann (Biostatistics Research Branch, NIH/NIAID) for performing statistical analyses. We thank B. Chesebro, G. S. Baron and S. J. Robertson for critiquing the manuscript. This research was supported in part by the Intramural Research Program of the NIH/NIAID. V.L.S. acknowledges support from the Alberta Heritage Foundation for Medical Research through a clinical fellowship award.
Footnotes
Author Contributions J.R.S. spearheaded the project, developed the critical methods and performed the PrPres disaggregation, fractionation and particle analyses. G.J.R. and A.G.H. purified PrPres and performed bioassays and other supporting experiments. R.E.R. provided bioassay standard curve data. V.L.S. and S.F.H. performed electron microscopy. B.C. helped with project design, data interpretation and writing (primarily with J.R.S.)
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
References
- 1.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 2.Kayed R, et al. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 3.Kocisko DA, et al. Cell-free formation of protease-resistant prion protein. Nature. 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- 4.Prusiner SB. Prions. Proc. Natl Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silveira JR, Caughey B, Baron GS. Prion protein and the molecular features of transmissible spongiform encephalopathy agents. Curr. Top. Microbiol. Immunol. 2004;284:1–50. doi: 10.1007/978-3-662-08441-0_1. [DOI] [PubMed] [Google Scholar]
- 6.Caughey B, Raymond GJ, Kocisko DA, Lansbury PT. Scrapie infectivity correlates with converting activity, protease resistance, and aggregation of scrapie-associated prion protein in guanidine denaturation studies. J. Virol. 1997;71:4107–4110. doi: 10.1128/jvi.71.5.4107-4110.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 8.Brown P, Liberski PP, Wolff A, Gajdusek DC. Conservation of infectivity in purified fibrillary extracts of scrapie-infected hamster brain after sequential enzymatic digestion or polyacrylamide gel electrophoresis. Proc. Natl Acad. Sci. USA. 1990;87:7240–7244. doi: 10.1073/pnas.87.18.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Safar J, et al. Molecular mass, biochemical composition, and physicochemical behaviour of the infectious form of the scrapie precursor protein monomer. Proc. Natl Acad. Sci. USA. 1990;87:6373–6377. doi: 10.1073/pnas.87.16.6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hope J. The nature of the scrapie agent: the evolution of the virino. Ann. NY Acad. Sci. 1994;724:282–289. doi: 10.1111/j.1749-6632.1994.tb38917.x. [DOI] [PubMed] [Google Scholar]
- 11.Morillas M, Vanik DL, Surewicz WK. On the mechanism of α-helix to β-sheet transition in the recombinant prion protein. Biochemistry. 2001;40:6982–6987. doi: 10.1021/bi010232q. [DOI] [PubMed] [Google Scholar]
- 12.Alper T, Haig DA, Clarke MC. The exceptionally small size of the scrapie agent. Biochem. Biophys. Res. Commun. 1966;22:278–284. doi: 10.1016/0006-291x(66)90478-5. [DOI] [PubMed] [Google Scholar]
- 13.Gibbs CJ, Gajdusek DC, Latarjet R. Unusual resistance to ionizing radiation of the viruses of kuru, Creutzfeldt-Jakob disease, and scrapie. Proc. Natl Acad. Sci. USA. 1978;75:6268–6270. doi: 10.1073/pnas.75.12.6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellinger-Kawahara CG, Kempner E, Groth D, Gabizon R, Prusiner SB. Scrapie prion liposomes and rods exhibit target sizes of 55,000 Da. Virology. 1988;164:537–541. doi: 10.1016/0042-6822(88)90569-7. [DOI] [PubMed] [Google Scholar]
- 15.Gabizon R, McKinley MP, Prusiner SB. Purified prion proteins and scrapie infectivity copartition into liposomes. Proc. Natl Acad. Sci. USA. 1987;84:4017–4021. doi: 10.1073/pnas.84.12.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raymond GJ, et al. Molecular assessment of the potential transmissibilities of BSE and scrapie to humans. Nature. 1997;388:285–288. doi: 10.1038/40876. [DOI] [PubMed] [Google Scholar]
- 17.Prusiner SB, et al. Molecular properties, partial purification, and assay by incubation period measurements of the hamster scrapie agent. Biochemistry. 1980;19:4883–4891. doi: 10.1021/bi00562a028. [DOI] [PubMed] [Google Scholar]
- 18.Gast K, et al. In: Laser Light Scattering in Biochemistry. Harding SE, Sattelle DB, Bloomfield VA, editors. Cambridge; The Royal Society of Chemistry: 1992. pp. 209–224. [DOI] [PubMed] [Google Scholar]
- 19.Masel J, Jansen VA, Nowak MA. Quantifying the kinetic parameters of prion replication. Biophys. Chem. 1999;77:139–152. doi: 10.1016/s0301-4622(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 20.Masel J, Genoud N, Aguzzi A. Efficient inhibition of prion replication by PrP-Fc2 suggests that the prion is a PrPSc oligomer. J. Mol. Biol. 2005;345:1243–1251. doi: 10.1016/j.jmb.2004.10.088. [DOI] [PubMed] [Google Scholar]
- 21.Tzaban S, et al. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry. 2002;41:12868–12875. doi: 10.1021/bi025958g. [DOI] [PubMed] [Google Scholar]
- 22.Riesner D, et al. Disruption of prion rods generates 10-nm spherical particles having high α-helical content and lacking scrapie infectivity. J. Virol. 1996;70:1714–1722. doi: 10.1128/jvi.70.3.1714-1722.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeffrey M, et al. Correlative light and electron microscopy studies of PrP localisation in 87V scrapie. Brain Res. 1994;656:329–343. doi: 10.1016/0006-8993(94)91477-x. [DOI] [PubMed] [Google Scholar]
- 24.Raymond GJ, Chabry J. In: Techniques in Prion Research. Lehmann S, Grassi J, editors. Birkhauser; Basel: 2004. pp. 16–26. [Google Scholar]
- 25.Wahlund K-G. In: Field-Flow Fractionation Handbook. Schimpf M, Caldwell K, Giddings JC, editors. New York; John Wiley & Sons: 2000. pp. 279–294. [Google Scholar]
- 26.Kocisko DA, et al. New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J. Virol. 2003;77:10288–10294. doi: 10.1128/JVI.77.19.10288-10294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maxson L, Wong C, Herrmann LM, Caughey B, Baron GS. A solid-phase assay for identification of modulators of prion protein interactions. Anal. Biochem. 2003;323:54–64. doi: 10.1016/j.ab.2003.07.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.