

Abstract
The absolute stereochemistry of the three unresolved structural components in neamphamide A (1) was determined to be (R)-β-methoxy-L-tyrosine, (2R,3R,4S)-4-amino-7-guanidino-2,3-dihydroxyheptanoic acid, and (2R,3R,4R)-3-hydroxy-2,4,6-trimethylheptanoic acid. Stereochemical assignments were made by chemical degradation of 1, derivatization of the resulting products, and then spectroscopic and chromatographic comparison of the derivatives with synthetically prepared standards. Using the same analytical protocol developed for 1, the β-methoxytyrosine residue in papuamide B (2) was found to be (R)-β-methoxy-D-tyrosine. This represents a rare example of divergent stereochemistry in an unusual amino acid residue that is present in two closely related classes of peptides.
Introduction
A series of potent HIV inhibitory cyclic depsipeptides have recently been described from a number of marine sponges. Peptides in this family include neamphamide A (1) from Neamphius huxleyi,1 the callipeltins from Callipelta sp.,2 and the papuamides from Theonella sponges.3 In addition to antiviral activity, these compounds have been reported to exhibit potent antifungal, cytotoxic, and sodium ionophore properties.4 Distinguishing structural characteristics of this family of peptides include a preponderance of unusual amino acid residues and unique N-terminal polyketide derived moieties. The novel structural features and diverse biological effects of these peptidic metabolites have generated considerable interest among synthetic chemistry groups. As a result, syntheses of structural subunits such as 3,4-dimethylglutamine (3,4-diMeGln),5 4-amido-7-guanidino-2,3-dihydroxyheptanoic acid (3, Agdha),6 -and 3-hydroxy-2,4,6-trimethylheptanoic acid (4, Htmha)7 have been described. These efforts provided essential building blocks for use in total synthesis of the parent peptides and they also helped to unambiguously define the absolute stereochemistry of several key chiral centers. However, synthetic efforts to prepare the intact natural products have been hampered by unresolved questions concerning the stereochemistry of the β-methoxytyrosine (5, βOMeTyr) residues which occur throughout this family of peptides. While stereoselective syntheses of all four diastereomers of 5 has been reported,8 the absolute stereochemistry of this residue in the natural peptides was never established. Decomposition of 5 during acid hydrolysis of the parent peptide prevented the successful application of standard techniques such as Marfey’s analysis.9 In the original report of neamphamide A (1), the chirality of the Agdha, Htmha and βOMeTyr residues was left undefined. Described herein is the first complete stereochemical assignment of neamphamide A (1) and a general method for determining the absolute stereochemistry of βOMeTyr (5) residues in other peptides. This technique was also used to unambiguously define the stereochemistry of the same residue in papuamide B (2), and the results of these analyses should help focus the synthetic efforts underway to prepare the parent peptides.
Results and Discussion
Our prior studies had established the absolute stereochemistry of 9 amino acid residues of neamphamide A (1) as D-Arg, L-Leu, D- and L-Asn, L-NMeGln, L-homoproline, D-allo-Thr (two residues), and (3S,4R)-3,4-diMe-L-Gln. In addition, C4 of the Agdha (3) moiety was assigned a S-configuration by chemical degradation involving diol-cleavage, oxidative work up, and acid hydrolysis, followed by Marfey’s analysis of the resulting arginine subunit.1 In the current study, a synthetic standard of 3 with defined stereochemistry was available for comparative purposes,6 but direct application of Marfey’s method to the peptide hydrolysate to determine the stereochemistry at C2 and C3 proved unsuitable. The L-FDAA derivative of 3 had poor retention on reversed-phase chromatography packings and unacceptable peak broadening in the LC-MS analysis. Accordingly, the γ-amino acid 3 was isolated from the hydrolysate of 1 and converted into cyclic derivatives appropriate for NMR based configurational analysis at C2, C3, and C4. Compound 1 was hydrolyzed in 6N HCl (107°C, 18.5 h), defatted with EtOAc, and the resulting aqueous hydrolysate was purified by anion- and then cation-exchange chromatography to give a mixture of 3 and Arg. While dissolved in CD3OD for NMR studies, compound 3 was gradually converted to the trideuteriomethyl ester derivative 6. This transformation was evidenced from 1H resonances at δ 4.03 (H3 of 3) and 4.37 (H2 of 3), which diminished in intensity while resonances at δ 4.03 (H3 of 6) and 4.35 (H4 of 6) emerged and grew with time (Supporting Information). Positive ion ESI-MS data obtained before and after the NMR experiments showed the appearance of a dominant ion peak at m/z 25210 and complete loss of the original peak at m/z 235 (Supporting Information), which supported the conversion of 3 to the methyl ester 6. Preparation of the acetonide of 6 with 2,2-dimethoxypropane and PPTS in DMF exclusively yielded the 2,3-O,O-isopropylidene derivative 7.11 In 1D gNOESY experiments, selective irradiation of one of the acetonide methyl signals at δ 1.36 excited the two oxymethine protons at δ 4.65 (H2) and δ 4.74 (H3). No NOE enhancement was observed with these protons upon irradiation of the other methyl group at δ 1.40, thus indicating H2 and H3 were on the same face of the dioxolane ring. To correlate the erythro-diol moiety at C2-C3 with the S-configuration of neighboring C4, 7 was treated with 2N HCl to recover 3, which was then refluxed in H2O for 60 h to achieve partial conversion into Agdha γ-lactam (8, 63% yield).12 The coupling constants observed between protons on the lactam ring of 8 were JH2,H3=4.7-4.9 Hz and JH3,H4=3.6 Hz. This combination of couplings was compared to the reported values for four similarly functionalized synthetic γ-lactams (Scheme 1),13 and they were only compatible with the (2R,3R,4S)-isomer. Further support for this stereochemical assignment was provided by mutual NOE interactions between H2 (δ 4.31) and H4 (δ 3.52) in a 1D gNOESY experiment. Thus, the configuration of 3 in the peptide 1 was determined to be (2R,3R,4S), which is consistent with the stereochemistry previously assigned to the Agdha residue in callipeltin A (9).2
Scheme 1.

aReagents and conditions: (a) 6 N HCl, 107 °C, 18.5 h; (b) solvent partitioning, 6 N HCl/EtOAc; (c) CD3OD, 65 h; (d) PPTS, 2,2-dimethoxypropane, DMF, 45 °C (30 min) -> 60 °C (1.5 h); (e) 2 N HCl in MeOH,60 °C, 15.5 h; (f) H O, reflux, 60 h. b bData reproduced from ref. 55.
The EtOAc extract of the acid hydrolysate of neamphamide A (1) contained free Htmha (4) as the major constituent. Careful interpretation of the 1H, gCOSY, and gHSQC spectra of the EtOAc extract allowed assignment of the NMR signals from 4, and these were compared with published data for four synthetic diastereomers of this fatty acid7d including (2R,3R,4R)-, (2S,3R,4R)-, (2S,3R,4S)-, and (2R,3R,4S)-4. 1H NMR chemical shifts for 4 showed the greatest similarity to those for the (2R,3R,4R)-isomer, which indicated that the hydroxy acid recovered from the hydrolysis of 1 had to have either (2R,3R,4R) or (2S,3S,4S)-stereochemistry. To establish the absolute stereochemistry of these chiral centers, 4 from 1 was condensed with (S)-(+)-phenylglycine methyl ester (PGME)14 and the product was subjected to LC-MS analysis. To prepare appropriate chromatographic standards, synthetic (2R,3R,4R)-4 was reacted either with (S)-(+)- or (R)-(-)-PGME to obtain (2R,3R,4R)-4 (S)-(+)-PGME amide and (2R,3R,4R)-4 (R)-(-)-PGME amide. The latter derivative is chromatographically equivalent to the (S)-(+)-PGME amide of enantiomeric (2S,3S,4S)-4 when chromatographed on an achiral support. The reversed-phase HPLC retention time for the PGME amide prepared from the natural product was identical to that for synthetic (S)-(+)-PGME amide, thus verifying a (2R,3R,4R) configuration for the subunit 4 in 1, which was the same as that established for callipeltin A (9).7c, 7d
The synthetic procedure we developed to obtain reference standards of the four diastereomers of βOMeTyr (5) was based on the stereoselective synthesis of β-hydroxytyrosine reported by Boger.15 An adaptation of Boger’s procedure (Scheme 2), in which a partially diastereoselective addition of a metallated Schöllkopf reagent16 (10) to 4-benzyloxybenzaldehyde formed the two key stereogenic centers, afforded roughly a 1:1 mixture of two epimeric carbinols (11a,b). Chromatographic separation afforded 11a and 11b in stereochemically pure form. Spectral correlation with Boger’s published spectra15 permitted the assignment of configuration for each isomer. Methylation of the hydroxyl group and hydrolysis of the bis-lactim ether using mild acid afforded stereochemically pure (S)- and (R)-O-benzyl-L-tyrosine methyl esters (13a and 13b, respectively). Transesterification with allyl alcohol mediated by titanium tetraisopropoxide afforded the allyl esters 14a and 14b in 15 and 17% overall yields, respectively, from 4-benzyloxybenzaldehyde. Repetition of this procedure using the enantiomeric Schöllkopf reagent ent-10 afforded ent-14a and ent-14b in stereochemically pure form and in comparable yields. Treatment of the allyl esters 14a, 14b, ent-14a, and ent-14b with 0.2 N LiOH and subsequent hydrogenolysis on Pd(OH)2 gave (S)-βOMe-L-Tyr, (R)-βOMe-L-Tyr, (R)-βOMe-D-Tyr, and (S)-βOMe-D-Tyr, respectively, in quantitative yields.
Scheme 2.

aReagents and conditions: (a) BuLi, 4-BnPhCHO, THF, -30 °C, 3 h; (b) NaI, CH3I, THF, 0 °C, 3 h; (c) 0.25 N HCl, THF-MeCN, 25 °C, 16 h; (d) Ti(Oi-Pr)4, allyl alcohol, 25 °C, 24 h; (e) 0.2 N LiOH-MeOH, rt, 2 days; (f) H2, Pd(OH)2/C, MeOH-4.4% HCOOH, rt, 4 h.
In a stability test of 5 using the (S)-βOMe-D-Tyr as a model, the amino acid tolerated 2 N HCl but partially decomposed when kept in 6 N HCl for 1.5 h at room temperature to give a burgundy-colored residue that was insoluble in water but soluble in MeOH. We presumed that acid catalyzed decomposition of the βOMeTyr residue might involve elimination of the β-methoxy group followed by a nucleophilic attack to form condensation products as illustrated in Scheme 3. In an initial effort to avoid decomposition of this residue during hydrolysis, 1 was fully protected with Cbz and then hydrolyzed in alkaline media, e.g. 1 N or 2.5 N KOH-MeOH, to liberate N-Cbz-βOMeTyr or N,O-bis(Cbz)-βOMeTyr. However, the reaction was inconsistent in yield and detection of the desired products by LC-MS was not successful. Development of an alternative approach to circumvent these problems was clearly warranted. Therefore, the authentic standards of 5 were converted into 3-methoxyaspartic acid (OMeAsp) by oxidation with RuCl3-NaIO4(CCl4-MeCN-0.1N sodium phosphate buffer=2:2:3, pH 7.6),17 and then isolated as their N-Cbz derivatives by treating the dried reaction mixtures with CbzCl followed by solvent-partitioning (Scheme 4). LC-MS analysis of the N-Cbz-OMeAsp products resulted in very broad, low intensity peaks, so the amino acid derivatives were methylated with TMS-diazomethane to give 15. While the peak shape and peak intensity of the dimethyl ester 15 was improved, it was not possible to resolve the four diastereotopic standards by HPLC, even when they were chromatographed on chiral supports (Regis Sumipax OA-3100 L-Val and Regis Pirkle D-phenylglycine). Successful separation of the four diastereomers was ultimately achieved by replacing the Cbz group with TFA (Scheme 4)18 and then analyzing the products by GC-MS on a Chirasil-L-Val capillary column. Although a molecular ion for N-TFA-OMeAsp dimethyl ester (16) was not observed under the GC-MS conditions, diagnostic fragment ions, e.g. m/z 196 [M- ·COOMe-MeOH]+ and m/z 103 [C(OMe)COOMe]+ (Figure 1), were used to identify each diastereomer which eluted in the order (2R,3R), (2S,3S), (2R,3S), and (2S,3R) with satisfactory peak separation (tR: 15.5, 15.8, 16.4, and 16.7 min, respectively).
Scheme 3.

Scheme 4.

aReagents and conditions: (a) RuCl3, NaIO4, CCl4-MeCN-0.1 M sodium phosphate buffer=2:2:3, pH 7.6, rt, 16 h; (b) CbzCl, 1,4-dioxane-1 N KOH, rt, 1h; (c) TMS-diazomethane, MeOH, rt, 1 h; (d) Pd(OH)2, 4.4% HCOOH in MeOH, H2, rt, 4 h; (e) 1.25 M HCl in MeOH,110 °C, 30 min; (f) TFAA, dichloromethane, 110 °C, 5 min.
Figure 1.

Fragmentation and the typical EI-MS spectrum of N-TFA-OMeAsp dimethyl ester (16).
With appropriate standards in hand, stereochemical assignments of the βOMeTyr residues in 1 and 2 were initiated. The peptides were oxidized with RuCl3-NaIO4, hydrolyzed in 6 N HCl (110 °C, 6 h), and the hydrolysates were successively derivatized with HCl-MeOH and TFAA to provide GC-MS samples. The extracted ion traces for the m/z 196 and m/z 103 fragment ions from N-TFA-OMeAsp dimethyl ester (16) both clearly showed the elution of (2S,3S)-16 (tR15.8 min) from the oxidative degradation product of 1 and (2R,3S)-16 (tR16.4 min) from that of 2 (Supporting Information). The complete oxidation, hydrolysis, derivatization and analysis sequence was repeated with both parent peptides, and the results were consistent each time. This allowed assignment of a (R)-βOMe-L-Tyr residue in 1 and a (R)-βOMe-D-Tyr residue in 2.

In conclusion, we have defined the absolute configuration of the hydroxy acid moiety and the two unusual amino acid residues in neamphamide A (1) as (2R,3R,4R)-Htmha, (2R,3R,4S)-Agdha and (R)-βOMe-L-Tyr, respectively. This is the first time the complete stereostructure has been assigned for a member of this family of anti-HIV sponge depsipeptides. In addition, a new analytical procedure was established to determine the absolute configuration of βOMeTyr residues in peptide natural products. Application of this technique to papuamide B (2) revealed the presence of (R)-βOMe-D-Tyr in 2, which is epimeric at Cα to the corresponding residue in 1. The stereochemical divergence of the βOMeTyr residues in 1 and 2 is somewhat surprising considering the structural similarities and comparable inhibitory activity against HIV for these two peptides. However, this finding is consistent with the observation that the βOMeTyr Cβ proton in 1 is a broadened singlet at δ5.03 while the same proton in 2 appears as a 9.3 Hz doublet at δ4.24. The differing stereochemistry at Cα suggests that during biosynthesis of 1 and 2 the βOMeTyr residues arise from incorporation of L-Tyr and D-Tyr, respectively, while the oxidative modification at Cβ occurs with conserved stereochemistry regardless of the configuration at Cα. Assignment of the complete absolute stereochemistry of neamphamide A (1) and the βOMeTyr residue in papuamide B (2) should now simplify the efforts required to synthesize these peptides. In addition, the method developed for analyzing the βOMeTyr residues should be applicable to other naturally occurring peptides that contain this amino acid derivative.
Experimental Section
GC-MS analysis. Chiral GC-MS analyses were executed with an Agilent 6890 Series/5973N gas chromatograph-mass selective detector system equipped with a Varian Chirasil-L-Val capillary column (25 m × 0.25 mm i.d., 0.16 μm film thickness), using helium as a carrier gas at a linear velocity of 29.2 cm/s. Temperatures were set to 250 °C at the injection port and 220 °C at the transfer line to the detector. Samples were injected splitless and chromatographed under the oven temperature program of 60 °C for 3 min, 5°C/min heat ramp to 200 °C, and the same temperature held for 10 min. Electron-impact ionization at 70 eV was employed for mass spectral detection and elution of N-TFA-OMeAsp dimethyl ester (16) was traced by diagnostic fragment ions at m/z 196 and m/z 103.
Conversion of βOMeTyr (5) into N-TFA-OMeAsp Dimethyl Ester (16). A solution of each deprotected amino acid standard (100 μg) in a mixture of MeCN (40 μL) and H2O (30 μL) was stirred with NaIO4(2.4 mg) until the salt crystals disappeared. To this solution was added CCl4(40 μL) and a 1mg/mL solution of RuCl3hydrate in 0.1 N sodium phosphate buffer (30 μL, pH 7.6) and the biphasic mixture was vigorously stirred at room temperature for 16 h. After removal of the solvent, a half portion of the oxidation product was reacted with CbzCl (20 μL) in a mixture of 1N KOH (50 μL) and freshly distilled 1,4-dioxane (50 μL). After stirring for an hour at ambient temperature, 100 μL of 1N KOH was added to the reaction mixture and the unreacted reagent was extracted with EtOAc (100 μL × 3). The aqueous layer was then acidified with 2 N HCl (150 μL) and extracted again with EtOAc (100 μL × 5), which yielded N-Cbz-OMeAsp. This material was further esterified with TMS-diazomethane (10 μL, 2M solution in n-hexane) in MeOH (50 μL) by stirring the reaction mixture for 1 h to give N-Cbz- OMeAsp dimethyl ester (15), followed by deprotection of the N-Cbz group by hydrogenolysis on 20% Pd(OH)2on carbon (1 mg suspension in 100 μL 4.4% HCOOH in MeOH) under a H2atmosphere at room temperature for 4 h. The glass-wool filtrate of the reaction suspension was concentrated and again methylated with 1.25 M HCl in MeOH (100 μL) at 110 °C for 20 min, followed by TFA-amidation with dichloromethane/TFA anhydride (1:1, 100 μL) at the same temperature for 5 min to give N-TFA- OMeAsp dimethyl ester (16). The derivatization product was dissolved in MeCN (15 μL) and a 1 μL aliquot was subjected to GC-MS analysis. Retention times (min): 15.5 for (2R,3R)-, 15.8 for (2S,3S)-, 16.4 for (2R,3S)-, and 16.7 for (2S,3R)-16.
Analysis of the βOMeTyr Residues in Neamphamide A (1) and Papuamide B (2). A 200-μg portion of each peptide was oxidized with RuCl3/NaIO4as described in the previous section. The dried reaction mixture was dissolved in 1% MeOH in H2O, applied to a short column of reversed-phase polymer (PolysorbTM MP-1, 100 mg), and desalted by washing with the same alcoholic water mixture. The peptide was recovered from the column by elution with MeOH and then hydrolyzed in degassed 6 N HCl (200 μL) at 110 °C for 6 h. Half of the hydrolysate was derivatized with HCl-MeOH followed by dichloromethane/TFA anhydride as described above, and a 1 μL aliquot of the product dissolved in MeCN (33 μL) was subjected to GC-MS analysis. Retention times (min): 15.8 for N-TFA-OMeAsp dimethyl ester (16) from 1 and 16.4 for 16 from 2.
Supplementary Material
Chart 1.

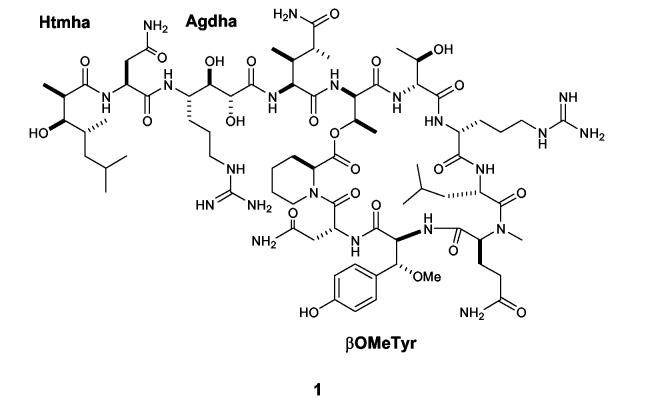
Structure of neamphamide A (1), papuamide B (2), Agdha (3), Htmha (4), and βOMeTyr (5).
Chart 2.

Structure of callipeltin A (9).
Acknowledgments
ACKNOWLEDGMENT. We thank Prof. M. V. D’Auria at Univ. Napoli “Federico II” for helpful discussions. M. A. L. gratefully acknowledges the financial support of the National Institutes of Health (AI-50888). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Footnotes
Supporting Information Available. Experimental procedures, 1H NMR spectra of 3 at 0 h and 24 h in CD3OD, positive ESI-MS spectra of 3 obtained before and after 60 h-NMR experiments, 1H NMR, gHSQC, and 1D gNOESY spectra of 7 in CD3OD, 1H NMR, gHSQC, and 1D gNOESY spectra of 8 in D2O, 1H NMR and gHSQC spectra of 4 from 1 and synthetic (2R,3R,4R)-4, GC-MS analysis of 16 derived from 1 and 2.
References and Notes
- (1).Oku N, Gustafson KR, Cartner LK, Wilson JA, Shigematsu N, Hess S, Pannell LK, Boyd MR, McMahon JB. J. Nat. Prod. 2004;67:1407–1411. doi: 10.1021/np040003f. [DOI] [PubMed] [Google Scholar]
- (2).(a) Zampella A, D’Auria MV, Paloma LG, Casapullo A, Minale L, Debitus C, Henin Y. J. Am. Chem. Soc. 1996;118:6202–6209. [Google Scholar]; (b) D’Auria MV, Zampella A, Paloma LG, Minale L, Debitus C, Roussakis C, Le Bert V. Tetrahedron. 1996;52:9589–9596. [Google Scholar]; (c) Zampella A, Randazzo A, Borbone N, Luciani S, Trevisi L, Debitus C, D’Auria MV. Tetrahedron Lett. 2002;43:6163–6166. [Google Scholar]
- (3).Ford PW, Gustafson KR, McKee TC, Shigematsu N, Maurizi LK, Pannell LK, Williams DE, de Silva ED, Lassota P, Allen TM, Van Soest R, Andersen RJ, Boyd MR. J. Am. Chem. Soc. 1999;121:5899–5909. [Google Scholar]
- (4).Trevisi L, Bova S, Cargnelli G, Danieli-Betto D, Floreani M, Germinario E, D’Auria MV, Luciani S. Biochem. Biophys. Res. Comm. 2000;279:219–222. doi: 10.1006/bbrc.2000.3906. [DOI] [PubMed] [Google Scholar]; (b) Trevisi L, Cargnelli G, Ceolotto G, Papparella I, Semplicini A, Zampella A, D’Auria MV, Luciani S. Biochem. Pharmacol. 2004;68:1331–1338. doi: 10.1016/j.bcp.2004.04.032. [DOI] [PubMed] [Google Scholar]
- (5).(a) Liang B, Carroll PJ, Joullie MM. Org. Lett. 2000;2:4157–4160. doi: 10.1021/ol006679t. [DOI] [PubMed] [Google Scholar]; (b) Okamoto N, Hara O, Makino K, Hamada Y. Tetrahedron: Asymmetry. 2001;12:1353–1358. [Google Scholar]; (c) Acevedo CM, Kogut EF, Lipton MA.Tetrahedron 2001576353–6359.18159221 [Google Scholar]
- (6).Thoen JC, Morales-Ramos ÁI, Lipton MA. Org. Lett. 2002;4:4455–4458. doi: 10.1021/ol0269852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Guerlavais V, Carroll PJ, Joullie MM. Tetrahedron: Asymmetry. 2002;13:675–680. [Google Scholar]; (b) Zampella A, Sorgente M, D’Auria MV. Tetrahedron: Asymmetry. 2002;13:681–685. [Google Scholar]; (c) Zampella A, D’Auria MV. Tetrahedron: Asymmetry. 2002;13:1237–1239. [Google Scholar]; (d) Turk JA, Visbal GS, Lipton MA. J. Org. Chem. 2003;68:7841–7844. doi: 10.1021/jo034738l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Okamoto N, Hara O, Makino K, Hamada Y. J. Org. Chem. 2002;67:9210–9215. doi: 10.1021/jo0258352. [DOI] [PubMed] [Google Scholar]; (b) Hansen DB, Wan X, Carroll PJ, Joullie MM. J. Org. Chem. 2005;70:3120. doi: 10.1021/jo047876z. [DOI] [PubMed] [Google Scholar]
- (9).Marphy P. Carlsberg Res. Comm. 1984;49:591–596. [Google Scholar]
- (10). In ref.1(a), the authors reported a molecular ion of Agdha at m/z 252 and attributed the increase of 17 mu to the formation of an NH3adduct. Although it was not mentioned if CD3OD was used in NMR experiments prior to the mass measurement, it is possible that the mass observed was the molecular ion of 16.
- (11).Meyers AI, Lawson JP. Tetrahedron Lett. 1982;23:4883–4886. [Google Scholar]
- (12).Hardy PM. Synthesis. 1978:290–291. [Google Scholar]
- (13).Huang Y, Dalton D, Carroll PJ. J. Org. Chem. 1997;62:372–376. doi: 10.1021/jo962028s. [DOI] [PubMed] [Google Scholar]
- (14).Nagai N, Kusumi T. Tetrahedron Lett. 1995;36:1853–1856. [Google Scholar]
- (15).Boger DL, Zhou J, Borzilleri RM, Nukui S, Castle SL. J. Org. Chem. 1997;62:2054–2069. doi: 10.1021/jo961346o. [DOI] [PubMed] [Google Scholar]
- (16).Schöllkopf U, Groth U, Deng C. Angew. Chem. Int. Ed. 1981;20:798–799. [Google Scholar]
- (17). Oxidation without using a buffered solution did not give OMeAsp.
-
(18). In a preliminary GC-MS experiment using D- and L-N-Cbz-Asp dimethyl esters as the models, both compounds eluted approximately 5 min after the thermal gradient reached the maximum temperature limit of the Chirasil column (200°C) with poor mutual separation. This suggested that other derivatization of the amino group instead of Cbz was required to improve the chromatographic behavior of the methylated OMeAsp derivatives.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.