

Abstract
The timing of mammalian circadian rhythm is determined by interlocking negative and positive transcriptional feedback loops that govern the cyclic expression of both clock regulators and output genes. In mammals, nuclear localization of the circadian regulators PER1–3 is controlled by multiple mechanisms, including multimerization with PER and CRY proteins. In addition, nuclear entry of mammalian PER1 (mPER1) can be regulated by a phosphorylation-dependent masking of its nuclear localization signal. Here we present evidence suggesting that nuclear localization of PER proteins is a dynamic process determined by both nuclear import and previously unrecognized nuclear export pathways. Examination of the subcellular localization of a series of truncated mPER1 proteins demonstrated that cytoplasmic localization is mediated by an 11-amino acid region with homology to leucine-rich nuclear export signals (NESs). Similar sequences were identified in mPER2 and mPER3 as well as in several insect PER proteins. The putative NESs from mPER1 and mPER2 were able to direct cytoplasmic accumulation when fused to a heterologous protein. Mutations in conserved NES residues and the nuclear export inhibitor leptomycin B each blocked the function of the NES. Full-length mPER1 was also exported from microinjected Xenopus laevis oocyte nuclei in an NES-dependent manner. The presence of a functional NES in mPER1 and mPER2 as well as related sequences in a variety of other PER proteins suggests that nuclear export may be a conserved and important feature of circadian regulators.
Circadian rhythm is a ubiquitous process that orders physiologic events in a 24-h day/night cycle (recently reviewed in Refs. 1–3). The timing of circadian clocks is established in a cell autonomous manner by interacting negative and positive transcription/translation-based negative feedback loops. The negative feedback loop begins by activating transcription of clock genes such as Period (Per) and Cryptochrome (Cry) family members. The products of these clock genes then negatively regulate their own expression, thus setting up the rhythmic oscillations of gene expression that drive the circadian clock. To produce stable oscillations of gene expression with a period of 24 h, there must also be a delay between the production and the action of the inhibitory clock gene products. Several mechanisms may contribute to delayed repression of the circadian promoter, including proteolysis of the repressors, alterations in repressor activity by post-translational modification (e.g. phosphorylation), and regulation of nuclear accumulation by alterations in rates of nuclear entry and/or nuclear egress.
Initial insights into the role of nuclear entry in the regulation of circadian rhythm came from studies on Drosophila. There, nuclear entry of the PERIOD (dPER)1 protein leads to repression of its own expression (4). A region of the dPER protein termed the cytoplasmic localization domain (CLD) (and recently recognized as a PAC domain often found carboxyl-terminal to PAS domains (5)) maintains monomeric dPER in the cytoplasm, where it is phosphorylated and degraded. When the Drosophila TIMELESS (TIM) protein accumulates to sufficient levels, it heterodimerizes with dPER, masking the CLD and allowing nuclear entry of the complex (6, 7). Nuclear dPER/Drosophila TIM represses its own expression. Delayed nuclear entry of dPER therefore generates a temporal separation between protein production and the onset of negative feedback that allows the feedback loop to oscillate on a 24-h time scale.
In mammals, PER function in circadian rhythm is less well understood. PER proteins are capable of partial repression of circadian promoters driven by the CLOCK/BMAL1 heterodimeric transcription factor, but the two CRYPTOCHROME proteins CRY1 and CRY2 appear to be much more potent repressors. Nonetheless, the PER proteins appear to be critical to proper clock function (8, 9). The Per genes are rhythmically expressed in the suprachiasmatic nucleus, and Per1 and Per2 mRNAs are induced by light pulses during night (10–12). Furthermore, mice lacking intact Per2 have a short period that rapidly becomes arrhythmic in total darkness. The critical biochemical function of the PER proteins remain unclear. One clue to their function is their intracellular localization. Several immunohistochemical studies have demonstrated that PER1 and PER2 accumulate in the nuclei of suprachiasmatic nucleus cells, suggesting a nuclear function (13, 14). However, conflicting results have been obtained in studies of PER proteins transiently expressed in tissue culture (reviewed in Ref. 15). The picture that has emerged is of multiple distinct pathways for PER proteins to enter the nucleus. We found that murine PER1 (mPER1) expressed alone in HEK 293 cells is a nuclear protein and that nuclear accumulation requires a functional nuclear localization signal (NLS) adjacent to the casein kinase Iε-binding site (16, 17). Phosphorylation of mPER1 by casein kinase Iε leads to cytoplasmic accumulation of the protein. In other cell types, e.g. COS-7 cells, mPER1 and mPER2 expressed alone are cytoplasmic (18, 19). Multiple mechanisms to relocate PER1 and PER2 to the nucleus have been described. CRYPTOCHROME proteins (CRY1 and CRY2) bind to the carboxyl terminus of mammalian PER proteins and facilitate their translocation into the nucleus (20).2 Additionally, mPER3 binding to mPER1 or mPER2 has been shown to contribute to nuclear localization of the heterodimer (19). However, neither of these mechanisms appears to fully explain the nuclear entry of PER proteins since mice lacking either mPER3 or both CRY proteins still have nuclear mPER1.
To better understand how the localization of PER proteins is determined, we examined the signals in the mPER1 protein that regulate its intracellular localization in HEK 293 cells. Domain analysis of mPER1 produced several unexpected results. First, in addition to the NLS of mPER1 at amino acids 824–851 (16), the amino terminus of mPER1 is also required for nuclear localization of the protein, as its removal led to cytoplasmic localization of mPER1. Removal of the region of mPER1 homologous to the CLD of dPER had no effect on localization of the protein. Rather, cytoplasmic localization of mPER1 was mediated by an 11-amino acid sequence immediately carboxyl-terminal to the CLD. This domain contains a leucine-rich sequence with homology to a nuclear export signal (NES) (21). The NESs of mPER1 and mPER2 are functional, as fusion of these regions to a nuclear localized yellow fluorescent protein (YFP-NLS) led to its cytoplasmic accumulation. Mutation of the NES and the inhibition of nuclear export by leptomycin B both prevented cytoplasmic localization. Nuclear export of PER proteins may be a general phenomenon since homologous sequences are present in mPER3 as well as in certain insect PER proteins. Finally, microinjection experiments using Xenopus laevis oocytes revealed that full-length mPER1, but not mPER1 with a mutant NES, was exported from nuclei following nuclear injection of the protein. The ability of PER proteins to regulate circadian rhythm may therefore be determined by combined rates of nuclear export, nuclear import, and degradation.
MATERIALS AND METHODS
Plasmid Construction
The expression constructs encoding Myc epitope-tagged full-length mPER1, Myc-tagged mPER1 protein fragment 496–1291 (Δ1–495), the tandem YFP, and the tandem YFP protein with the NLS (amino acids 709–921 from mPER1) fused to the carboxyl terminus of the second YFP (YFP-NLS) have been previously described (16). Plasmids encoding Myc tagged-mPER1 protein fragments 206–1291 (Δ1–205) and 485–1291 (Δ1–484) were constructed using QuikChange (Stratagene) to create an EcoRI site in the DNA sequence that encodes either amino acids 204 and 205 or amino acid 483, respectively, in the full-length mPER1 construct. These mutagenized constructs were cut with EcoRI to excise the DNA fragment encoding either amino acids 1–205 or 1–484, respectively, and religated. The CLD/NES regions of mPER1 and mPER2 were amplified by PCR and cloned into the YFP-NLS construct between BspEI and HindIII. These new CLD/NES constructs were then mutagenized with QuikChange to create the various NES mutants. The YFP-NLS construct containing only the NES of mPER1 was made by introducing an EcoRV site into the +P1-(411–498) (where P1 indicates the mPER1 protein) construct with QuikChange, followed by EcoRV digestion and religation of the vector. All of the constructs described above were sequenced to ensure that the introduced mutations or sequences were correct. In addition, all of the mPER1 constructs were expressed in in vitro transcription/translation reactions (TnT, Promega) to confirm expression of a protein of the expected size.
Cell Line Maintenance and Transfections
HEK 293 cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal calf serum and 1× penicillin/streptomycin (Invitrogen) in 5% CO2. The cells were transfected when they reached 70–90% confluence in six-well dishes with 0.5–1 μg of total plasmid DNA using Opti-MEM and Lipo-fectAMINE Plus reagent (Invitrogen) according to the manufacturer’s instructions. Transfections were allowed to proceed for 3 h. Twenty-four hours after transfection, the cells were replated on poly-l-lysine-treated glass coverslips.
Immunofluorescence
Twenty-four hours after transfection, cells were replated on glass coverslips, washed three times with phosphate-buffered saline (PBS), and then fixed with 4% paraformaldehyde for 10 min. After three more washes with PBS, the cells were permeabilized with 0.1% Triton X-100 for 5 min and washed again with PBS with 1% goat serum. The cells that were transfected with the various YFP constructs were incubated with Hoechst 33258, washed three times with PBS with 1% goat serum, and mounted onto slides with Antifade (Molecular Probes, Inc.). The cells that were transfected with the various mPER constructs were incubated with an anti-Myc monoclonal antibody conjugated to Alexa 488 dye (Molecular Probes, Inc.) and Hoechst 33258 for 1 h in PBS with 1% goat serum. Cells were visualized using an Olympus BX50 fluorescence microscope (magnification × 60), and images were captured using mFISH software (Vysis, Inc.). Images were assembled using Photoshop Version 4.0 (Adobe) and Canvas Version 6.01 (Deneba Software). Excel 98 (Microsoft) was used for graphical analysis of the data.
Cell Treatments with the Nuclear Export Inhibitor Leptomycin B
Forty-eight hours after transfection, cells were treated with either 10 ng/ml leptomycin B (LMB) or vehicle for 4 h prior to immunofluorescence microscopy. LMB was a generous gift from Minoru Yoshida (University of Tokyo).
X. laevis Oocyte Nuclear Export Assay
The proteins indicated in Fig. 5 were synthesized in programmed rabbit reticulocyte lysates (TnT, Promega) in the presence of [35S]methionine according to the manufacturer’s instructions. The various mPER1 proteins were mixed with the luciferase protein, and 5–7 nl of the protein mixture was injected into the nuclei of X. laevis oocytes kept at 4 °C. Following the injections, the oocytes were incubated at 18 °C for the indicated times. At the end of the incubation period, the nuclei were removed from individual oocytes by manual microdissection as previously described (22), except that harvest buffer volumes were reduced, and the acetone precipitation was omitted. Individual nuclear and cytoplasmic fractions were then mixed with SDS-PAGE sample buffer and analyzed by SDS-PAGE. The amount of each 35S-labeled protein was quantitated using a PhosphorImager and ImageQuant Version 1.11 (Molecular Dynamics, Inc.). Each experiment was performed in duplicate on at least three separate occasions.
Fig. 5. mPER1 is exported from the nuclei of X. laevis oocytes.

A, nuclear export of mPER1 in X. laevis oocytes requires a functional NES. The indicated 35S-labeled proteins were injected into the nuclei of X. laevis oocytes. At the indicated times following injection, the nuclei were separated from the cytoplasm, and the nuclear and cytoplasmic fractions were analyzed separately by SDS-PAGE. Each lane contains the proteins from the nucleus or cytoplasm of one oocyte, respectively. In all experiments, luciferase was co-injected with wild-type (WT mPER1) or mutant mPER1 (NES mut mPER1), but only the luciferase co-injected with wt mPER1 is shown. The NES mutant mPER1 protein has Ile-493 and Leu-496 mutated to alanine. These mutations are in the context of the full-length mPER1 protein. This experiment was repeated several times with similar results. B, quantitation of the amount of mPER1 protein found in the cytoplasm at various times post-injection. The amount of mPER1 protein in the oocyte cytoplasm at the indicated times following nuclear injection is expressed as a percentage of the total amount of mPER1 in the oocyte (nuclear fraction + cytoplasmic fraction) at each time point. C, the rate of cytoplasmic accumulation is controlled in part by the amino terminus of mPER1. The indicated 35S-labeled proteins were injected into the nuclei of X. laevis oocytes, and the experiment was carried out as described for A. The Δ 1–205 mPER1 protein is the same as the protein shown in Fig. 1A. This experiment was repeated several times with similar results. N, nuclear fraction; C, cytoplasmic fraction.
RESULTS
The CLD Is Not Required for Cytoplasmic Localization of mPER1
Previous analysis of the subcellular localization of mPER1 transiently expressed in HEK 293 cells indicated that the protein is localized predominantly in the nucleus. Further analysis revealed that casein kinase Iε can alter the subcellular localization of mPER1 through a phosphorylation-dependent masking of the NLS and suggested that the NLS is the predominant regulator of mPER1 subcellular localization (16). A similarly located NLS has been identified in mPER3 (19). However, all three PER proteins also contain a conserved CLD related in sequence to the functionally identified dPER CLD. To determine the role of additional domains within mPER1 in its subcellular localization, several amino-terminal truncations of mPER1 were analyzed (Fig. 1A). Although full-length mPER1 was predominantly nuclear when expressed in HEK 293 cells, deletion of the first 205 amino acids (Δ1–205) resulted in a protein that was localized predominantly in the cytoplasm (Fig. 1B). This suggests that there is a functional NLS in this region. Computer-aided inspection of the amino acid sequence in this region identified a potential SV40 large T antigen-like NLS between amino acids 150 and 158. However, mutation of key basic residues in this putative NLS sequence had no effect on the nuclear localization of the mPER1 protein (data not shown). Furthermore, fusion of this putative NLS sequence to a green fluorescent protein-β-galactosidase carrier protein did not drive nuclear localization of the fusion protein (data not shown). Taken together, these data imply that amino acids 150–158 do not encode a functional NLS, but that region 1–205 is important for the nuclear localization of mPER1 through an as yet unidentified mechanism.
Fig. 1. Successive amino-terminal truncations of mPER1 reveal that two distinct regions are important for mediating the subcellular localization of mPER1.

A, diagram of the mPER1 truncation mutants. The truncated forms of mPER1 used in B are drawn, with previously described domains within mPER1 indicated by boxes in varying shades of gray and the putative NES indicated by hatched boxes. The PAS A and B domains and CLD (10, 28) and the NLS, casein kinase Iε (CKIε)-binding region, and NLS-masking domain (16) have been described. B, the amino-terminal region of mPER1 (amino acids 1–205) is important for the nuclear localization of mPER1, whereas amino acids 485–495 are important for cytoplasmic localization. HEK 293 cells were transfected with the indicated Myc-mPER1 constructs, and the subcellular location of the proteins was determined with immunofluorescence. The Myc-tagged mPER1 proteins were detected with an anti-Myc monoclonal antibody directly coupled to Alexa 488, and nuclei were visualized by Hoechst staining. Representative micrographs are shown. The subcellular localization of the various mPER proteins depicted in the micrographs was observed in >95% of the cells. Experiments were repeated at least twice, and >100 cells were counted for each transfected construct.
Further amino-terminal truncations of mPER1 that deleted the PAS A domain (Δ1–328) or the PAS A and B domains (Δ1–416) had no further effect on the subcellular localization of mPER1 (data not shown). Likewise, deletion of the PAS A and B domains and the CLD (Δ1–484) (Fig. 1A) also produced a protein that was predominantly localized to the cytoplasm (Fig. 1B). This result was unexpected considering that the CLD is required for the cytoplasmic localization of dPER in S2 cells (6) and mPER3 in COS-7 cells (19). However, further truncation of the protein by 11 amino acids (Δ1–495) (Fig. 1A) resulted in a protein that accumulated predominantly in the nucleus (Fig. 1B). These data suggest that unlike the dPER protein, the CLD is not required for the subcellular localization of mPER1. Instead, the region between amino acids 485 and 495 of mPER1 appears to be required for the cytoplasmic localization of this protein, although in the context of the full-length protein, the effect of this region is overridden by sequences in the amino terminus.
PER Proteins Contain Sequences Resembling NESs in the Region Flanking the CLD
An analysis of the sequence around region 485–495 that is required for mPER1 cytoplasmic localization revealed a leucine-rich stretch of amino acids homologous to an NES motif between amino acids 486 and 498 (Fig. 2) (21). NESs bind to the CRM1/exportin-1 nuclear export receptor and mediate CRM1-dependent nuclear export of NES-containing proteins (23). The presence of this putative NES within mPER1 could explain why this region is required for the cytoplasmic localization of mPER1. Similar hydrophobic regions are also present in other PER proteins, including mammalian PER1–3 and possibly Drosophila and silkmoth (Antheraea pernyi) PER proteins as well (Fig. 2).
Fig. 2. Numerous PER proteins contain a putative NES carboxyl-terminal to the CLD.

The positions and sequences of the putative NES regions from mPER1, mPER2, mPER3, dPER, and A. pernyi (ap) PER are shown in relation to a diagram of the full-length mPER1 protein. The hydrophobic residues known to be important for NES function in other proteins are shown in boldface. The PER sequences are aligned based on the position of these hydrophobic residues. Numbers indicate the locations of the amino acids within the respective proteins. For comparison, a consensus sequence of the leucine-rich NES is shown (29). Although originally defined as “leucine-rich,” other hydrophobic residues (Ile, Phe, Val, and Met) have been shown to substitute for the second through fourth leucines in functional NES sequences of various proteins (26, 29). Within the consensus sequence, X denotes any amino acid. CKIε, casein kinase Iε.
The NES of mPER1 Can Mediate the Nuclear Export of a Heterologous Protein
The ability of the putative NES of mPER1 to direct nuclear export of a heterologous protein was tested using fragments of mPER1 fused to a nuclear targeted tandem YFP (YFP-NLS) (16). Fusion of the CLD and NES of mPER1 (+P1-(411–498)) re-localized YFP-NLS from the nucleus to the cytoplasm in >90% of the cells (Fig. 3, A, B, and D). To determine whether the putative NES alone is sufficient for directing the cytoplasmic localization of the YFP protein, amino acids 485–498 alone were fused to YFP-NLS (Fig. 3, A, B, and D). Again, this region alone was sufficient to re-localize YFP-NLS from the nucleus to the cytoplasm. As a further test of the role of the NES in PER1 nuclear export, three conserved hydrophobic residues were mutated to either alanine or lysine (Fig. 3A). Mutation of the NES (+P1-(411–498)mut NES) prevented the cytoplasmic accumulation of the YFP fusion protein, whereas the protein with the wild-type NES (+P1-(411–498)) was localized predominantly in the cytoplasm (Fig. 3, B and D). These data support the hypothesis that the putative NES (and not the CLD) mediates cytoplasmic localization of mPER1 and are consistent with the results of the deletion studies in mPER1 (Fig. 1).
Fig. 3. Fusion of the CLD/NES region of mPER1 (amino acids 411–498) to a YFP carrier protein promotes nuclear export.
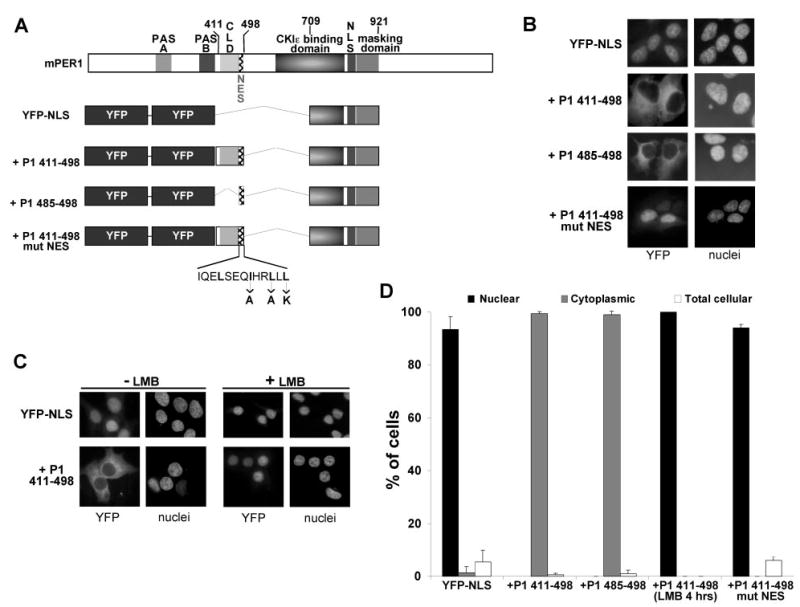
A, diagrams of the YFP carrier protein (YFP-NLS) and the YFP-CLD/NES fusion proteins. The YFP fusion proteins used in B–D are drawn below a full-length mPER1 diagram, and the specific mutations made in the NES are indicated below the NES sequence. The regions of mPER1 fused to the YFP-NLS protein are indicated by +P1 plus the corresponding amino acid numbers. All regions of mPER1 fused to YFP are aligned with the respective domains of full-length mPER1. B, the mPER1 NES alone controls the cytoplasmic localization of YFP. HEK 293 cells were transfected with the indicated Myc-mPER1 constructs, and the subcellular location of the proteins was determined by autofluorescence of YFP. Nuclei were visualized by Hoechst staining. Representative micrographs are shown. C, the cytoplasmic localization of the YFP-CLD/NES fusion proteins can be inhibited by LMB. HEK 293 cells were transfected with the indicated YFP fusion constructs and treated with vehicle (*minus; LMB) or with LMB (+ LMB) for 4 h. Following the treatment, the subcellular location of the proteins was determined by autofluorescence of YFP, and nuclei were visualized by Hoechst staining. Representative micrographs are shown. D, quantitation of the experiments illustrated above. Black bars represent predominantly nuclear localization; gray bars represent predominantly cytoplasmic localization; and white bars represent equal localization in the cytoplasm and nucleus. All experiments were performed at least twice, and >100 cells were counted for each condition tested. The lack of error bars for the LMB data indicates that replicate experiments produced identical results.
Cytoplasmic accumulation of NES-containing proteins is due to nuclear export, frequently mediated by the nuclear export receptor CRM1. To determine whether the cytoplasmic localization of the YFP-CLD/NES fusion protein is mediated by CRM1-dependent nuclear export, cells expressing either the nuclear localized YFP protein (YFP-NLS) or the YFP-CLD/NES fusion protein (+P1-(411–498)) were treated with the CRM1-specific nuclear export inhibitor LMB (24). As expected, LMB treatment had no effect on the nuclear localization of the YFP-NLS protein (Fig. 3C, upper right panels). However, LMB prevented the cytoplasmic accumulation of the YFP-CLD/NES fusion protein (Fig. 3, C, lower right panels; and D). These data suggest that the CRM1-dependent nuclear export pathway is either directly or indirectly involved in mediating the cytoplasmic localization of this YFP-CLD/NES fusion protein.
The NES Sequence from mPER2 Can Also Mediate the Nuclear Export of a Heterologous Protein
Similar to mPER1, putative NES sequences were identified in a region carboxyl-terminal to the CLDs in mPER2 and mPER3 (Fig. 2). However, the putative NES sequence in mPER2 differs from that of mPER1 and the consensus sequence due to the presence of a methionine instead of a leucine in the last position of the sequence. To test whether the related mPER2 sequences could direct the cytoplasmic localization of a heterologous protein, the CLD/NES region (amino acids 382–469) of mPER2 was fused to the YFP-NLS protein (Fig. 4A). Similar to the results seen with the NES of mPER1, the CLD/NES of mPER2 (+P2-(382–469), where P2 indicates the mPER2 protein) also directed YFP-NLS to the cytoplasm. Furthermore, both the triple-point mutant within the NES sequence (+P2-(382–469)mut NES) and LMB treatment similarly prevented cytoplasmic localization of the YFP-NLS protein (Fig. 4B). Interestingly, a single-point mutation in the mPER2 NES sequence that changed the terminal methionine to lysine (+P2-(382–469)M224K) also had a significant effect on the subcellular localization of YFP-NLS (Fig. 4B). The corresponding single-amino acid substitution in the mPER1 NES (L224K) had very little effect on the subcellular localization of the YFP-NLS protein (data not shown). These data indicate that a methionine in the terminal position of the NES sequence functions as well as a leucine and that this terminal residue in the sequence is more important for the function of the mPER2 NES than it may be in the mPER1 NES, at least in the context we utilized. Furthermore, the ability of this NES sequence to function as an NES appears to be conserved in other mammalian PER proteins. For example, the CLD/NES region of mPER3 (amino acids 321–407) was similarly able to drive cytoplasmic accumulation when fused to YFP (data not shown).
Fig. 4. Fusion of the CLD/NES region of mPER2 (amino acids 382–469) to a YFP carrier protein promotes nuclear export.

A, diagrams of the YFP carrier protein (YFP-NLS) and the YFP-CLD/NES fusion proteins. The YFP fusion proteins used in B are drawn. + P2 indicates the mPER2 protein and is followed by the corresponding amino acid numbers used in the fusion with YFP-NLS. The specific mutations made in the NES are indicated below each NES sequence. The region of mPER1 fused to YFP to promote nuclear localization is indicated above that region. B, fusion of the CLD/NES region of mPER2 to YFP promotes the cytoplasmic localization of YFP and can be inhibited by LMB or mutation of the NES sequence. HEK 293 cells were transfected with the indicated YFP fusion constructs, and one set of cells expressing the +P2-(382–469) construct was treated with LMB for 4 h. The subcellular location of the proteins was determined by autofluorescence of YFP, and nuclei were visualized by Hoechst staining. Black bars represent predominantly nuclear localization; gray bars represent predominantly cytoplasmic localization; and white bars represent equal localization in the cytoplasm and nucleus. All experiments were performed at least twice, and >100 cells were counted for each condition tested.
Full-length mPER1 Is Exported from the Nuclei of Xenopus Oocytes
The results described above established that there is a domain in mPER1 and mPER2 capable of promoting nuclear export in protein fragments and when fused to a heterologous reporter. To determine whether the full-length mPER1 protein undergoes nuclear export in vivo, [35S]methionine-labeled mPER1 was microinjected into the nuclei of X. laevis oocytes. [35S]Methionine/luciferase was co-injected as an internal control for localization of the initial injection. At specific time points following microinjection, nuclear and cytoplasmic fractions from individual oocytes were isolated and analyzed for the presence of mPER1 and luciferase (Fig. 5). Nuclear injection of luciferase (~60 kDa) resulted in predominantly nuclear luciferase at both 2 and 4 h post-injection, indicating appropriate targeting of the protein and minimal diffusion from the nucleus (Fig. 5A). In contrast, wild-type mPER1 (~180 kDa) was exported from the nucleus, with half the protein cytoplasmic within 2 h. Notably, at 4 h post-injection, there was no further increase in the amount of mPER1 in the cytoplasm. Mutation of two conserved hydrophobic residues within the NES (I493A and L496A) prevented the nuclear export of mPER1 (Fig. 5, A and B). These data indicate that mPER1 is actively exported from the nucleus utilizing an NES.
The fact that only half of the wild-type mPER1 protein was found in the cytoplasm suggests either that it may be rapidly re-imported into the nucleus or that a fraction of the mPER1 protein molecules in the nucleus are actively retained there through interaction with another protein (see below). To test whether PER1 cytoplasmic localization is influenced by nuclear re-import, mPER1 lacking the amino-terminal nuclear localization domain (Δ1–205) (Fig. 1) was similarly tested for nuclear export. As shown in Fig. 5C, removal of the first 205 amino acids of mPER1 allowed ~85% of the mPER1 protein to be exported to the cytoplasm compared with ~50% of the full-length protein. Since this amino-terminal truncation removes a potent NLS, these results are consistent with full-length mPER1 undergoing cycles of export and re-import.
Our previous study demonstrated that phosphorylation of mPER1 by casein kinase Iε promotes its cytoplasmic accumulation by masking an NLS (16). To test whether phosphorylation of mPER1 can accelerate nuclear export in addition to inhibiting nuclear import, 35S-labeled mPER1 was incubated with recombinant casein kinase Iε in the reticulocyte extracts prior to oocyte nuclear injection. We were unable to detect any effect of casein kinase Iε on the rate of mPER1 nuclear export (data not shown).
DISCUSSION
Previous models of circadian machinery have envisioned a unidirectional flow of circadian regulators from the cytoplasm to the nucleus, followed by degradation in an unknown compartment. In this study, we have identified a novel function of the mammalian PER proteins, that of nuclear export, and show that in both mPER1 fragments and intact protein, nuclear export is directed by a leucine-rich NES similar in sequence to those found in diverse cellular regulatory proteins. One caveat of this study is that HEK 293 cells may not accurately reflect the intracellular environment seen in suprachiasmatic nucleus cells. In fact, PER proteins accumulate in different compartments in different cell lines. For this reason, we also tested the function of the NES in an unrelated cell system. We found that the NES directed nuclear export of mPER1 in two distinct systems, HEK 293 cells and Xenopus oocytes. The data therefore suggest that the NES is generally important in regulating the subcellular localization of mPER1 in diverse cells.
Previous studies identified a sequence in dPER (termed the CLD) as a key regulatory of PER cytoplasmic retention (6). This domain has been recently recognized as a conserved sequence (termed the PAC domain) found carboxyl-terminal to multiple PAS domains and proposed to contribute to proper structure of the PAS domain (5). Yagita et al. (19) found that deletion of the mPER3 CLD (a deletion that apparently did not impinge on the immediately adjacent NES) led to nuclear localization in COS-7 cells. However, we found that truncation of the CLD from mPER1 had no effect on its cytoplasmic localization. What then is the function of the CLD in the cytoplasmic retention of the PER proteins? One possibility is that the CLD is a structural element of the protein; and in specific constructs, its deletion can alter the structure and hence the function of the adjacent NES. If PER localization is in fact determined by competing rates of nuclear import and export, minor changes in NES function and consequently nuclear export rates could have a substantial impact on the global localization of the protein. The discrepant results of PER localization seen after expression in diverse cell lines may therefore simply reflect differences in the relative rates of PER nuclear import and export.
The finding that PER localization is dynamic rather than unidirectional may explain a finding in the silkmoth. In this system, PER is predominantly a cytoplasmic protein in clock neurons, while still undergoing circadian variations in mRNA and protein abundance (25). Silkmoth and Drosophila PER proteins share most (but not all) of the NES hydrophobic residues identified in mPER1 and mPER2 (Fig. 2). It may be that silkmoth PER is in fact actively shuttling between the cytoplasm and the nucleus in these cells, with the more readily visualized protein in the cytoplasm, but with functional PER also present in the nucleus.
Nucleocytoplasmic shuttling provides the opportunity for regulation of protein function by moving it from one compartment to another. For example, phosphorylation-regulated binding of the Mdm2 protein to p53 facilitates translocation of p53 from the nucleus to the cytoplasm, where it is degraded (26). The lymphoid transcription factor NF-AT is sequestered in the cytoplasm by phosphorylation and relocated to the nucleus by calcineurin-mediated dephosphorylation (27). In the case of PER shuttling, immunohistochemistry suggests that the majority of PER protein is located in the nucleus. It is possible that there is a pool of cytoplasmic PER proteins not well visualized by immunohistochemistry that is in equilibrium with the more concentrated and hence readily visualized nuclear protein. Certainly, immunolocalization of PER proteins may miss a significant fraction of the protein localized to the cytoplasm. There are several (nonexclusive) potential functions of PER nuclear export. Nuclear export of the PER protein may be required for its degradation in the cytoplasm. Alternatively, PER nuclear export may serve to expedite transport of other PER-binding proteins such as CRY1, CRY2, and casein kinase Iε to the cytoplasm before PER is re-imported into the nucleus. Last, PER is a modest inhibitor of circadian transcription. Nuclear export of PER may serve to up-regulate CLOCK/BMAL1 transcription, perhaps in response to photic input. Clues to the role of PER export may come when external stimuli that alter its localization are identified in intact animals. The ultimate test of the importance of nuclear export in the regulation of circadian rhythm will be to assess the effect of targeted mutations of the various PER nuclear export sequences on circadian rhythm in mice.
Acknowledgments
We thank Bob Schackmann for synthesizing the oligonucleotides used in this work.
Footnotes
This work was supported by National Institutes of Health Grant R01 CA71074 (to D. M. V.) and by the Huntsman Cancer Foundation. Oligonucleotide synthesis was supported in part by National Institutes of Health Grant 3P30 CA42014.
The abbreviations used are: dPER, Drosophila PER; mPER, murine PER; CLD, cytoplasmic localization domain; HEK, human embryonic kidney; NLS, nuclear localization signal; NES, nuclear export signal; YFP, yellow fluorescent protein; PBS, phosphate-buffered saline; LMB, leptomycin B.
E. J. Eide and D. M. Virshup, manuscript in preparation.
References
- 1.Dunlap JC. Cell. 1999;96:271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 2.Reppert SM, Weaver DR. Annu Rev Physiol. 2001;63:647–676. doi: 10.1146/annurev.physiol.63.1.647. [DOI] [PubMed] [Google Scholar]
- 3.Lowrey PL, Takahashi JS. Annu Rev Genet. 2000;34:533–562. doi: 10.1146/annurev.genet.34.1.533. [DOI] [PubMed] [Google Scholar]
- 4.Curtin KD, Huang ZJ, Rosbash M. Neuron. 1995;14:365–372. doi: 10.1016/0896-6273(95)90292-9. [DOI] [PubMed] [Google Scholar]
- 5.Ponting CP, Aravind L. Curr Biol. 1997;7:R674–R677. doi: 10.1016/s0960-9822(06)00352-6. [DOI] [PubMed] [Google Scholar]
- 6.Saez L, Young MW. Neuron. 1996;17:911–920. doi: 10.1016/s0896-6273(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 7.Sehgal A, Rothenfluh-Hilfiker A, Hunter-Ensor M, Chen Y, Myers MP, Young MW. Science. 1995;270:808–810. doi: 10.1126/science.270.5237.808. [DOI] [PubMed] [Google Scholar]
- 8.Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, Weaver DR. Neuron. 2001;30:525–536. doi: 10.1016/s0896-6273(01)00302-6. [DOI] [PubMed] [Google Scholar]
- 9.Zheng B, Albrecht U, Kaasik K, Sage M, Lu W, Vaishnav S, Li Q, Sun ZS, Eichele G, Bradley A, Lee CC. Cell. 2001;105:683–694. doi: 10.1016/s0092-8674(01)00380-4. [DOI] [PubMed] [Google Scholar]
- 10.Albrecht U, Sun ZS, Eichele G, Lee CC. Cell. 1997;91:1055–1064. doi: 10.1016/s0092-8674(00)80495-x. [DOI] [PubMed] [Google Scholar]
- 11.Shearman LP, Zylka MJ, Weaver DR, Kolakowski LF, Jr, Reppert SM. Neuron. 1997;19:1261–1269. doi: 10.1016/s0896-6273(00)80417-1. [DOI] [PubMed] [Google Scholar]
- 12.Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, Okamura H. Cell. 1997;91:1043–1053. doi: 10.1016/s0092-8674(00)80494-8. [DOI] [PubMed] [Google Scholar]
- 13.Field MD, Maywood ES, O’Brien JA, Weaver DR, Reppert SM, Hastings MH. Neuron. 2000;25:437–447. doi: 10.1016/s0896-6273(00)80906-x. [DOI] [PubMed] [Google Scholar]
- 14.Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, van der Horst GT, Hastings MH, Reppert SM. Science. 2000;288:1013–1019. doi: 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- 15.Eide EJ, Virshup DM. Chronobiol Int. 2001;18:389–398. doi: 10.1081/cbi-100103963. [DOI] [PubMed] [Google Scholar]
- 16.Vielhaber E, Eide E, Rivers A, Gao ZH, Virshup DM. Mol Cell Biol. 2000;20:4888–4899. doi: 10.1128/mcb.20.13.4888-4899.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vielhaber E, Virshup DM. IUBMB Life. 2001;51:73–78. doi: 10.1080/15216540117461. [DOI] [PubMed] [Google Scholar]
- 18.Takano A, Shimizu K, Kani S, Buijs RM, Okada M, Nagai K. FEBS Lett. 2000;477:106–112. doi: 10.1016/s0014-5793(00)01755-5. [DOI] [PubMed] [Google Scholar]
- 19.Yagita K, Yamaguchi S, Tamanini F, van der Horst GT, Hoeijmakers JH, Yasui A, Loros JJ, Dunlap JC, Okamura H. Genes Dev. 2000;14:1353–1363. [PMC free article] [PubMed] [Google Scholar]
- 20.Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- 21.Gerace L. Cell. 1995;82:341–344. doi: 10.1016/0092-8674(95)90420-4. [DOI] [PubMed] [Google Scholar]
- 22.Ullman KS, Shah S, Powers MA, Forbes DJ. Mol Biol Cell. 1999;10:649–664. doi: 10.1091/mbc.10.3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ullman KS, Powers MA, Forbes DJ. Cell. 1997;90:967–970. doi: 10.1016/s0092-8674(00)80361-x. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. Nature. 1997;390:308–311. doi: 10.1038/36894. [DOI] [PubMed] [Google Scholar]
- 25.Sauman I, Reppert SM. Neuron. 1996;17:889–900. doi: 10.1016/s0896-6273(00)80220-2. [DOI] [PubMed] [Google Scholar]
- 26.Roth J, Dobbelstein M, Freedman DA, Shenk T, Levine AJ. EMBO J. 1998;17:554–564. doi: 10.1093/emboj/17.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okamura H, Aramburu J, Garcia-Rodriguez C, Viola JP, Raghavan A, Tahiliani M, Zhang X, Qin J, Hogan PG, Rao A. Mol Cell. 2000;6:539–550. doi: 10.1016/s1097-2765(00)00053-8. [DOI] [PubMed] [Google Scholar]
- 28.Takumi T, Taguchi K, Miyake S, Sakakida Y, Takashima N, Matsubara C, Maebayashi Y, Okumura K, Takekida S, Yamamoto S, Yagita K, Yan L, Young MW, Okamura H. EMBO J. 1998;17:4753–4759. doi: 10.1093/emboj/17.16.4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mowen K, David M. Mol Cell Biol. 2000;20:7273–7281. doi: 10.1128/mcb.20.19.7273-7281.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]