

Abstract
The serine/threonine protein kinase casein kinase I ε (CKIε) is a key regulator of metazoan circadian rhythm. Genetic and biochemical data suggest that CKIε binds to and phosphorylates the PERIOD proteins. However, the PERIOD proteins interact with a variety of circadian regulators, suggesting the possibility that CKIε may interact with and phosphorylate additional clock components as well. We find that CRY1 and BMAL1 are phosphoproteins in cultured cells. Mammalian PERIOD proteins act as a scaffold with distinct domains that simultaneously bind CKIε and mCRY1 and mCRY2 (mCRY). mCRY is phosphorylated by CKIε only when both proteins are bound to mammalian PERIOD proteins. BMAL1 is also a substrate for CKIε in vitro, and CKIε kinase activity positively regulates BMAL1-dependent transcription from circadian promoters in reporter assays. We conclude that CKIε phosphorylates multiple circadian substrates and may exert its effects on circadian rhythm in part by a direct effect on BMAL1-dependent transcription.
Circadian rhythms allow organisms to optimize their metabolic and physiologic behavior in response to the 24 h day. The rhythm is generated by cell-autonomous transcription-translation feedback loops highly conserved in outline if not in detail in most eukaryotes studied to date (recently reviewed in Refs. 1–4). Extensive genetic investigation coupled with molecular studies has uncovered many of the essential elements of the metazoan clock. The central feature of the clock is transcription, regulated by a heterodimeric transcription factor that drives expression of genes whose protein products are negative regulators of their own transcription. This negative feedback loop establishes stable molecular oscillations with a period of ~24 h. In mammals, the transcription factors are the PAS domain-containing proteins CLK1 and BMAL1, whereas the negative factors include the PERIOD proteins PER1 and PER2 and the cryptochromes CRY1 and CRY2. The stability and period of the circadian oscillations are likely to be dependent on multiple factors, including the rate at which these regulators accumulate in the cell (i.e. the sum of their synthesis and degradation rates) and the rate at which they accumulate in the nucleus. Finally, the activities of circadian regulators are modified by post-translational modifications.
Analysis of animals and humans with altered circadian rhythms demonstrates the importance of phosphorylation in the regulation of the molecular clock. Mutations in the casein kinase Iε (CKIε) homolog double-time (dbt) in Drosophila markedly lengthen or shorten the circadian period, depending on the allele (5–8). In hamsters, a point mutation in CKIε that decreases kinase activity causes the semidominant short period tau phenotype (9). The functional role of CKIε within the circadian clock has not been fully elucidated (1, 10). A number of studies indicate the PER proteins are critical targets of CKIε. dbt-mutant flies have altered patterns of PER phosphorylation and accumulation (6, 8, 11). Mutations in a CKIε phosphorylation site in human PER2 have been identified in a family with advanced sleep phase syndrome (FASPS), a dominantly inherited short circadian period disorder (12). Overexpression of CKIε leads to decreased mPER1 protein half-life and alters the nucleocytoplasmic localization of the proteins (13–15). Whether CKIε can phosphorylate other circadian regulatory proteins has not yet been explored.
One central feature of the circadian regulatory machinery is the multiple interactions between the clock proteins. CKIε forms stable complexes with PER proteins. PER proteins heterodimerize with each other and interact with CRY1 and CRY2 (16–18). CRY1 and CRY2 also interact with the heterodimeric transcription factor CLK/BMAL1 both functionally and in yeast two-hybrid assays (19, 20). Hence, the potential for higher order complexes exists. Given the changing abundance of these factors over time, different complexes are likely to form at different times of day.
In the present study, we examined the ability of CKIε to interact with and phosphorylate other clock proteins in vitro and in cell culture, both directly and when in multiprotein complexes. We find that mCRY1 and mCRY2 are substrates of CKIε but only when both CRY and kinase are bound to PER1 or PER2. Furthermore, mCRY1 can overcome the CKIε-dependent cytoplasmic retention of mPER1 seen in human embryonic kidney 293 (HEK 293) cells and relocalize the entire multimeric complex to the nucleus. The ability of CKIε to phosphorylate CLK and BMAL1 was examined as well. BMAL1 but not CLK is a substrate of CKIε in vitro. BMAL1 phosphorylation by CKIε is neither dependent on nor stimulated by the presence of PER or CRY proteins. BMAL1 is also a phosphoprotein when expressed in HEK 293 cells, and co-expression of a dominant-negative CKIε inhibits BMAL1 phosphorylation. Inhibition of CKIε activity using dominant-negative CKIε or dsRNA-mediated interference also led to decreased CLK/BMAL1-dependent transcription. The data suggest CKIε has multiple functional substrates in the circadian rhythm regulatory apparatus.
MATERIALS AND METHODS
Cell Line Maintenance and Transfection
HEK 293 cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal calf serum in a humidified incubator at 37 °C and 5% CO2. Cells were transfected using LipofectAMINE PLUS (Invitrogen) with between 0.3 and 0.7 μg of expression constructs, as indicated in the figures, with the total amount of transfected DNA brought up to 1 μg with the addition of empty vector.
Immunoprecipitations and Immunofluorescence
HEK 293 cells were transfected with the indicated expression constructs and harvested 18 h later in HTS buffer (10 mm HEPES, pH 7.5, 0.1% Triton X-100, 100 mm NaCl, 2 mm EDTA, 2 mm EGTA, and 2 mm DTT, supplemented with Complete™ (Roche Molecular Biochemicals) protease inhibitors). Myc epitope-tagged proteins were immunoprecipitated with 1 μg of 9E10 anti-Myc monoclonal antibodies and protein A-agarose beads. The beads were then washed three times in HTS buffer, and proteins were eluted with SDS-PAGE loading buffer. After SDS-PAGE, the proteins were transferred to a nitrocellulose membrane (Hybond-C extra, Amersham Biosciences) and further analyzed by immunoblotting using the indicated antibodies. For immunofluorescence, HEK 293 cells were transiently transfected as indicated in Fig. 3. 18 h after transfection, the cells were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. Anti-Myc (9E10) and anti-influenza hemagglutinin epitope (12CA5) monoclonal antibodies were directly conjugated to Alexa 488 (green) or Alexa 594 (red) according to the manufacturer’s directions (Molecular Probes). The antibodies and 4,6-diamidino-2-phenylindole nuclear stain were incubated with the cells for 1 h at room temperature. The epitope-tagged proteins were then visualized using an Olympus BX50 fluorescence microscope equipped with a cooled charge-coupled device camera (Photometrics Ltd.) at 60× magnification. Images were acquired using the mFISH (Vysis, Inc) software package and further analyzed with PhotoShop 5.0 (Adobe).
Fig. 3. mCRY1 promotes nuclear localization of the mPER1/CKIε complex.
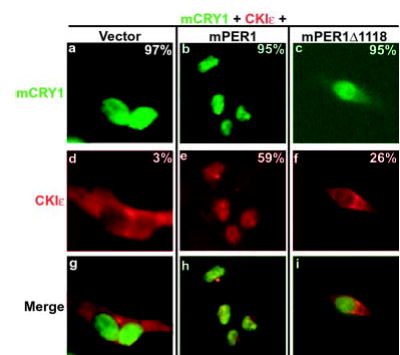
CKIε is localized to the nucleus through interaction with the PER·CRY complex. Myc-tagged mCRY1 and hemagglutinin epitope-tagged CKIε were transiently expressed in HEK 293 cells in the presence of vector (a, d, and g), full-length mPER1 (b, e, and h), or truncated mPER1 (Δ1118, lacking the CRY interaction domain) (c, f, and i) as indicated. Protein localization was visualized by staining the cells with anti-Myc antibodies conjugated to Alexa 488 (green) and anti-hemagglutinin antibodies conjugated to Alexa 594 (red). The percent of transfected cells exhibiting nuclear localization of the indicated protein is shown in each panel.
Kinase Assays
HEK 293 cells were transiently transfected with expression constructs as indicated in the figures. Eighteen hours after transfection, Myc epitope-tagged proteins were immunoprecipitated from 300 μg of soluble cell free extract with 1 μg of 9E10 anti-Myc monoclonal antibodies as was done above. Beads were kept on ice and washed several times with HMB buffer (30 mm HEPES, pH 8, 7 mm MgCl2, 100 μg/ml bovine serum albumin, and 25 μm ATP). After washing, the beads were split three ways. To one bead fraction, HMB, [γ-32P]ATP, and 50 ng of recombinant CKIεΔC320 was added to a final volume of 20 μl. This truncated form of CKIε was used to avoid the auto-inhibition seen with full-length CKIε in vitro (21). Another bead fraction was treated identically except for the omission of recombinant kinase as a negative control. The beads were then incubated at 37° for 15 min. The reactions were stopped by washing the beads three times with HM buffer (30 mm HEPES, pH 8, 7 mm MgCl2). Proteins were eluted from the beads with Laemmli sample buffer and separated by SDS-PAGE, and dried gels were analyzed by PhosphorImager (Amersham Biosciences). To confirm the presence of proteins in the immunoprecipitate, the third bead fraction was also run out on SDS-PAGE and analyzed by immunoblotting as above.
For in vitro kinase assays (Fig. 5A), V5 epitope-tagged CLK and BMAL1 were synthesized in rabbit reticulocyte lysate using the TNT Coupled Reticulocyte System (Promega). The proteins were immunoprecipitated from 90 μl of each synthesis reaction with 3 μg of anti-V5 antibodies. Bead washes, in vitro kinase assays, and Western blotting were performed as described above.
Fig. 5. BMAL1 phosphorylation is regulated by CKIε in vitro and in intact cells.
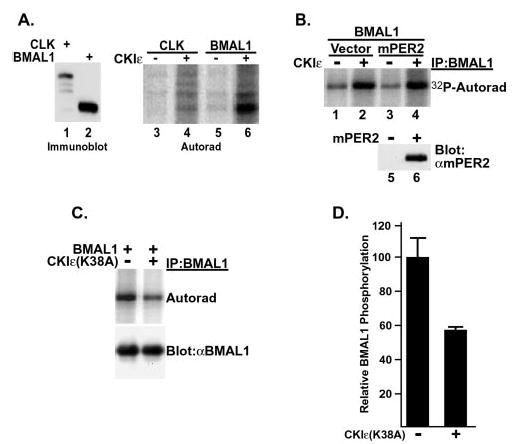
A, BMAL1 but not CLK is a substrate of CKI. V5 epitope-tagged CLK (lanes 1, 3, and 4) and BMAL1 (lanes 2, 5, and 6) were expressed in rabbit reticulocyte lysates and immunoprecipitated, and the beads were split three ways. One fraction was probed with anti-V5 antibodies to verify the presence of protein in the pellet (lanes 1 and 2), and the remaining fractions were subjected to an in vitro kinase reaction in the absence (lanes 3 and 5) or presence (lanes 4 and 6) of added recombinant CKIε (ΔC320) as indicated. B, BMAL1 was phosphorylated by CKIε in vitro independent of mPER2. HEK 293 cells were transiently transfected with plasmids encoding Myc-tagged BMAL1 along with FLAG-tagged mPER2 or empty vector. The Myc-tagged BMAL1 was immunoprecipitated and incubated with [γ-32P]ATP and without (−) or with (+) recombinant CKIε (ΔC320). Co-immunoprecipitation of mPER2 with BMAL1 was confirmed by immunoblotting a fraction of the immunoprecipitates with anti-FLAG antibodies (lanes 5 and 6). The presence of co-immunoprecipitating mPER2 did not stimulate phosphorylation of BMAL1. C, BMAL phosphorylation in intact cells is reduced by co-expression of dominant-negative CKIε. HEK 293 cells were transiently transfected with plasmids encoding Myc epitope-tagged BMAL1 and either vector (−) or a kinase-inactive form of CKIε (K38A) (+). At 18 h post-transfection, cells were labeled with [32P]orthophosphoric acid for 3 h, and then the BMAL1 protein was immunoprecipitated (IP) from lysates with anti-Myc monoclonal antibodies. 32P incorporation into BMAL1 was assessed by SDS-PAGE and phosphorimaging analysis. BMAL1 protein expression was determined by immunoblot of cell free lysate. The data shown is a representative result of three separate experiments. D, Quantitation of BMAL1 phosphorylation without (−) or with (+) co-expression of CKIε (K38A). The results shown are the average of three independent experiments.
Metabolic Labeling of Cells
HEK 293 cells were transiently transfected as indicated in Fig. 4, A and B. Eighteen hours after transfection, cells were labeled for 3 h with [32P]orthophosphoric acid (2 mCi/ml) in phosphate-free Dulbecco’s modified Eagle’s medium. After labeling, cells were washed and then lysed in radioimmune precipitation buffer (1% Nonidet P-40, 0.5% deoxycholic acid, 0.1% sodium dodecyl sulfate, 150 mm NaCl, 150 mm Tris, pH 8.0) supplemented with 2 mm dithiothreitol, 10 mm sodium fluoride, 10 mm β-glycerol phosphate, 200 nm okadaic acid, and Complete™ (Roche Molecular Biochemicals) protease inhibitors). Myc epitope-tagged CRY or BMAL1 was immunoprecipitated from 1 mg of soluble lysate with 8 μg of 9E10 anti-Myc antibodies and protein A-agarose beads. After washing, bound proteins were eluted with Laemmli sample buffer and examined by SDS-PAGE and phosphorimaging analysis.
Fig. 4. mPER-dependent mCRY1 phosphorylation.
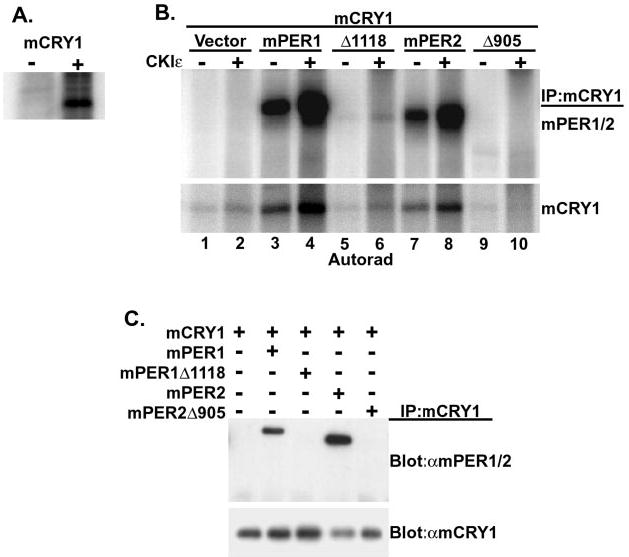
A, mCRY1 is a phosphoprotein in intact cells. HEK 293 cells were transiently transfected with either empty vector (−) or with a plasmid encoding Myc-tagged mCRY1 (+). 18 h after transfection, cells were metabolically labeled with [32P]orthophosphoric acid for 3 h, at which time cell free lysates were prepared and subjected to immunoprecipitation with anti-Myc antibody. 32P incorporation into mCRY1 was assessed by SDS-PAGE and phosphorimaging analysis. B, mCRY1 is phosphorylated by CKIε in a CKIε· PER·mCRY1 complex in vitro. HEK 293 cells were transiently transfected with mCRY1 and PER expression plasmids as indicated. mCRY1 protein was then immunoprecipitated (IP) with anti-Myc antibodies, and the immunoprecipitates were split three ways. Two of the three fractions were subjected to an in vitro kinase assay in the presence of [γ-32P]ATP and without (−) or with (+) added recombinant CKIε (ΔC320). mCRY1 phosphorylation was then visualized by SDS-PAGE and phosphorimaging. C, the third bead fraction was subjected to immunoblotting after SDS-PAGE to confirm the presence of CRY and PER proteins in the immunoprecipitates as indicated in the figure.
Transcription Assays and RNA Interference
HEK 293 cells seeded in a single well of a six-well dish were transiently transfected with between 25 and 250 ng of luciferase reporter plasmid using LipofectAMINE Plus (Invitrogen) according to the manufacturer’s directions. Where indicated, 250 ng each of CLK and BMAL1 and 100 ng of CKIε or CKIε (K38A) expression constructs were also co-transfected. In all cases 25 ng of a plasmid encoding β-galactosidase was added, and the total amount of DNA transfected was brought up to 1 μg with the addition of empty vector. After 24 h, cell-free extracts were made by the addition of lysis buffer (100 mm potassium phosphate, pH 7.8, 0.2% Triton X-100, and 0.5 mm DTT). Luciferase activity was detected using the Tropix Dual-Light assay system (PE Biosystems) and measured using a microtiter plate luminometer and the Revelation MLX software package (Dynex Technologies). β-Galactosidase activity was measured as a transfection efficiency control and was used to normalize the luciferase data.
Endogenous CKIε and CKIδ mRNA was targeted in HEK 293 cells by the addition of a 21-nucleotide duplex small interfering RNA (siRNA). This duplex RNA targets nucleotides 520 –538 of the CKIε-coding sequence and was constructed using the ribooligonucleotide (r) pair with the sequences 5′-rCUGGGGAAGAAGGGCAACCdTT-3′ and 5′-rGGUUGCCCUUCUUCCCCAGdTT-3′. The identical nucleotide sequence is present in CKIδ as well. As a control for the specificity of this duplex, a ribooligonucleotide pair with the inverse sequence was used with the sequences 5′-rCCAACGGGAAGAAGGGGUCdTT-3′ and 5′-rGACCCCUUCUUCCCGUUGGdTT. The oligonucleotides were annealed essentially as described in Elbashir et al. (22). 4.5 μg of each duplex was then introduced into 6-well tissue culture plate using Oligofectamine according to the manufacturer’s directions (Invitrogen). After 48 h, the transfection mixture was removed, and the cells were transfected with mPer1-luc reporter, CLK, and BMAL1 expression constructs as described above. Twenty-four hours after transfection, luciferase activity was detected and normalized to β-galactosidase activity. CKIδ and CKIε abundance was assessed by immunoblot using UT31, a rabbit polyclonal antibody that recognizes a shared amino-terminal epitope on CKIε and CKIδ (23).
RESULTS
mCRY Binds a Conserved Domain in the Carboxyl Terminus of PER
Genetic, biochemical, and immunofluorescence data suggest there is a physical interaction between mCRY and mPER (17, 18, 20). To map the region of mPER1 and mPER2 that bind mCRY1 and mCRY2, amino- and carboxyl-terminal truncations of mPER1 and mPER2 were co-expressed with full-length mCRY1 or mCRY2, and the mPER protein was immunoprecipitated. As expected, mPER1 efficiently immunoprecipitated full-length mCRY1 (Fig. 1A, lane 1′). mCRY1 also binds mPER1Δ1219, a truncation of mPER1 lacking the carboxyl-terminal 72 residues (Fig. 1A, lane 5′). However, when mPER1 was further truncated by an additional 100 residues (mPER1Δ1118), all detectable mCRY binding was lost (Fig. 1A, lane 6′). These results indicate that mCRY1 binds to mPER1 in its carboxyl-terminal region bound by amino acids 1117 and 1219, outside the CKIε binding domain. Supporting this, a fragment of mPER1 encompassing amino acids 1027 and 1291, mPER1(1027–1291), also binds mCRY1 (Fig. 1A, lane 4′). The efficiency of the mCRY1 interaction with mPER1(1027–1291) and mPER1Δ1219 is less than that seen with full-length mPER1, suggesting that additional contacts between CRY and PER proteins may occur in the full-length protein (Fig. 1A, lanes 4′ and 5′). As reported by others, mCRY does not require the PAS domains of mPER for stable interaction (Fig. 1A, lane 2′) (20). The mCRY1 binding domain of mPER2 similarly maps to between residues 1127 and 1225 (Fig. 1B). mCRY2 interacts with mPER1 and mPER2 in the same domain as does mCRY1 (data not shown). This CRY binding region of mPER1 and mPER2 is conserved among vertebrate PER proteins but is absent in insect PER and other proteins in GenBank™ (data not shown).
Fig. 1. mCRY interacts with a conserved domain in the carboxyl terminus of mPER1 and mPER2.
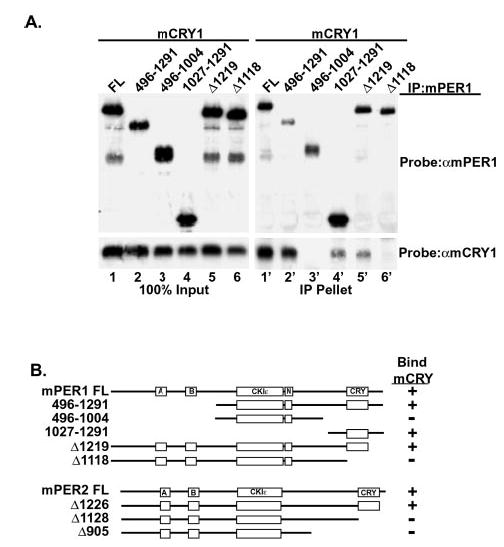
A, HEK 293 cells were transiently transfected with plasmids expressing full-length V5 epitope-tagged mCRY1 and the indicated truncations of Myc epitope-tagged mPER1 or mPER2. mPER was immunoprecipitated (IP) with anti-Myc antibodies and the presence of mCRY in the immunoprecipitate was assessed by immunoblotting with anti-V5 antibodies. B, schematic representation of mCRY·mPER binding results, with selected domains on mPER indicated. Boxes labeled A and B indicate PAS domains, whereas CKIε indicates the CKIε/δ binding site, and N indicates one of the nuclear localization signals. FL, full length.
mPER Is a Scaffold That Brings mCRY1 and CKIε into a Multimeric Complex
mPER1 and mPER2 bind to both endogenous and overexpressed CKIε using a conserved domain in the approximate center of PER (e.g. mPER1 amino acids 496 –815) (12, 13). We therefore tested whether CKIε and CRY could simultaneously bind to PER, forming a ternary complex in intact cells. Full-length mCRY1 was expressed with mPER1 or mPER2 as indicated in Fig. 2. mCRY was immunoprecipitated, and the presence of mPER in the pellet was probed by Western blotting. mCRY1 efficiently interacted with both full-length mPER1 and mPER2 (Fig. 2, lanes 2′ and 4′) but not mPER1Δ1118 or mPER2Δ905 (Fig. 2, lanes 3′ and 5′), confirming that the carboxyl terminus of mPER is required for interaction. Immunoprecipitation of mCRY1 alone in the absence of mPER1 did not pull down endogenous CKIε (Fig. 2, lane 1′). However, when either mPER1 or mPER2 was co-expressed, mCRY1 consistently co-immunoprecipitated both mPER and CKIε (Fig. 2, lanes 2′ and 4′ and data not shown). Consistent with this result, when mPER1Δ1118 (deficient in mCRY binding) was co-expressed with mCRY1, neither truncated mPER1Δ1118 nor CKIε co-immunoprecipitated with mCRY1 (Fig. 2, lane 3′). CKIε also failed to immunoprecipitate with mCRY1 when mPER2Δ905 was expressed (Fig. 2, lane 5′). Taken together, these results indicate that CKIε and mCRY1 are able to interact with each other only indirectly and that they require the presence of mPER to bring them into a trimeric complex. Thus, the mPER protein acts as a scaffold that allows formation of a mCRY1·CKIε· mPER multimeric complex.
Fig. 2. CKIε interaction with mCRY1 requires intact PER protein.
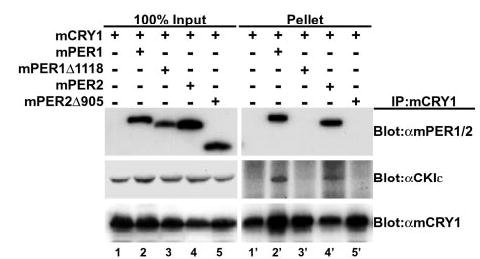
Endogenous CKIε co-immunoprecipitates (IP) with mCRY1 only in the presence of full-length mPER proteins. Myc epitope-tagged mCRY1 and FLAG epitope-tagged mPER were transiently expressed in HEK 293 cells as indicated. mCRY was immunoprecipitated with anti-Myc antibodies, and the presence of co-precipitating mPER was assessed by SDS-PAGE and immunoblotting with anti FLAG antibodies. The blot was sequentially stripped and reprobed with an anti-CKIε polyclonal antibody to detect the presence of co-immunoprecipitating endogenous CKIε and with an anti-Myc antibody to confirm the presence of mCRY1 in the immunoprecipitate pellet. An equal amount of input cell lysates is shown on the left (lanes 1–5), whereas the proteins present in the immunoprecipitate are shown on the right (lanes 1′–5′).
mCRY1 Brings the Cytoplasmic mPER1·CKIε Complex into the Nucleus
PER subcellular localization is a dynamic process regulated by at least two nuclear localization signals, a nuclear export signal, protein-protein interactions, and protein phosphorylation (11, 15, 24). CKIε kinase activity can influence mPER1 subcellular localization by either sequestering it in the cytoplasm in HEK 293 cells (13) or driving it to the nuclear compartment in COS7 cells (15). mCRY1 protein is predominately localized to the nuclear compartment in transfected cells, where it inhibits CLK/BMAL1-dependent transcription (18). mCRY1 also binds and promotes nuclear translocation of mPER1 and mPER2 proteins (18). These observations, when taken together with the fact that mCRY1, mPER1, and CKIε form a multiprotein complex suggest at least two possibilities with respect to subcellular localization. One, mCRY1 may translocate the CKIε·mPER1 complex to the nucleus, overcoming the CKIε-dependent mPER1 cytoplasmic localization seen in HEK 293 cells. Conversely, CKIε may promote mCRY1 cytoplasmic localization that is dependent on its interaction with mPER1.
To distinguish between these two possibilities, epitope-tagged CKIε and mCRY1 were transiently expressed in HEK 293 cells with empty vector and either full-length or truncated mPER1 (Fig. 3), and their intracellular localization was examined by direct immunofluorescence microscopy. In the absence of mPER1, mCRY1 was nuclear in 97% of cells examined, whereas in the same cells, CKIε was predominantly cytoplasmic (only nuclear in 3% of cells) (Fig. 3, panels a, d, and g). However, when mPER1 was co-expressed with CKIε and mCRY1, CKIε localization changed from predominantly cytoplasmic to nuclear (Fig. 3, panels b, e, and h). Thus, mPER1 allows mCRY1 to bring CKIε to the nucleus despite the CKIε kinase activity that otherwise promotes mPER1 cytoplasmic localization in HEK 293 cells (13). Consistent with this model, when the mCRY1 binding-deficient form of mPER1 (mPER1Δ1118) was co-expressed, CKIε remained predominantly in the cytoplasm (Fig. 3, panels c, f, and i).
These results, when taken together with the immunoprecipitation data (Fig. 2), suggest that mPER1 must interact with both CKIε and mCRY1 in order for mCRY1 to bring CKIε into the nucleus. We note that there was some accumulation of CKIε in the nucleus when mPER1Δ1118 was expressed (compare Fig. 3, panels d–f), again suggesting there is some additional interaction between mPER1Δ1118 and mCRY1 (Fig. 2).
mCRY1 Requires mPER1 or mPER2 to Be Phosphorylated by CKIε in Vitro
The ability of CKIε to phosphorylate PER1 requires that the kinase first bind to the substrate, presumably ensuring a high local concentration and/or appropriate positioning of the kinase (Ref. 13 and data not shown). Because mCRY1 can be found in a multimeric complex with PER and CKIε, mCRY1 present in this complex might similarly be phosphorylated by CKIε. We first assessed whether mCRY1 was in fact a phosphoprotein in intact cells. Epitope-tagged mCRY1 was transiently expressed in 32P-metabolically labeled HEK 293 cells, immunoprecipitated, and analyzed by autoradiography (Fig. 4A). mCRY1 incorporated significant amounts of 32P during a 3-h labeling, indicating it is in fact phosphorylated in cultured cells.
To test whether CKIε could phosphorylate mCRY1, mCRY1 alone and mCRY1·mPER complexes were assembled by co-expression in HEK 293 cells, and the proteins were recovered by immunoprecipitation of mCRY1 (Fig. 4C). When mCRY1, expressed alone, was immunoprecipitated and incubated with recombinant CKIε, there was minimal phosphorylation of mCRY1 (Fig. 4B, lane 2). However, when full-length mPER1 or mPER2 was co-expressed with mCRY1, mCRY1 was efficiently phosphorylated by CKIε (Fig. 4B, lanes 4 and 8). Phosphorylation of mCRY1 seen without added CKIε may be due to endogenous CKIε and/or CKIδ co-precipitating with PER1 and PER2 (Fig. 4B, lanes 3 and 7). The interaction of CRY with mPER was required for CRY phosphorylation by CKIε, since mCRY1 phosphorylation fell back to background levels when truncated mPER proteins that do not interact with mCRY1 were co-expressed with mCRY1 (Fig. 4B, lanes 6 and 10). mCRY2 was similarly phosphorylated by CKIε only in the presence of PER containing both CKIε and CRY binding sites (data not shown). These results suggest that PER acts as a scaffold that brings mCRY and CKIε into close proximity with each other. This effectively raises the local concentration of CKIε around mCRY, allowing for efficient phosphorylation of mCRY by CKIε.
To test the potential biological function of mCRY phosphorylation by CKIε, we tested the effect of CKIε co-expression on the ability of CRY1 and CRY2 to repress transcription from the mPer1 promoter (18). No consistent effect of active or dominant-negative CKIε on CRY activity was found (data not shown). Similarly, no effect of dominant-negative CKIε (K38A) on the level of CRY1 phosphorylation in transfected cells was detectable. Thus, although CKIε can clearly phosphorylate mCRY in a multimeric complex in vitro, the physiological significance of this phosphorylation remains to be determined.
BMAL1 Phosphorylation Is Regulated by CKIε
CKIε is found both in the nucleus and cytoplasm depending on cell and tissue type (13, 15, 25). Depending on its subcellular localization CKIε may phosphorylate distinct sets of substrates. Because CKIε forms intermolecular complexes with and phosphorylates clock proteins such as mPER1, mPER2, and mCRY1, we next asked if CKIε could phosphorylate other circadian regulators. CKIε was able to phosphorylate BMAL1 immunoprecipitated from programmed reticulocyte lysates and from transfected cells (Fig. 5, A and B). Immunoprecipitated CLK was not phosphorylated by CKIε (Fig. 5A). We did not observe a reproducible, robust interaction between CKIε and BMAL1 by co-immunoprecipitation assays. Unlike the stimulation of mCRY phosphorylation seen in the presence of mPER, BMAL1 phosphorylation by CKIε was not appreciably altered by the formation of a BMAL1·mPER2 complex (Fig. 5B and data not shown).
BMAL1 was found by metabolic labeling to be a phosphoprotein in cells (Fig. 5C). Because CKIε was able to directly phosphorylate BMAL1 in vitro, the effect of overexpression of a dominant-negative form of CKIε on BMAL1 phosphorylation in intact cells was also examined. CKIε (K38A) co-expression reduced BMAL1 phosphorylation by about 40% (Fig. 5, C and D). The decrease in BMAL1 phosphorylation was not due to a decrease in BMAL1 protein levels, which were unchanged regardless of the presence or absence of CKIε (K38A) (Fig. 5C, lower panel). The partial reduction in BMAL1 phosphorylation could be due to complete inhibition of phosphorylation of a subset of sites or a partial reduction in CKIε activity. Phosphopeptide mapping of BMAL1 expressed without and with CKIε (K38A) demonstrated that there was a global decrease in BMAL1 phosphorylation (data not shown), consistent with CKIε (K38A) partially blocking the endogenous kinases phosphorylating BMAL1.
CKIε Regulates BMAL1-dependent Transcription
Phosphorylation regulates the activity of multiple transcription factors (26, 27). Because BMAL1 is phosphorylated by CKIε in vitro, and expression of a dominant-negative form of CKIε decreases BMAL1 phosphorylation in intact cells, we examined whether CKIε regulated the activity of a CLK/BMAL1-dependent promoter. Transcriptional activity was assessed by the CLK/BMAL1-dependent expression of luciferase from the mPer1 promoter (Fig. 6A). Co-expression of active CKIε had no effect on luciferase expression, whereas expression of dominant-negative CKIε, CKIε (K38A), caused a 40% decrease in luciferase activity. This change in luciferase expression is similar in magnitude to the decrease in BMAL1 phosphorylation seen with CKIε (K38A) co-expression. CKIε may regulate the expression of multiple genes, as it has been implicated in processes as diverse as Wnt/β-catenin signaling and NFAT (nuclear factor of activated T cells) nuclear localization (28, 29). However, CKIε (K38A) is not a global transcriptional repressor, as it had no effect on luciferase expression from actin and cdc2 promoters (Fig. 6, B and C).
Fig. 6. CKIε regulates BMAL1 driven transcription.
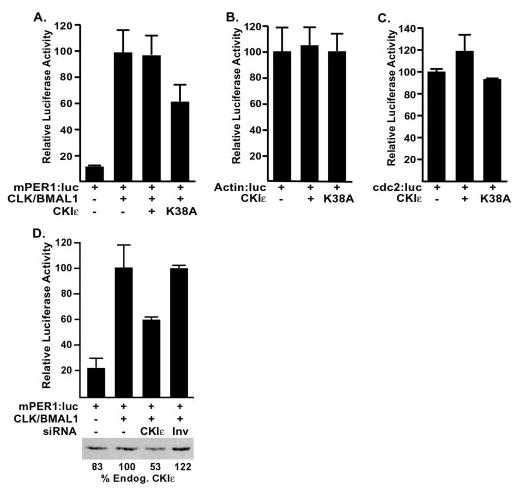
A, overexpression of dominant-negative CKIε leads to decreased BMAL1-driven transcription. HEK 293 cells were transiently transfected with a CLK·BMAL1-responsive promoter (mPer1:luc) and CLK, BMAL1, and CKIε expression constructs or empty vector as indicated. After 18 h, cell free extracts were prepared, and the luciferase activity was analyzed. B and C, CKIε (K38A) has minimal effect on other promoters. HEK 293 cells were transiently transfected with plasmids encoding either CKIε or dominant-negative CKIε (K38A) and a reporter with the actin promoter (B) or the cdc2 promoter (C) driving luciferase expression. 18 h after transfection, luciferase activity was analyzed as in A. D, dsRNA-mediated interference leads to depletion of CKIε/δ and inhibition of CLK/BMAL1-driven gene expression. Endogenous CKIε/δ was partially eliminated in HEK 293 cells by transfection of a 21-nucleotide RNA duplex (siRNA) directed against the CKIε/δ sequence. As a control, the sequence was introduced in the inverted orientation (Inv). After 48 h, the transfection mixture was removed, and plasmids encoding both CLK and BMAL1 (where indicated) along with a reporter containing luciferase behind the mPer1 promoter (18) were introduced. After an additional 24 h, whole cell lysates were prepared, and luciferase activity analyzed as described in A. The lower panel shows a representative immunoblot of CKIε. The relative amount of endogenous CKIε was analyzed using NIH Image software and the quantitation shown beneath.
To further assess the role of CKIε in regulation of circadian E box-containing promoters, we attempted to eliminate CKIε and CKIδ expression using siRNA (22). A 21-base pair RNA duplex (CKIε nucleotides 520 –538, a sequence conserved in CKIδ) was transiently transfected into HEK 293 cells. A dsRNA with the same sequence but in reverse order (denoted Inv) was similarly transfected in control experiments. The effect of siRNA on protein expression was assessed by immunoblot of cell extracts (Fig. 6D). Under these conditions, the dsRNA targeting CKIε and CKIδ reduced their abundance by almost 50%. As Fig. 6D illustrates, treatment of cells with the CKIε/δ dsRNA led to a 40% decrease in luciferase expression driven by the mPer1 promoter compared with mock-treated and control-treated cells. Hence, both dominant-negative CKIε and siRNA-mediated inhibition of CKI activity led to similar decreases in BMAL1-dependent transcriptional activity.
DISCUSSION
CKIε is firmly established as a critical component of the circadian clock. Studies in flies and mammals suggest that the stability and localization of the PERIOD proteins are regulated by CKIε-dependent phosphorylation. In this study, we present evidence that CKIε acts on additional circadian substrates, including a PER-dependent phosphorylation of CRY and direct phosphorylation of BMAL1. Inhibition of CKI activity by either expression of dominant-negative CKIε or use of small interfering RNAs reduced expression from the mPer1 but not the cdc2 nor actin promoters. Although the data cannot exclude the possibility that additional CKI substrates may influence transcription from the mPer1 promoter, they are consistent with a model in which direct phosphorylation of BMAL1 by CKIε positively regulates the activity of BMAL1 in transcription.
The PER protein acts as a scaffold, bringing CKIε and CRY proteins into close proximity. Both CRY proteins interact with the carboxyl termini of mPER1 and mPER2 in a region of high sequence homology between the two proteins and distant from the previously defined PAS domain and CKIε binding site. Identification of a similar CRY binding site on PER has been reported recently by others (30). Notably, the CRY binding domain has no homologous region in insect PER, consistent with a distinct role for cryptochromes as light-sensing proteins in non-vertebrates. The presence of distinct CRY and CKIε binding sites on PER allows formation of a ternary complex between CKIε, CRY, and PER proteins. Formation of this complex appears to have at least two consequences. First, CRY can overcome the CKIε-mediated PER1 cytoplasmic localization. This is consistent with immunolocalization studies showing nuclear CKIε in neurons within the suprachiasmatic nucleus (25, 31) and positions CKIε to phosphorylate nuclear circadian regulators. This conclusion is supported by the recent report of Lee et al. (32), who found CKIε and CKIδ in a complex with both PER and CRY in rat and hamster liver and additionally found circadian regulation of CKI nuclear localization. Second, CRY present in this complex can be efficiently phosphorylated by CKIε in vitro. The functional consequence of CKIε-dependent CRY phosphorylation is the subject of ongoing investigation, but it does not appear to modify the inhibitory effect of CRY upon circadian promoters (data not shown).
We also examined whether or not BMAL1 was a substrate for CKIε. Given that CRY requires the formation of a CRY·PER·CKIε multimeric complex for its phosphorylation by CKIε, it was unexpected that CKIε can in fact directly phosphorylate BMAL1. This phosphorylation appears to be physiologically relevant, since overexpression of dominant-negative CKIε led to both a decrease in BMAL1 phosphorylation and a decrease in BMAL1-dependent transcription from E-box-containing promoters. Similarly, a dsRNA-mediated decrease in CKIε protein in intact cells led to a decrease in BMAL1 transcriptional activation function. That the CKIε with the R178C mutation found in the tau hamster is likely to exhibit a dominant-negative phenotype (i.e. bind to substrates and block access of active kinases) (1, 9) suggests that decreased phosphorylation of BMAL1 may contribute to the tau short period phenotype. An alternative hypothesis not excluded here is that in the cell CKIε may regulate a pathway that then regulates BMAL1 phosphorylation and activity. Although in these studies we used CKIε, the closely related CKIδ that can similarly bind to and phosphorylate PER proteins is likely to similarly phosphorylate BMAL1 (13, 25). Recent work from two groups suggests both BMAL1 and CLK are phosphorylated in intact animals. Both proteins have phosphorylation-dependent electrophoretic mobility shifts that change through the circadian day (32). Finally, we note that Sanada and co-workers (33) recently show that mitogen-activated protein kinase phosphorylates BMAL1 on several sites and modestly inhibits the activity of BMAL1 in transcription assays. Mitogen-activated protein kinase and CKIε are likely to phosphorylate distinct sites on BMAL1. BMAL1 may therefore be the target of several distinct signaling pathways, with divergent effects on its activity.
Immunolocalization studies of CKIε have produced conflicted results, dependent on cell type and co-expression of additional circadian proteins. The finding of a multimeric CKIε·PER·CRY complex that can relocalize CKIε from cytoplasm to nucleus implies that some of the variability in CKIε localization seen in cultured cells is due to co-expression of circadian regulators. For example, PER and CRY levels may be higher in COS7 than in HEK 293 cells and, hence, target CKIε to the nucleus more readily in those cells. These results also suggest that one function of the PER·CRY complex in vivo may be to relocalize CKIε to the nucleus during periods of CRY abundance, facilitating phosphorylation of BMAL1 and perhaps additional nuclear circadian regulators. What remains to be resolved is how the stimulatory effect of CKIε phosphorylation and the inhibitory effect of CRY on BMAL1 activity are coordinated to produce stable oscillations in expression from circadian promoters.
Acknowledgments
We thank R. Schackmann for oligonucleotide synthesis, M. Morgan for assistance with immunofluorescence microscopy, and S. Reppert and A. Schönthal for plasmids.
Footnotes
These studies were funded in part by National Institutes of Health Grants R01CA71074 and P30CA42014 and by the Huntsman Cancer Foundation.
The abbreviations used are: CLK, CLOCK; mCRY1 and mCRY2, murine cryptochrome 1 and 2, respectively; mCRY, both mCRY1 and mCRY2; CKIε, casein kinase I epsilon; PER, mammalian PERIOD proteins; HEK 293 cells, human embryonic kidney 293; DTT, dithiothreitol; siRNA, small interfering RNA; dsDNA, double-stranded DNA.
References
- 1.Eide EJ, Virshup DM. Chronobiol Int. 2001;18:389 –398. doi: 10.1081/cbi-100103963. [DOI] [PubMed] [Google Scholar]
- 2.Reppert SM, Weaver DR. Annu Rev Physiol. 2001;63:647–676. doi: 10.1146/annurev.physiol.63.1.647. [DOI] [PubMed] [Google Scholar]
- 3.Lowrey PL, Takahashi JS. Annu Rev Genet. 2000;34:533–562. doi: 10.1146/annurev.genet.34.1.533. [DOI] [PubMed] [Google Scholar]
- 4.Allada R, Emery P, Takahashi JS, Rosbash M. Annu Rev Neurosci. 2001;24:1091–1119. doi: 10.1146/annurev.neuro.24.1.1091. [DOI] [PubMed] [Google Scholar]
- 5.Kloss B, Price JL, Saez L, Blau J, Rothenfluh A, Wesley CS, Young MW. Cell. 1998;94:97–107. doi: 10.1016/s0092-8674(00)81225-8. [DOI] [PubMed] [Google Scholar]
- 6.Price JL, Blau J, Rothenfluh A, Abodeely M, Kloss B, Young MW. Cell. 1998;94:83–95. doi: 10.1016/s0092-8674(00)81224-6. [DOI] [PubMed] [Google Scholar]
- 7.Rothenfluh A, Abodeely M, Young MW. Curr Biol. 2000;10:1399 –1402. doi: 10.1016/s0960-9822(00)00786-7. [DOI] [PubMed] [Google Scholar]
- 8.Suri V, Hall JC, Rosbash M. J Neurosci. 2000;20:7547–7555. doi: 10.1523/JNEUROSCI.20-20-07547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, Menaker M, Takahashi JS. Science. 2000;288:483–492. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vielhaber EL, Virshup DM. Int Union Biochem Mol Biol Life. 2001;51:273–278. [Google Scholar]
- 11.Kloss B, Rothenfluh A, Young MW, Saez L. Neuron. 2001;30:699 –706. doi: 10.1016/s0896-6273(01)00320-8. [DOI] [PubMed] [Google Scholar]
- 12.Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, Fu YH. Science. 2001;291:1040 –1043. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- 13.Vielhaber E, Eide E, Rivers A, Gao ZH, Virshup DM. Mol Cell Biol. 2000;20:4888 –4899. doi: 10.1128/mcb.20.13.4888-4899.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keesler GA, Camacho F, Guo Y, Virshup D, Mondadori C, Yao Z. Neuroreport. 2000;11:951–955. doi: 10.1097/00001756-200004070-00011. [DOI] [PubMed] [Google Scholar]
- 15.Takano A, Shimizu K, Kani S, Buijs RM, Okada M, Nagai K. FEBS Lett. 2000;477:106 –112. doi: 10.1016/s0014-5793(00)01755-5. [DOI] [PubMed] [Google Scholar]
- 16.Yagita K, Yamaguchi S, Tamanini F, van Der Horst GT, Hoeijmakers JH, Yasui A, Loros JJ, Dunlap JC, Okamura H. Genes Dev. 2000;14:1353–1363. [PMC free article] [PubMed] [Google Scholar]
- 17.Field MD, Maywood ES, O’Brien JA, Weaver DR, Reppert SM, Hastings MH. Neuron. 2000;25:437–447. doi: 10.1016/s0896-6273(00)80906-x. [DOI] [PubMed] [Google Scholar]
- 18.Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- 19.Griffin EA, Jr, Staknis D, Weitz CJ. Science. 1999;286:768 –771. doi: 10.1126/science.286.5440.768. [DOI] [PubMed] [Google Scholar]
- 20.Shearman LP, Sriram S, Weaver DR, Maywood ES, Chaves I, Zheng B, Kume K, Lee CC, van der Horst GT, Hastings MH, Reppert SM. Science. 2000;288:1013–1019. doi: 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- 21.Cegielska A, Gietzen KF, Rivers A, Virshup DM. J Biol Chem. 1998;273:1357–1364. doi: 10.1074/jbc.273.3.1357. [DOI] [PubMed] [Google Scholar]
- 22.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Nature. 2001;411:494 –498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 23.Fish K, Cegielska A, Getman M, Landes G, Virshup DM. J Biol Chem. 1995;270:14875–14883. doi: 10.1074/jbc.270.25.14875. [DOI] [PubMed] [Google Scholar]
- 24.Vielhaber EL, Duricka D, Ullman KS, Virshup DM. J Biol Chem. 2001;276:45921–45927. doi: 10.1074/jbc.M107726200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Camacho F, Cilio M, Guo Y, Virshup D, Patel K, Khorkova O, Styren S, Morse B, Yao Z, Keesler GA. FEBS Lett. 2001;489:159 –165. doi: 10.1016/s0014-5793(00)02434-0. [DOI] [PubMed] [Google Scholar]
- 26.Hunter T, Karin M. Cell. 1992;70:375–387. doi: 10.1016/0092-8674(92)90162-6. [DOI] [PubMed] [Google Scholar]
- 27.Karin M, Hunter T. Curr Biol. 1995;5:747–757. doi: 10.1016/s0960-9822(95)00151-5. [DOI] [PubMed] [Google Scholar]
- 28.Peters JM, McKay RM, McKay JP, Graff JM. Nature. 1999;401:345–350. doi: 10.1038/43830. [DOI] [PubMed] [Google Scholar]
- 29.Sakanaka C, Leong P, Xu L, Harrison SD, Williams LT. Proc Natl Acad Sci U S A. 1999;96:12548 –12552. doi: 10.1073/pnas.96.22.12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyazaki K, Mesaki M, Ishida N. Mol Cell Biol. 2001;21:6651–6659. doi: 10.1128/MCB.21.19.6651-6659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishida Y, Yagita K, Fukuyama T, Nishimura M, Nagano M, Shigeyoshi Y, Yamaguchi S, Komori T, Okamura H. J Neurosci Res. 2001;64:612–616. doi: 10.1002/jnr.1114. [DOI] [PubMed] [Google Scholar]
- 32.Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- 33.Sanada K, Okano T, Fukada Y. J Biol Chem. 2002;277:267–271. doi: 10.1074/jbc.M107850200. [DOI] [PubMed] [Google Scholar]