

Abstract
Earlier studies have implicated BDNF in stress and in the mechanism of action of antidepressants. It has been shown that antidepressants upregulate, whereas corticosterone downregulates, BDNF expression in rat brain. Whether various classes of antidepressants reverse corticosterone-mediated downregulation of BDNF is unclear. Also not known is how antidepressants or corticosterone regulate BDNF expression. To clarify this, we examined the effects of various classes of antidepressants and corticosterone, alone and in combination, on the mRNA expression of total BDNF and of individual BDNF exons, in rat brain. Normal or corticosterone pellet-implanted (100 mg, 21 days) rats were injected with different classes of antidepressants, fluoxetine, desipramine, or phenelzine, intraperitoneally for 21 days and sacrificed 2 h after the last injection. mRNA expression of total BDNF and of exons I-IV was measured in frontal cortex and hippocampus. Given to normal rats, fluoxetine increased total BDNF mRNA only in hippocampus, whereas desipramine or phenelzine increased BDNF mRNA in both frontal cortex and hippocampus. When specifc exons were examined, desipramine increased expression of exons I and III in both brain areas, whereas phenelzine increased exon I in both frontal cortex and hippocampus but exon IV only in hippocampus. On the other hand, fluoxetine increased only exon II in hippocampus. Corticosterone treatment of normal rats decreased expression of total BDNF mRNA in both brain areas, specifically decreasing exons II and IV. Treatment with desipramine or phenelzine of corticosterone pellet-implanted rats reversed the corticosterone-induced decrease in total BDNF expression in both brain areas; however, fluoxetine reversed the decrease only partially in hippocampus. Interestingly, antidepressant treatment of corticosterone pellet-implanted rats increased only those specific exons that are increased during treatment of normal rats with each particular antidepressant. We found that although corticosterone and antidepressants both modulate BDNF expression, and antidepressants reverse the corticosterone-induced BDNF decrease, antidepressants and corticosterone differ in how they regulate the expression of BDNF exon(s).
Keywords: BDNF, exons, depression, antidepressants, mRNA, stress, glucocorticoid
INTRODUCTION
Corticosteroids released in response to a hyperactive limbic hypothalamic-pituitary-adrenal axis (HPA) system cause many functional changes in neurons, including neuronal loss (Sapolsky, 1999), hippocampal atrophy (Saposky, 2000), dendritic remodeling (Reagen and McEwen, 1997), cell differentiation (Slotkin et al., 1998), reduced neurogenesis (Gould et al., 1997; Gass et al., 2003), and reduced neural plasticity (Magarinos et al., 1996; McEwen, 1999). Adrenal steroids also play an important role in modulating many aspects of central nervous functions, including regulation of mood, emotion, behavior, and learning (McEwen, 1987). In this regard, a hyperactive HPA axis, with the consequent hypercortisolism, observed in depressed patients, represents one of the most consistent findings in biological psychiatry (Gold et al., 1988). A number of studies show higher cortisol levels in plasma and increased levels of corticotropin-releasing hormone and adrenocorticotropic hormone (Carrol et al., 1976; Murphy et al., 1991; Holsboer, 2001; Young et al., 2003; Pariante, 2003) in depressed patients. As occurs with corticosterone in rats, depressed patients also show dysfunctions in the cortico-limbic system, including abnormal glial and neuronal pathology (Sheline et al., 1996; Ongur et al., 1998; Rajkowska et al., 1999; Drevets et al., 2000).
Interestingly, many of the beneficial effects of antidepressants in depressed patients are exerted through their action on the HPA system. For example, antidepressant treatment is associated with a reduction of activity in basal and stress-induced hyperactive HPA axis (Pepin et al., 1992a, 1992b; Peiffer et al., 1991; Montkowski et al., 1995; Pariante and Miller, 2001; Budziszewska, 2002; Barden, 2004). The normalization of the hyperactive HPA axis and the mood-stabilizing effects occur simultaneously during antidepressant treatment, which suggests that these two effects are directly or indirectly related. How antidepressants reverse a hyperactive HPA axis is unclear. One of the proposed mechanisms is that antidepressants cause effects on cellular corticosteroid receptor levels rendering the HPA axis more susceptible to feedback inhibition by cortisol (reviewed in Barden, 2004); although there are reports suggesting that glucocorticoid receptor upregulation is not a prerequisite for antidepressant action on the HPA axis (reviewed in Pariante and Miller, 2004).
The role of brain-derived neurotrophic factor (BDNF), one of the most prevalent neurotrophic factors in adult brain, in stress, depression, and in the mechanism of action of antidepressants is well appreciated. It has been shown that BDNF expression is decreased after corticosterone treatment (Chao and McEwen, 1994; Smith et al., 1995; Schaaf et al., 1997, 1998, 2000), during stress (Smith et al., 1995; Nibuya et al., 1995; 1999; Ueyama et al., 1997; Rasmussen et al., 2002), and in depressed patients (Karege et al., 2002; Dwivedi et al., 2003; Shimuzu et al., 2003). On the other hand, several classes of antidepressants increase expression of BDNF in rat brain (Nibuya et al., 1995; Lindeforts et al., 1995; Russo-Neustadt et al., 2000; Coppel et al., 2003) as well as in depressed patients (Chen et al., 2001; Dwivedi et al., 2003; Shimuzu et al., 2003; Karege et al., 2005; Gervasoni et al., 2005). These studies have concluded that stress decreases BDNF expression and that antidepressants increase its expression. However, it is unclear whether antidepressant treatment reverses corticosterone-induced changes in BDNF expression, and if so what would be the mechanism(s)?
On a molecular level, BDNF is highly regulated. The rat BDNF gene contains four separate promoters that are linked to four main transcript forms (Timmusk et al., 1993; Nakayama et al., 1994). Each transcript has four short 5′ noncoding exons (I-IV) containing separate promoters and one shared 3′ exon (exon V) encoding the mature BDNF protein. Although the biological significance of these BDNF transcripts is not clear, it appears that these transcripts can facilitate multi-level regulation of BDNF expression and may determine the tissue-specific expression. The various BDNF transcripts are differentially expressed in different tissues, with exons I-III primarily expressed in brain, and exon IV more abundant in peripheral tissues (Timmusk et al., 1993). As mentioned above stress/corticosterone downregulates BDNF expression, but which promoter (s) is involved in the effects of corticosterone is not known. In addition, it is imperative to examine whether antidepressants reverse these corticosterone-induced changes in BDNF expression and whether these effects are related to altered expression of a specific BDNF transcript(s).
Therefore, the present study was undertaken to examine in rat brain: 1) whether corticosterone causes specific changes in BDNF transcript (s), 2) whether different classes of antidepressants reverse corticosterone-induced changes in BDNF transcript (s), 3) whether treatment with these antidepressants of normal rats causes specific changes in BDNF transcript (s), and 4) whether the reversal of BDNF transcript changes by antidepressants is associated with changes in the same or different BDNF transcript(s) affected as those by corticosterone. In order to examine whether antidepressants share a common or different effects on the expression of BDNF and its various exons, we chose antidepressants that act via different mechanisms of actions, such as desipramine (norepinephrine uptake inhibitor), fluoxetine (selective serotonin reuptake inhibitor), and phenelzine (monoamine oxidase inhibitor).
EXPERIMENTAL PROCEDURES
Animals
Virus-free Sprague-Dawley male rats initially weighing 220–250 g were used. Rats were housed in groups of three under standard laboratory conditions (temperature 21 ± 1°C, humidity 55 ± 5%, 12-hr light/dark cycle). Animals were provided free access to food. Rats were acclimatized for one week before the start of experiments. Each treatment group contained 6 rats.
Corticosterone treatment
All the experiments were carried out in accordance with the National Institute of Health Guide for the care and use of laboratory animals. The Animal Care Committee of the University of Illinois at Chicago approved this study. The procedure for corticosterone treatment has been described in our earlier publications (Dwivedi and Pandey, 2000, Dwivedi et al., 2000). Rats under light halothane anesthesia were subcutaneously implanted with corticosterone pellets containing 100 mg of corticosterone in a cholesterol base (Innovative Research of America, Sarasota, FL). These corticosterone pellets can maintain a physiological serum concentration of corticosterone for 21 days. The release of CORT after implantation of a 100-mg corticosterone pellet is 4.76 mg per day. Control rats underwent an identical surgery procedure with implantation of a cholesterol base pellet or underwent no treatment; these two types of treatment did not differ in their results in the final determinations. Rats were decapitated 21 days after pellet implantation.
Antidepressant treatment
Intraperitoneal injections of desipramine (10 mg/kg), fluoxetine (5 mg/kg) or phenelzine (10 mg/kg) were given once daily for 21 days to normal rats. The normal control rats were given intraperitoneal injections of an equal volume of normal saline (0.9% w/v). In a separate experiment, corticosterone-treated rats were given intraperitoneal injections of desipramine (10 mg/kg), fluoxetine (5 mg/kg) or phenelzine (10 mg/kg) once daily for 21 days. Sham rats were given an equal volume of normal saline (0.9% w/v). Animals were sacrificed 2 h after the last drug treatment. The doses of the drugs were based on their free-base weight. The selection of the doses was based on our previous studies (Pandey et al., 1991; Dwivedi et al., 1994; Dwivedi and Pandey, 1997; Dwivedi et al., 2002) and on reports in the literature (Okada et al., 1988; Ozawa and Rasenick, 1991; Ulrichsen et al., 1992; Trouvin et al., 1993; Todd et al., 1995; Mann et al., 1995).
The trunk blood was collected on ice and was centrifuged, and then the serum was stored at −80°C until the assays were performed. Serum corticosterone levels were measured by a commercially available radioimmunoassay kit (ICN Biomedical, Inc., Cleveland, OH). Brains were removed quickly after the blood was taken. Frontal cortex and hippocampus were dissected out and immediately stored at −80°C until analyzed. For both experimental protocols, rats were decapitated between 09.00 hr and 11.00 hr, corresponding to 3–5 hr after lights on.
RNA isolation
Frontal cortices and hippocampi were homogenized in 4 M guanidine isothiocyanate, 50 mM Tris/HCl (pH = 7.4), and 25 mM EDTA, and the total RNA was isolated by CsCl2 ultracentrifugation. The yield of total RNA was determined by measuring the absorbance of an aliquot of the resuspended precipitated RNA stock at a wavelength of 260/280 nm, and samples showing a ratio of ≥ 1.7 were used. The extent of degradation of mRNA was also assessed by evaluating the sharpness of 28S and 18S ribosomal RNA bands. None of the samples used in this study showed detectable signs of degradation. To check for possible DNA contamination, after each extraction, samples were run by RT-PCR without adding the reverse transcriptase enzyme.
Oligonucleotides
The primer pairs were designed to allow amplification of 219–524 base pairs (bp): forward, 5′ GGATGAGGACCAGAAGGTTCG, and reverse, 5′ GATACCGGGACTTTCTCCAGG (GenBank accession # M61175). Each primer contained comparable G/C content to minimize variability in hybridization efficiency at the annealing temperature. The specificity of the BDNF mRNA product were checked by sequencing the amplified areas with the Sequenase Version 2.0 DNA Sequencing Kit using HindIII and EcoRI, which produced fragments of the expected size.
Synthesis and cloning of internal standards
Internal standard targeted by the same primers used to amplify the canonic sequence was generated by site-directed mutagenesis to introduce a Bgl II restriction site between the amplification primers so that the digestion of the amplicon would generate two fragments of approximately equal molecular size. The internal primer sequence for BDNF was 5′ATGATGCC GCAAAGATCTCTATGAGGG (359–382 bp). The underlined bases indicate the Bgl II restriction site, whereas bold and italicized bases indicate the mutation sites. The single-strand internal primers were designed and synthesized so that the restriction site was introduced with only a minimal number of base substitutions, and also such that there was a 24-bp overlap of the primary PCR products. Each of the internal standards was synthesized in two PCR steps starting with a cDNA template reverse transcribed from the total RNA. The internal standard templates were first cloned into a pGEM4Z vector and then amplified using M13 primers. The cRNA corresponding to the sense strand was synthesized from an M13 amplified template using Sp6 RNA polymerase by means of an in-vitro transcription kit.
Quantitative analyses of BDNF mRNA by competitive RT-PCR
Quantitation of BDNF mRNA was determined using internal standards as described earlier (Dwivedi et al., 2003). Decreasing concentrations of BDNF internal standard cRNA (800–50 pg) were added to 1 μg of total RNA. The RNA/cRNA mixtures were denatured at 80°C for 6 min and then reverse transcribed with cloned Moloney murine leukemia virus (M-MLV) reverse transcriptase (200 U; Life Technologies, Grand Island, NY, USA) in RT buffer containing 50 mM Tris/HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, 10 mM DTT, and 0.2mM dNTPs using random hexamers (2.5 mM) and ribonuclease inhibitor (HPRI) (8.3 U) in a volume of 20 μl. The RT mixture was incubated at 37°C for 60 min to promote cDNA synthesis. The reaction was terminated by heating the samples at 98°C for 5 min. In all assays, one RT reaction was performed in the absence of RNA as a control.
Competitive PCR amplification
After the termination of the RT reaction, cDNA aliquots containing reverse-transcribed material were amplified with Hot Tub DNA polymerase in the Thermal Cycler (9600; Perkin Elmer, Norwalk, CT, USA). The amplification mixture contained cDNA, 0.5 μM specific primer pairs, 1.5 mM MgCl 2, 50 mM Tris/HCl (pH 9.0), 20 mM ammonium sulfate, 15 mM KCl, and 0.5 U of Hot Tub DNA polymerase in a 100-μl volume. Trace amounts of [32P]dCTP (0.5–1 μCi/sample) were included during the PCR step for subsequent quantification. The PCR mixture was amplified for 27 cycles with denaturation (94°C, 15 s), annealing (60°C, 30 s), and elongation (72°C, 30 s) amplification steps. The reaction was terminated with a 5-min final elongation step. Following amplification, aliquots were digested with Bgl II in triplicate and run by 1.5% agarose gel electrophoresis.
To quantitate the amount of product corresponding to the reverse-transcribed and amplified mRNA, the ethidium bromide-stained bands were excised and counted. The results were calculated as the counts incorporated into the amplified cRNA standard divided by the counts incorporated into the corresponding mRNA amplification product versus the known amount of internal standard (cRNA) added to the test sample. The results are expressed as attomoles/μg of total RNA.
RT-PCR assay for determination of various Exon-containing BDNF transcript expression
Primers for specific BDNF exons were designed according to Altieri et al. (2004). The following primer sequences were used: exon I: forward, 5′GCTGGTGCAGGAA AGCAAC, reverse, 5′CCAGGTAAGAAAAGCTTCGCC (amplified from bases 543–618; GenBank accession # X67106); exon II: forward, 5′GGC TGGAATAGACTCTTG GCA, reverse, 5′CCGGTGGCTAGATCCTGGA (amplified from bases 2118–2207; GenBank accession # X67106); exon III: forward, 5′CACTGAAGGCGTGCGAG TATT, reverse, 5′TGTACTCCTGTTCTTCAGCAAGAA (amplified from bases 750–832; GenBank accession # X67107), exon IV: forward, 5′GGCGCAG GGACCAGG, reverse, 5′TCAGGGTCCACACAAAGCTC (amplified from bases 2006–2076; GenBank accession # X67107). β-Actin was used as an internal control. The sequence for β-Actin was: forward, 5′TGAACCCTAAGGCCAACCGTG, reverse, 5′CTCATAGCTCTT CTCCAGGG (GenBank accession # V01217), which was amplified from bases 1659–2053 cRNA. One microgram of RNA was used for the RT-PCR assay. The procedure for RT-PCR was similar as described above. Four to five independent RT-PCRs were performed for each primer pair. We initially standardized the linearity of PCR amplification and used the appropriate number of PCR cycles so that signals were not saturated. The radioactivity count (cpm) obtained from BDNF exon amplification was normalized with respect to β-Actin. The ratio is expressed as a percentage of control ± standard deviation (SD).
Statistics
Data were analyzed using the SPSS 9.0 (Chicago, IL) statistical software package. All values are given as the mean ± SD. One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons were used to examine the effects of corticosterone or antidepressant treatment on total and individual exons of BDNF. An α value lower than 0.05 was considered significant.
RESULTS
Serum corticosterone levels
Serum corticosterone levels were measured in sham, corticosterone- treated and corticosterone-treated rats given antidepressants. We also measured serum corticosterone levels in normal and antidepressant-treated rats. The serum corticosterone levels are given in Table 1. As in our earlier studies, we found that corticosterone level was significantly higher in corticosterone pellet-implanted rats (Dwivedi and Pandey, 2000; Dwivedi et al., 2000), and that desipramine or phenelzine normalized the increased level of corticosterone, whereas fluoxetine had no significant effects on the level of corticosterone.
Table 1.
Serum corticosterone levels in different groups
| Groups (n = 6) | Corticosterone level (ng/ml) |
|---|---|
| Sham | 46.8 ± 9.9 |
| Corticosterone | 116.2 ± 30.5a |
| Corticosterone + fluoxetine | 104.2 ± 35.7b |
| Corticosterone + desipramine | 60.2 ± 10.4c |
| Corticosterone + phenelzine | 47.8 ± 12.2d |
Values are the mean ± S.D. Overall group difference: df = 4, F = 12.7, P < 0.001.
Different treatment groups were compared with sham.
P < 0.001,
P < 0.001,
P = 0.32,
P = 0.94.
Effects of antidepressant treatment of normal rats on total BDNF mRNA level in frontal cortex and hippocampus
To examine the effects of antidepressants on mRNA level of BDNF, initially we determined total BDNF mRNA expression level in frontal cortex and hippopcampus of antidepressant-treated rats. A representative gel electrophoresis showing competitive RT-PCR for BDNF in the frontal cortex is given in Figure 1A., whereas representative graph showing the quantitation of BDNF mRNA is given in Figure 1B. The amplification product for BDNF from the mRNA template was at 306 bp, whereas the corresponding digestion product from cRNA was at 152+154 bp. It was observed that mRNA level of BDNF was about five folds higher in hippocampus than frontal cortex (Figure 2). When BDNF mRNA level was compared between antidepressant-treated and normal rats, it was observed that desipramine or phenelzine significantly increased expression of BDNF in both frontal cortex (Figure 2A) and hippocampus (Figure 2B). On the other hand, fluoxetine caused a small but significant increase in BDNF level only in hippocampus (Figure 2B) but not in frontal cortex (Figure 2A).
Figure 1.

(A) Representative gel electrophoreses showing competitive PCR-analysis for BDNF mRNA content in frontal cortex obtained from one sham rat. Decreasing concentrations of standard cRNA (50-3.12 pg) were added to a constant amount (1 μg) of total RNA isolated from frontal cortex. The mixtures were reverse transcribed and PCR-amplified in the presence of trace amounts of [32P]dCPT; aliquots were digested by Bgl II and electrophoresed on 1.5% agarose gel. The higher molecular size band corresponds to the amplification product arising from the mRNA, whereas the lower bands arise from cRNA generated from the internal standard digested by Bgl II. (B) Data derived from the agarose gels are plotted as the counts incorporated into the amplified cRNA standard divided by the counts incorporated into the corresponding mRNA amplification product versus the known amount of internal standard cRNA added to the test sample. The point of equivalence represents the amount of BDNF mRNA.
Figure 2.
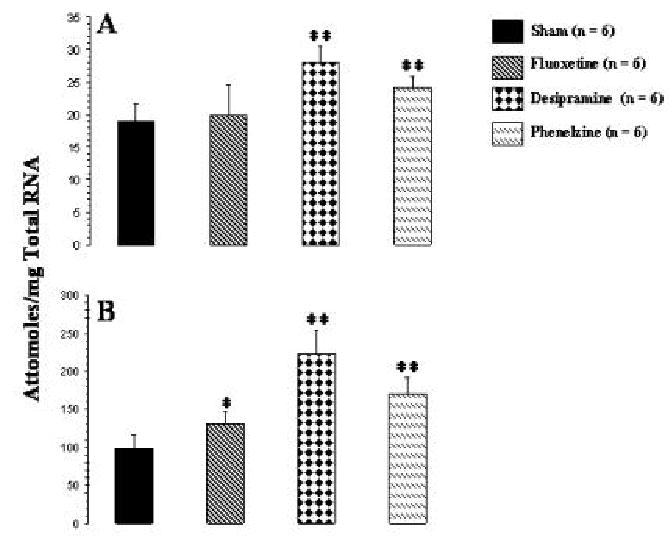
mRNA level of total BDNF in frontal cortex (A) and hippocampus (B) of rats given i.p. injections of antidepressants for 21 days. Animals were sacrificed 2 h after the last antidepressant injection. Values are the mean ± S.D. from 6 rats. Antidepressant-treated rats were compared with sham rats. Overall group differences were: frontal cortex, df = 3, 20, F = 12.0, P <0.001; hippocampus, df = 3,20, F = 35.7, P <0.001. *P <0.001, ** P <0.007.
Effects of corticosterone pellet implantation and of antidepressant treatment of corticosterone pellet-implanted rats on total BDNF mRNA level in frontal cortex and hippocampus
Corticosterone pellet implantation caused a significant decrease in mRNA levels of BDNF in both frontal cortex (Figure 3A) and hippocampus (Figure 3B) as compared with sham rats. We also observed that treatment with desipramine or phenelzine of corticosterone pellet-implanted rats significantly reversed the corticosterone-induced decrease in BDNF mRNA levels in frontal cortex (Figure 3A) and hippocampus (Figure 3B). Desipramine caused complete reversal of the decrease in mRNA level of BDNF in both frontal cortex and hippocampus, whereas phenelzine reduced it partially in frontal cortex but almost completely in hippocampus. fluoxetine, on the other hand was not able to significantly reverse the corticosterone mediated decrease in BDNF mRNA expression in frontal cortex (Figure 3A) but was able to reverse it partially but significantly in hipocampus (Figure 3B).
Figure 3.
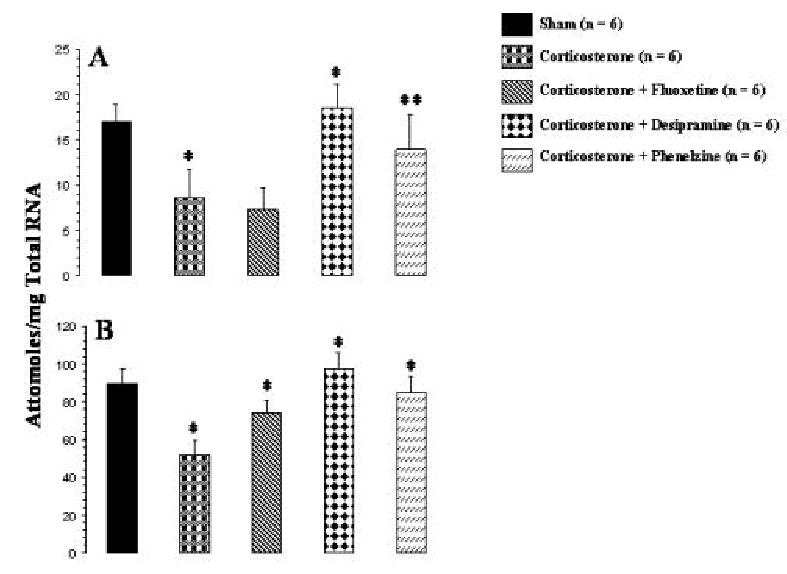
Effects of corticosterone and of antidepressant treatment of corticosterone-treated rats on mRNA levels of total BDNF in rat frontal cortex (A) and hippocampus (B). Corticosterone pellet was implanted for 21 days and i.p. injections of an antidepressant were simultaneously started and continued for 21 days. Animals were sacrificed 2 hr after the last antidepressant injection. Values are the mean ± S.D. from 6 rats. Overall group differences were: frontal cortex, df = 4, 25, F = 17.6, P <0.001; hippocampus, df = 4,25, F = 29.8, P <0.001. Corticosterone-treated group was compared with sham group, whereas antidepressant-treated group was compared with corticosterone-treated group. *P <0.004, **P <0.001.
Effects of antidepressant treatment of normal rats on expression of BDNF transcripts in frontal cortex and hippocampus
Comparison of the expression levels of the various BDNF exons shows that the hippocampus had higher expression of all the exons [frontal cortex (cpm): exon I, 1317 ± 175; exon II, 2263 ± 244; exon III, 4110 ± 158, exon IV, 1645 ± 121; hippocampus (cpm): exon I, 2203 ± 346; exon II, 8208 ± 465; exon III, 25141 ± 3127; exon IV, 5027 ± 250]; although the ratio of the expression in both hippocampus and frontal cortex was similar, i.e., exon III>II>IV>I. The effects of various antidepressants on the levels of exon-specific BDNF transcripts were evaluated by RT-PCR. The results of the expression of BDNF transcripts after antidepressant treatment in rat frontal cortex and hippocampus are given in Figures 4 and 5, respectively. Treatment with desipramine selectively and significantly increased expression of exons I and III in both frontal cortex (Figure 4) and hippocampus (Figure 5). In both brain areas, the increase in expression of exon III by desipramine was more pronounced than in exon I. No significant effects of desipramine treatment on expression of exons II or IV were observed, either in frontal cortex or in hippocampus. Phenelzine increased expression of exon I in both frontal cortex (Figure 4) and hippocampus, and exon IV only in hippocampus (Figure 5). In both brain areas, the increase in expression of exon IV was greater than in exon I. On the other hand, fluoxetine treatment caused a small but significant increase in expression of only the exon II-containing BDNF transcript in hippocampus (Figure 5) but had no significant effects on expression of other exon-containing transcripts in this brain area. Fluoxetine also did not cause any significant change in expression of any of the BDNF transcripts in frontal cortex (Figure 4).
Figure 4.
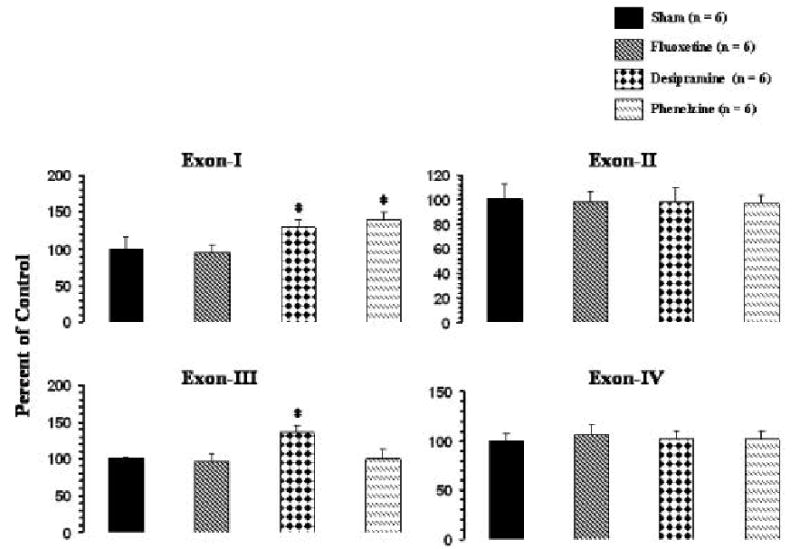
mRNA levels of BDNF transcripts in frontal cortex of rats treated with antidepressants. Rats were given i.p. injections of an antidepressant for 21 days. Animals were sacrificed 2 h after the last antidepressant injection. Values are the mean ± S.D. from 6 rats. Antidepressant-treated group was compared with sham group. Overall group differences were: exon I, df = 3,20, F = 20.3, P <0.001; exon II, df = 3,20, F = 0.12, P = 0.94; exon III, df = 3,20, F = 24.7, P <0.001; exon IV, df = 3,20, F = 0.58, P = 0.63. *P <0.001.
Figure 5.
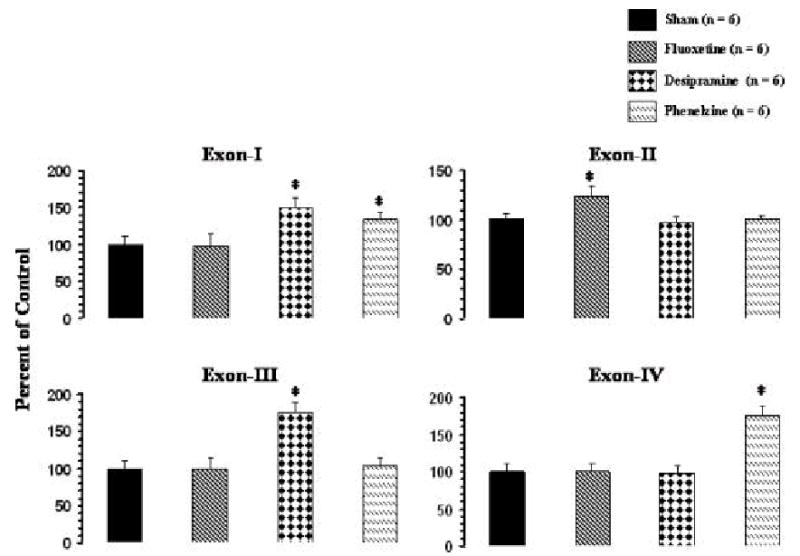
mRNA levels of BDNF transcripts in hippocampus of rats treated with antidepressants. Rats given i.p. injections of an antidepressant for 21 days. Animals were sacrificed 2 h after the last antidepressant injection. Values are the mean ± S.D. from 6 rats. Antidepressant-treated group was compared with sham group. Overall group differences were: exon I, df = 3,20, F = 25.1, P <0.001; exon II, df = 3,20, F = 15.4, P <0.001; exon III, df = 3,20, F = 53.7, P <0.001; exon IV, df = 3,20, F = 68.4, P <0.004. * P <0.001.
Effects of corticosterone pellet implantation and of antidepressant treatment of corticosterone pellet-implanted rats on expression of BDNF transcripts in frontal cortex and hippocampus
Effects of corticosterone pellet implantation on expression of BDNF transcripts containing exons I-IV in frontal cortex and hippocampus are given in Figures 6 and 7, respectively. Corticosterone significantly decreased the levels of exons II and IV in both frontal cortex (Figure 6) and hippocampus (Figure 7) but had no significant effects on exons I or III, either in frontal cortex or in hippocampus.
Figure 6.
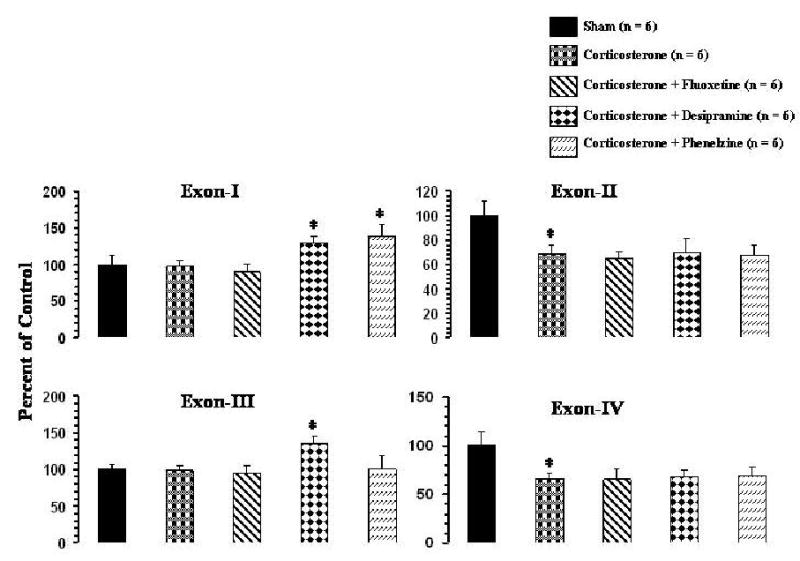
Effects of corticosterone and of antidepressant treatment of corticosterone-treated rats on mRNA levels of BDNF transcripts in rat frontal cortex. Corticosterone pellet was implanted for 21 days and i.p. injections of an antidepressant were simultaneously started and continued for 21 days. Animals were sacrificed 2 hr after the last antidepressant injection. Values are the mean ± S.D. from 6 rats. Overall group differences were: exon I, df = 4,25, F = 19.82, P <0.001; exon II, df = 4,25, F = 14.2, P <0.001; exon III, df = 4,25, F = 12.2, P <0.001; exon IV, df = 4,25, F = 15.1, P <0.001. * P <0.001.
Figure 7.
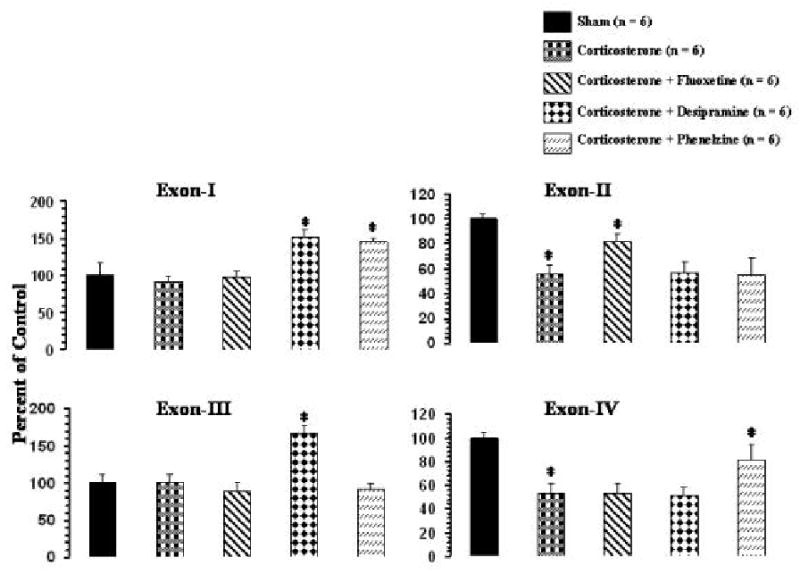
Effects of corticosterone and of antidepressant treatment of corticosterone-treated rats on mRNA levels of BDNF transcripts in rat hippocampus. Corticosterone pellet was implanted for 21 days and i.p. injections of an antidepressant were simultaneously started and continued for 21 days. Animals were sacrificed 2 hr after the last antidepressant injection. Values are the mean ± S.D. from 6 rats. Overall group differences were: exon I, df = 4,25, F = 44.1, P <0.001; exon II, df = 4,25, F = 34.2, P <0.001; exon III, df = 4,25, F = 60.0, P <0.001; exon IV, df = 4,25, F = 35.4, P <0.001. * P <0.001.
Treatment with fluoxetine did not have any effects on the corticosterone-mediated decrease in expression levels of exons II and IV in frontal cortex (Figure 6). As was the case with fluoxetine treatment of normal rats, fluoxetine also did not cause any significant changes in expression of exons I or III in frontal cortex (Figure 6). In hippocampus, however, fluoxetine partially reversed the corticosterone-induced decrease in expression of exon II but was not effective in reversing the decrease of exon IV (Figure 7). Fluoxetine did not cause significant effects on the levels of exons I or III in hippocampus (Figure 7).
Treatment with desipramine of corticosterone-treated rats showed that the levels of exons I and III were significantly increased, whereas the corticosterone-induced decreases in the levels of exons II and IV were not significantly reversed in either frontal cortex (Figure 6) or hippocampus (Figure 7).
In frontal cortex, treatment with phenelzine increased expression level of exon I but did not affect the corticosterone-mediated decrease in exons II or IV. Also, phenelzine did not cause a significant change in expression of exon III (Figure 6). On the other hand, as in the frontal cortex, phenelzine significantly increased the level of exon I in hippocampus (Figure 7). In addition, phenelzine partially normalized the corticosterone-mediated decrease in expression of exon IV in hippocampus but did not affect the corticosterone-mediated decrease in exon II. Phenelzine also did not cause any significant change in expression of the exon III-containing BDNF transcript in hippocampus (Figure 7).
DISCUSSION
This comprehensive examination of the effects of corticosterone or antidepressants, and of antidepressant effects of corticosterone on BDNF in rat brain, which showed deceptively simple results of decrease versus increase in total BDNF, revealed complex regulation processes when the effects on individual BDNF exons were examined.
Corticosterone and BDNF
Our study shows that the level of BDNF expression, as measured with primers to exon V mRNA, which detects all BDNF transcripts, was lower in hippocampus of rats implanted with a corticosterone pellet (100 mg) for 21 days. This is in agreement with earlier reports that exogenous corticosterone causes a decrease in mRNA levels of BDNF in hippocampus (Chao and McEwen, 1994; Smith et al., 1995; Schaff et al., 1997, 1998). In addition, we observed that the level of BDNF mRNA was also decreased in frontal cortex, which suggests that the effects of glucocorticoids on BDNF are not limited to the hippocampus. To examine whether the decrease in BDNF is due to specific transcript(s), we determined mRNA levels of exons I-IV, which are linked to four separate promoters and result in four main transcript forms, each containing a 5′ exon and the BDNF protein encoding exon V (Timusek et al., 1993; Nakayama et al., 1994). We observed that corticosterone selectively decreased the expression of transcripts II and IV, but not transcripts I or III, in both frontal cortex and hippocampus. Our results thus suggest that the decrease of BDNF mRNA expression by glucocorticoids may be due to a decrease in expression of the specific BDNF transcripts that contain exons II and IV.
A few studies suggest that stress, which activates the HPA axis and elevates glucocorticoids, decreases mRNA expression of BDNF in rat hippocampus (Smith et al., 1995; Nibuya et al., 1995; Ueyama et al., 1997). Two recent studies suggest that longer times of immobilization stress decrease total BDNF expression, along with a specific decrease in exon IV in hippocampus (Marmigere et al., 2003) and hypothalamus (Rage et al., 2002). As do the stress studies, our study also shows a decrease in exon IV; however, we found a decrease in an additional exon, i.e., exon II, by corticosterone. The reason for the differences between our and other studies is not clear, but the possibility that different levels of circulating glucocorticoids may affect the expression of various exons differently cannot be ruled out. It is pertinent to mention that only a high dose of corticosterone decreases BDNF, which is not affected by a low dose (Schaff et al., 1997). In our study, we used a high dose of glucocorticoids, with a resulting plasma level of 116 ± 30.5 ng/ml. With this dose, we had earlier reported robust effects of increased levels of corticosterone on various signaling molecules in normal rat brain, and that this dose was effective in normalizing an adrenalectomy-induced decrease in the levels of corticosterone, as well as in reversing adrenalectomy-induced changes in these signaling molecules (Dwivedi and Pandey, 2000; Dwivedi et al., 2000).
Antidepressants and BDNF
A number of studies suggest that dysfunction of the HPA axis is corrected by clinically effective antidepressants (Heuser et al., 1996; Holsboer and Bardern, 1996). Antidepressants increase mRNA levels of mineralocorticoid receptors (MR) and glucocorticoid receptors (GR), and their hormone binding activities (Kitayama et al., 1988; Seckl and Fink, 1992; Reul et al., 1993, 1994). Also, GR promoter activity is increased by antidepressants, both in vitro (Pepin et al., 1992) and in vivo (Peiffer et al., 1991). More recent studies demonstrate that antidepressants inhibit GR-mediated gene transcription (Budziszewska et al., 2000). Thus, antidepressants increase GR level and enhance the GR-mediated feedback inhibition, which may lead to a lowering of corticosterone level and inhibit corticosterone-mediated transcription of genes. In addition to effects on corticosteroid receptors, antidepressants can also influence the HPA axis system through other mechanisms. For example, it has been shown that chronic but not acute treatment with certain classes of antidepressants increases the stress-induced decrease in the expression of BDNF in rat hippocampus (Nibuya et al., 1995). This effect appears to be mediated by corticosterone (Smith et al., 1995). It is not known, however, whether antidepressants directly affect corticosterone-mediated changes in the expression of BDNF, and if so, what would be the mechanism? For this purpose, we administered different classes of antidepressants (serotonin uptake blocker, fluoxetine, norepinephrine blocker, desipramine, or monoamine oxidase inhibitor, phenelzine) to normal rats and to rats implanted with a corticosterone pellet for 21 days. We observed that treatment with desipramine or phenelzine of normal rats increased mRNA levels of total BDNF in both frontal cortex and hippocampus, whereas fluoxetine increased mRNA level of BDNF only in hippocampus. An interesting observation was noted when we examined the effects of antidepressants on the expression of individual exons containing BDNF transcripts. We found that desipramine specifically increased exons I and III in both frontal cortex and hippocampus; fluoxetine increased only exon II in hippocampus; whereas phenelzine effectively increased exons I and IV in hippocampus but only exon I in frontal cortex. Our findings thus suggest that different classes of antidepressants regulate the expression of different exons in a specific manner. Thus it appears that there is no unified mechanism for the regulation of BDNF exons by antidepressants and that various classes of antidepressants may affect BDNF exon expression differently. Also, there is a brain region specificity of the regulation of BDNF exons by various classes of antidepressants.
In a recent study, Dias et al. (2003) examined the effects of chronic antidepressants on BDNF transcript levels in rat hippocampus, amygdala, and cortex. They observed that desipramine increased exon III in different cortical areas, whereas fluoxetine had no significant effects on BDNF exons in any of the brain areas studied. Another recent study by Altieri et al. (2004) also showed no effect of chronic fluoxetine treatment on BDNF transcripts in hippocampus. Our observation of increased exon III by desipramine is similar to the findings of Dias et al. (2003), but we also noted an increase in expression of exon I. In addition, contrary to reports by Dais et al. (2003) and Altieri et al. (2004), we found a selective increase in exon II by fluoxetine in both frontal cortex and hippocampus. The discrepancy between our results and those reported by these investigators is not clear. In fact, conflicting results have been reported in support of (Nibuya et al., 1995; 1996; Russo-Neustadt and Cotman, 1999; Russo-Neustadt et al., 1999; Russo-Neustadt et al., 2000; Coppell et al., 2003; Xu et al., 2003; DeFoubert et al., 2004) or against (Russo-Neustadt et al., 1999; Miro et al., 2002; Conti et al., 2002;) increased mRNA levels of total BDNF by antidepressants. The differences in the effects of antidepressants on BDNF transcripts between our results and those reported by Dias et al. (2003) and Altieri et al. (2004) could be attributed to different doses, route of administration, or the time elapsed between the last dose of drug administration and sacrifice. The dose of fluoxetine used in the present study and those used by Dias et al. (2003) and Altieri et al. (2004) were similar (5 mg/kg); however, the route of administration in our study was different from that of Altieri et al. (2004) (oral vs. i.p.). Similarly, the dose of desipramine was different in our study (10 mg/kg) from that of Dias et al. (2003) (15 mg/kg). Despite these differences, our study clearly demonstrates that chronic fluoxetine, desipramine, or phenelzine regulate expression of BDNF transcripts in a specific manner, which is also brain region specific.
Corticosterone, antidepressants and BDNF
When we examined the effects of antidepressants on the corticosterone-mediated decrease in total BDNF, it was observed that chronic treatment with desipramine completely reversed the corticosterone-induced decrease in BDNF in both frontal cortex and hippocampus. Fluoxetine was able to partially reverse the changes in hippocampal BDNF, but did not cause any change in frontal cortex. phenelzine, on the other hand, reversed the corticosterone-induced decrease in BDNF partially in frontal cortex and completely in hippocampus. Interesting results were noted when individual BDNF transcripts were examined after antidepressant treatment of corticosterone-implanted rats: the antidepressants were able to increase mRNA levels of only those BDNF transcripts that were affected when the respective antidepressant was given to normal rats without corticosterone pellet implantation. Thus, desipramine increased exons I and III in frontal cortex and hippocampus; fluoxetine increased exon II in hippocampus; and phenelzine increased exon I in frontal cortex and exons I and IV in hippocampus. Surprisingly, except for the changes in exon II by fluoxetine in frontal cortex and in exon IV by phenelzine in hippocampus, the corticosterone-mediated decrease in exons II and IV persisted even after antidepressant treatment. But, as mentioned earlier, the overall observation was that all the antidepressants increased the level of total BDNF mRNA in brain of corticosterone-treated rats. Although it is difficult to asses the extent of involvement of a particular exon in regulation of overall BDNF expression, it is interesting to note that we see complete reversal by desipramine in both frontal cortex and hippocampus since the increase in exon III was very robust in these brain areas. On the other hand, in hippocampus, fluoxetine was able to reverse the corticosterone-mediated decrease of only exon II, but not exon IV; therefore, the reversal was partial. But no effect of fluoxetine on total BDNF expression was observed in frontal cortex, since fluoxetine was not able to increase either exon II or exon IV in frontal cortex. On the other hand, phenelzine was partially effective in frontal cortex because of its effects on exon II, but complete reversal was noted in hippocampus because phenelzine increased the levels of both corticosterone-decreased exons II and IV. Thus, it appears that apparently antidepressants are effective in causing an increase in total BDNF expression in corticosterone-treated rats; however, the mechanisms for the downregulation of BDNF transcripts by corticosterone and those that affect their upregulation by antidepressants are quite different.
The mechanisms of regulation of specific exons by corticosterone or antidepressants are not clear at the present time. Interestingly, Smith et al. (1995) reported that stress decreases the expression of BDNF which is not blocked by adrenalectomy, suggesting that factors other than adrenal-glucocorticoids mediate this effect. On the other hand, Schaaf et al. (1998) reported that a high-dose corticosterone treatment of adrenalectomized rats decreased BDNF in hippocampus, and concluded that this decrease was mediated by GR. The presence of the glucocorticoid response element (GRE) on the promoter region of exons may inhibit transcription via a negative GRE mechanism. Although no classical GRE have been found in promoter regions on the BDNF genes, a putative GRF in BDNF promoter IV has been reported (Benraiss et al., 2001). Lauterborn et al. (1998) showed that removal of circulating adrenal hormones enhances activity-induced expression of transcripts I and II, although no GRE binding sites have been found on these promoters. Other factors may also contribute to the decrease in expression of BDNF by corticosterone, such as that GR may downregulate BDNF expression by interacting with other transcription factors. Several AP-1 binding sites have been reported on the BDNF promoter (Hayes et al., 1997), and GR suppresses the activity of AP-1 (Unlap and Jope, 1994). In addition, all BDNF exons are regulated in an activity-dependent manner (Falkenberg et al., 1992; Timmusk et al., 1993; Timmusk and Metsis et al., 1994). Promoter I is activated by a mechanism involving calcium influx (Murray et al., 1998). In addition, Calcium calmodulin (CaM) dependent-kinase II has been demonstrated to specifically mediate promoter IV activation by Ca2+influx (Takeuchi et al., 2000). BDNF promoter III interacts with a calcium responsive transcription factor (Tao et al., 1998; 2002). BDNF promoter III is also regulated by CaM kinase IV and transcription factor cyclic AMP response element binding protein (CREB) (Shieh et al., 1998; Tao et al., 1998). It has been demonstrated that CREB is repressed by GR (Stauber et al., 1992; Imai et al., 1993) and that antidepressants increase expression and functions of CREB (Nibuya et al., 1996; Thome et al., 2000; Conti et al., 2002). Since corticosterone does not affect exon III and since antidepressants increase other BDNF transcripts in addition to exon III, there must be additional mechanisms operating in the regulation of these exons by corticosterone or antidepressants.
In conclusion, our study for the first time reveals the regulation of selective BDNF exons by corticosterone and also suggests that there is a complex interaction between antidepressants and corticosterone in regulating BDNF exons. We observed that the various classes of antidepressants administered chronically to rats increase different exons, which suggests that there is no unified mechanism of regulation of BDNF by antidepressants. In addition, antidepressants also regulate BDNF exons other than those affected by corticosterone. Although there was an overall increase in BDNF by antidepressants in corticosterone-treated rats, the mechanisms appear to be quite different.
Table 2.
Summary of the regulation of total BDNF and BDNF exon expression by corticosterone and antidepressants
| Measures | |||||
|---|---|---|---|---|---|
| Treatment | Total BDNF | Exon I | Exon II | Exon III | Exon IV |
| Frontal Cortex | |||||
| Antidepressants | |||||
| Fluoxetine | NC | NC | NC | NC | NC |
| Desipramine | ↑ | ↑ | NC | ↑ | NC |
| Phenelzine | ↑ | ↑ | NC | NC | NC |
| Corticosterone | ↓ | NC | ↓ | NC | ↓ |
| Corticosterone + | |||||
| Fluoxetine | NC | NC | NC | NC | NC |
| Desipramine | - | - | NC | - | NC |
| Phenelzine | - | - | NC | NC | NC |
| Hippocampus | |||||
| Antidepressants | |||||
| Fluoxetine | ↑ | NC | ↑ | NC | NC |
| Desipramine | ↑ | ↑ | NC | ↑ | NC |
| Phenelzine | ↑ | ↑ | NC | NC | ↑ |
| Corticosterone | ↓ | NC | ↓ | NC | ↓ |
| Corticosterone + | |||||
| Fluoxetine | - | NC | - | NC | NC |
| Desipramine | - | - | NC | - | NC |
| Phenelzine | - | - | NC | NC | - |
NC, No change, ↑increase; ↓decrease, - Reversal
Acknowledgments
This work was supported by RO1MH68777 from the National Institute of Mental Health and a Career Development Award (KO1MH 01836) from the National Institute of Mental Health to Dr. Yogesh Dwivedi. Technical assistance provided by Ms. Miljana Petkovic is acknowledged.
Footnotes
Section Editor: Dr. Werner Sieghart
References
- Altieri M, Marini F, Arban R, Vitulli G, Jansson BO. Expression analysis of brain-derived neurotrophin factor (BDNF) mRNA isoforms after chronic and acute antidepressant treatment. Brain Res. 2004;1000:148–155. doi: 10.1016/j.brainres.2003.12.028. [DOI] [PubMed] [Google Scholar]
- Barden N. Implication of the hypothalamic-pituitary-adrenal axis in the physiopathology of depression. J Psychiatry Neurosci. 2004;29:185–193. [PMC free article] [PubMed] [Google Scholar]
- Benraiss A, Chmielnicki E, Lerner K, Roh D, Goldman SA. Adenoviral brain-derived neurotrophic factor induces both neostriatal and olfactory neuronal recruitment from endogenous progenitor cells in the adult forebrain. J Neurosci. 2001;21:6718–6731. doi: 10.1523/JNEUROSCI.21-17-06718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budziszewska B. Effect of antidepressant drugs on the hypothalamic-pituitary-adrenal axis activity and glucocorticoid receptor function. Pol J Pharmacol. 2002;54:343–349. [PubMed] [Google Scholar]
- Budziszewska B, Jaworska-Feil L, Kajta M, Lason W. Antidepressant drugs inhibit glucocorticoid receptor-mediated gene transcription – a possible mechanism. Br J Pharmacol. 2000;130:1385–1393. doi: 10.1038/sj.bjp.0703445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll BJ, Curtis GC, Mendels J. Cerebrospinal fluid and plasma free cortisol concentrations in depression. Psychol Med. 1976;6:235–244. doi: 10.1017/s0033291700013775. [DOI] [PubMed] [Google Scholar]
- Chao HM, McEwen BS. Glucocorticoids and the expression of mRNAs for neurotrophins, their receptors and GAP-43 in the rat hippocampus. Brain Res Mol Brain Res. 1994;26:271–276. doi: 10.1016/0169-328x(94)90099-x. [DOI] [PubMed] [Google Scholar]
- Chen B, Dowlatshahi D, MacQueen GM, Wang J-F, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. 2001;50:260–265. doi: 10.1016/s0006-3223(01)01083-6. [DOI] [PubMed] [Google Scholar]
- Conti AC, Cryan JF, Dalvi A, Lucki I, Blendy JA. cAMP response element-binding protein is essential for the upregulation of brain-derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepressant drugs. J Neurosci. 2002;22:3262–3268. doi: 10.1523/JNEUROSCI.22-08-03262.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppell A, Pei Q, Zetterston TSC. Bi-phasic change in BDNF gene expression following antidepressant drug treatment. Neuropharmacology. 2003;44:903–910. doi: 10.1016/s0028-3908(03)00077-7. [DOI] [PubMed] [Google Scholar]
- De Foubert G, Carney SL, Robinson CS, Destexhe EJ, Tomlinson R, Hicks CA, Murray TK, Gaillard JP, Deville C, Xhenseval V, Thomas CE, O’Neill MJ, Zetterstrom TSC. Fluoxetine-induced change in rat brain expression of brain-derived neurotrophic factor varies depending on length of treatment. Neurosci. 2004;128:597–604. doi: 10.1016/j.neuroscience.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Dias BG, Banerjee SB, Duman RS, Vaidya VA. Differential regulation of brain-derived neurotrophin factor transcripts by antidepressant treatments in the adult rat brain. Neuropharmacology. 2003;45:553–563. doi: 10.1016/s0028-3908(03)00198-9. [DOI] [PubMed] [Google Scholar]
- Drevets WC. Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog Brain Res. 2000a;126:413–431. doi: 10.1016/S0079-6123(00)26027-5. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Agrawal AK, Rizavi HS, Pandey GN. Antidepressants reduce phosphoinositide-specific phospholipase C (PI-PLC) activity and the mRNA and protein expression of selective PLC β1 isozyme in rat brain. Neuropharmacology. 2002;43:1269–1279. doi: 10.1016/s0028-3908(02)00253-8. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Pandey GN. Effects of subchronic administration of antidepressants and anxiolytics on levels of the α subunits of G proteins in the rat brain. J Neural Transm. 1997;104:747–760. doi: 10.1007/BF01291891. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Pandey GN. Adrenal glucocorticoids modulate [3H]cyclic AMP binding to protein kinase A (PKA), cyclic AMP-dependent PKA activity, and protein levels of selective regulatory and catalytic subunit isoforms of PKA in rat brain. JPET. 2000a;294:103–116. [PubMed] [Google Scholar]
- Dwivedi Y, Pandey SC, Ren X, Tueting P, Pandey GN. Effect of desipramine, lithium, and adinazolam on serotonin receptor subtypes in rat brain. Soc Neurosci Abstr. 1994;20:1161. [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2003;60:804–815. doi: 10.1001/archpsyc.60.8.804. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Rao JS, Pandey GN. Modifications in the phosphoinositide signaling pathway by adrenal glucocorticoids in rat brain: focus on phosphoinositide-specific phospholipase C and inositol 1,4,5-trisphosphate. JPET. 2000b;295:244–254. [PubMed] [Google Scholar]
- Falkenberg T, Metsis M, Timmusk T, Lindefors N. Entorhinal cortex regulation of multiple brain-derived neurotrophic factor promoters in the rat hippocampus. Neurosci. 1992b;57:891–896. doi: 10.1016/0306-4522(93)90034-d. [DOI] [PubMed] [Google Scholar]
- Gass P, Kretz O, Wolfer DP, Berger S, Tronche F, Reinchardt HM, Kellendonk C, Lipp HP, Schmid W, Schutz G. Genetic disruption of mineralocorticoid receptor leads to impaired neurogenesis and granule cell degeneration in the hippocampus of adult mice. EMBO Rep. 2003;1:447–451. doi: 10.1093/embo-reports/kvd088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervasoni N, Aubry JM, Bondolfi G, Osiek C, Schwald M, Bertschy G, Karege F. Partial normalization of serum brain-derived neurotrophic factor in remitted patients after a major depressive episode. Neuropsychobiology. 2005;51:234–238. doi: 10.1159/000085725. [DOI] [PubMed] [Google Scholar]
- Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestations of depression. Relation to neurobiology of stress (Part II) N Eng J Med. 1988b;6:413–420. doi: 10.1056/NEJM198808183190706. [DOI] [PubMed] [Google Scholar]
- Gould E, McEwen B, Tanapat P, Galea L, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci. 1997;17:2492–2498. doi: 10.1523/JNEUROSCI.17-07-02492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Shimizu E, Iyo M. Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res Rev. 2004;45:104–114. doi: 10.1016/j.brainresrev.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Hayes VY, Towner MD, Isackson PJ. Organization, sequence and functional analysis of a mouse BDNF promoter. Mol Brain Res. 1997;45:189–198. doi: 10.1016/s0169-328x(96)00254-9. [DOI] [PubMed] [Google Scholar]
- Heuser IJE, Schweiger U, Gotthardt U, Schmider J, Lammers CH, Dettling M, Yassouridis A, Holsboer F. Pituitary-adrenal system regulation and psychopathology during amitriptyline treatment in elderly depressed patients and normal comparison subjects. Amer J Psychiat. 1996;153:93–99. doi: 10.1176/ajp.153.1.93. [DOI] [PubMed] [Google Scholar]
- Holsboer F. Stress, hypercortisolism and corticosteroid receptors in depression: implicatons for therapy - Review Article. J Affect Disord. 2001;62:77–91. doi: 10.1016/s0165-0327(00)00352-9. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Barden N. Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr Rev. 1996;17:187–205. doi: 10.1210/edrv-17-2-187. [DOI] [PubMed] [Google Scholar]
- Imai E, Miner JN, Mitchell JA, Yamamoto KR, Granner DK. Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J Biol Chem. 1993;268:5353–5356. [PubMed] [Google Scholar]
- Karege F, Bondolfi G, Gervasoni N, Schwald M, Aubry JM, Bertschy G. Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol Psychiatry. 2005;57:1068–1072. doi: 10.1016/j.biopsych.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry J-M. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psych Res. 2002;109:143–148. doi: 10.1016/s0165-1781(02)00005-7. [DOI] [PubMed] [Google Scholar]
- Kitayama L, Janson AM, Cintra A, Fuxe K, Agnati LF, Ogren SO, Harfstrand A, Eneroth P, Gustafsson JA. Effects of chronic imipramine treatment on gluocorticoid receptor immunoreactivity in various regions of the rat brain. J Neural Transm. 1988;73:191–203. doi: 10.1007/BF01250136. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Poulsen FR, Stinis CT, Isackson PJ, Gail CM. Transcript-specific effects of adrenalectomy on seizure-induced BDNF expression in rat hippocampus. Mol Brain Res. 1998;55:81–91. doi: 10.1016/s0169-328x(97)00368-9. [DOI] [PubMed] [Google Scholar]
- Lindeforts N, Brodin E, Metsis M. Spatiotemporal selective effects on brain-derived neurotrophic factor and trkB messenger RNA in rat hippocampus by electroconvulsive shock. Neurosci. 1995;65:661–670. doi: 10.1016/0306-4522(94)00550-o. [DOI] [PubMed] [Google Scholar]
- Magarinos AM, McEwen BS, Flugge G, Fuchs E. Chronic psychosocial stress causes apical dendritic atrophy of hippocampal CA3 pyramidal neurons in subordinate tree shrews. J Neurosci. 1996;16:3534–3540. doi: 10.1523/JNEUROSCI.16-10-03534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann CD, Bich VT, Hrdina PD. Protein kinase C in rat brain cortex and hippocampus: effects of repeated administration of fluoxetine and desipramine. Br J Pharmacol. 1995;115:595–600. doi: 10.1111/j.1476-5381.1995.tb14973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmigere F, Givalois L, Rage F, Arancibia S, Tapia-Arancibia L. Rapid induction of BDNF expression in the hippocampus during immobilization stress challenge in adult rats. Hippocampus. 2003;13:646–655. doi: 10.1002/hipo.10109. [DOI] [PubMed] [Google Scholar]
- McEwen Bs. Glucocorticoid-biogenic amine interactions in reaction in mood and behavior. Biochem Pharmacol. 1987;36:1755–1763. doi: 10.1016/0006-2952(87)90234-6. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress and hippocampal plasticity. Ann Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- Miro X, Perez-Torres F, Artigas P, Puigdomenech JM, Palacios G, Mengod Regulation of cAMP phosphodiesterase mRNAs expression in rat brain by acute and chronic fluoxetine treatment. An in situ hybridization study. Neuropharmacology. 2002;43:1148–1157. doi: 10.1016/s0028-3908(02)00220-4. [DOI] [PubMed] [Google Scholar]
- Montkowski A, Barden N, Wotjak C, Stec I, Ganster J, Meaney M, Engelmann M, Reul JMHM, Landgraf R, Holsboer F. Long-term antidepressant treatment reduces behavioral deficits in transgenic mice with impaired glucocorticoid receptor function. J Neuroendocrinol. 1995;7:841–845. doi: 10.1111/j.1365-2826.1995.tb00724.x. [DOI] [PubMed] [Google Scholar]
- Murphy BEF. Steriods and depression. J Steroid Biochem Mol Biol. 1991;38:537–539. doi: 10.1016/0960-0760(91)90312-s. [DOI] [PubMed] [Google Scholar]
- Murray KD, Hayes VY, Gall CM, Isackson PJ. Attenuation of the seizure-induced expression of BDNF mRNA in adult rat brain by an inhibitor of calium/calmodulin-dependent protein kinases. Eur J Neurosci. 1998;10:377–387. doi: 10.1046/j.1460-9568.1998.00019.x. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Gahara Y, Kitamura T, Ohara O. Distinctive four promoters collectively direct expression of brain-derived neurotrophic factor gene. Mol Brain Res. 1994;21:206–218. doi: 10.1016/0169-328x(94)90251-8. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP-response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16:2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Takahashi M, Russell DS, Duman RS. Chronic stress increases catalytic trkB mRNA in rat hippocampus. Neurosci Lett. 1999;267:81–84. doi: 10.1016/s0304-3940(99)00335-3. [DOI] [PubMed] [Google Scholar]
- Okada F, Tokumitsu Y, Ui M. Possible involvement of pertussis toxin substrates (Gi, Go) in desipramine-induced refractoriness of adenylate cyclase in cerebral cortices of rates. J Neurochem. 1988;51:194–199. doi: 10.1111/j.1471-4159.1988.tb04855.x. [DOI] [PubMed] [Google Scholar]
- Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci USA. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa H, Rasenick MD. Chronic electroconvulsive treatment augments coupling of the GTP-binding protein Gs to the catalytic moiety of adenylyl cyclase in a manner similar to that seen with chronic antidepressant drugs. J Neurochem. 1991;56:330–338. doi: 10.1111/j.1471-4159.1991.tb02599.x. [DOI] [PubMed] [Google Scholar]
- Pandey GN, Pandey SC, Davis JM. Effect of desipramine on inositol phosphate formation and inositol phospholipids in rat brain and human platelets. Psychopharmacol Bull. 1991;27:255–261. [PubMed] [Google Scholar]
- Pariante CM. Depression, stress and the adrenal axis. J Neuroendocrinol. 2003;15:811–812. doi: 10.1046/j.1365-2826.2003.01058.x. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Thomas SA, Lovestone S, Makoff A, Kerwin RW. Do antidepressants regulate how cortisol affects the brain? Psychoneuroendocrinology. 2004;29:423–447. doi: 10.1016/j.psyneuen.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Peiffer A, Veilleux S, Barden N. Antidepressant and other centrally acting drugs regulate glucocorticoid receptor mRNA levels in rat brains. Psychoneuroendocrinology. 1991;16:505–515. doi: 10.1016/0306-4530(91)90034-q. [DOI] [PubMed] [Google Scholar]
- Pepin MC, Govindan MV, Barden N. Increased glucocorticoid receptor gene promoter activity following antidepressant treatment. Mol Pharmacol. 1992a;41:1016–1022. [PubMed] [Google Scholar]
- Pepin M-C, Pothier F, Barden N. Antidepressant drug action in a transgenic mouse model of the endocrine changes seen in depression. Mol Pharmacol. 1992b;42:991–995. [PubMed] [Google Scholar]
- Rage F, Givalois L, Marmigere F, Tapia-Arancibia L, Arancibia S. Immobilization stress rapidly modulates BDNF mRNA expression in the hypothalamus of adult male rats. Neurosci. 2002;112:309–318. doi: 10.1016/s0306-4522(02)00072-6. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1083–1084. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- Rasmussen A, Shi L, Duman RS. Downregulation of BDNF mRNA in the hippocampal dentate gyrus after re-exposure to cues previously associated with footshock. Neuropsychopharmacology. 2002;27:133–142. doi: 10.1016/S0893-133X(02)00286-5. [DOI] [PubMed] [Google Scholar]
- Reagen L, McEwen B. Controversies surrounding glucocorticoid-mediated cell death in the hippocampus. J Chem Neuroanat. 1997;13:148–158. doi: 10.1016/s0891-0618(97)00031-8. [DOI] [PubMed] [Google Scholar]
- Reul JMHM, Labeur MS, Grigoriadis DE, De Souza EB, Holsboer F. Hypothalamic-pituitary-adrenocortical axis changes in the rat after long-term treatment with the reversible monoamine oxidase-A inhibitor moclobemide. Neuroendocrinology. 1994;60:509–519. doi: 10.1159/000126788. [DOI] [PubMed] [Google Scholar]
- Reul JMHM, Stec I, Soder M, Holsboer F. Chronic treatment of rats with the antidepressant amitriptyline attenuates the activity of the hypothalamic-pituitary-adrenocortical system. Endocrinology. 1993;133:312–320. doi: 10.1210/endo.133.1.8391426. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A, Beard RC, Cotman CW. Exercise, antidepressant medications, and enhanced brain-derived neurotrophic factor expression. Neuropsychopharmacology. 1999;21:679. doi: 10.1016/S0893-133X(99)00059-7. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt AA, Beard RC, Huang YM, Cotman CW. Physical activity and antidepressant treatment potentiate the expression of specific brain-derived neurotrophic factor transcripts in the rat hippocampus. Neurosci. 2000;101:305–312. doi: 10.1016/s0306-4522(00)00349-3. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt ABR, Cotman CW. Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology. 1999;21:679–682. doi: 10.1016/S0893-133X(99)00059-7. [DOI] [PubMed] [Google Scholar]
- Sapolsky R. Glucocorticoids, stress, and their adverse neurological effects: relevance to aging. Exp Gerontol. 1999;34:721–729. doi: 10.1016/s0531-5565(99)00047-9. [DOI] [PubMed] [Google Scholar]
- Sapolsky R. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry. 2000;57:925–935. doi: 10.1001/archpsyc.57.10.925. [DOI] [PubMed] [Google Scholar]
- Schaaf MJM, de Jong J, de Kloet ER, Vreugdenhil E. Downregulation of BDNF mRNA and protein in the rat hippocampus by corticosterone. Brain Res. 1998;813:112–120. doi: 10.1016/s0006-8993(98)01010-5. [DOI] [PubMed] [Google Scholar]
- Schaaf MJM, de Kloet ER, Vreugdenhil E. Corticosterone effects on BDNF expression in the hippocampus. Implications for memory formation. Stress. 2000;30:201–208. doi: 10.3109/10253890009001124. [DOI] [PubMed] [Google Scholar]
- Schaaf MJM, Hoetelmans RWM, de Kloet ER, Vreugdenhil E. Corticosterone regulates expression of BDNF and trkB but not NT–3 and trkC mRNA in the rat hippocampus. J Neurosci Res. 1997;48:334–341. [PubMed] [Google Scholar]
- Secki JR, Fink G. Antidepressants increase glucocorticoid and mineralacorticoid receptor mRNA expression in rat hippocampus in vivo. Neuroendocr. 1992;55:621–626. doi: 10.1159/000126180. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Shimizu E, Hashimoto K, Okamura N, Koike K, Komatsu N, Kumakiri C, Nakazato M, Watanabe H, Shinoda N, Okada S-I, Iyo M. Alterations of serum levels of brain-derived neurotrophin factor (BDNF) in depressed patients with or without antidepressants. Biol Psychiatry. 2003;54:70–75. doi: 10.1016/s0006-3223(03)00181-1. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Zhang J, McCook EC, Seidler JF. Glucocorticoid administration alters nuclear transcription factors in fetal rat brain: implications for the use of antenatal steroids. Brain Res Dev Brain Res. 1998;111:11–24. doi: 10.1016/s0165-3806(98)00115-1. [DOI] [PubMed] [Google Scholar]
- Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15:1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauber C, Altschmied J, Akerblom IE, Marrow JL, Mellon PL. Mutual cross-interference between glucocorticoid receptor and CREB inhibits transactivation in placental cells. New Biol. 1992;4:527–40. [PubMed] [Google Scholar]
- Takeuchi Y, Yamamoto H, Miyakawa T, Miyamoto E. Increase of brain-derived neurotrophic factor gene expression in NG108-15 cells by the nuclear isoforms of Ca2+/calmodulin-dependent protein kinase II. J Neurochem. 2000;74:1913–1922. doi: 10.1046/j.1471-4159.2000.0741913.x. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Tao X, West AE, Chen WG, Corfas G, Greenberg ME. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron. 2002;33:383–395. doi: 10.1016/s0896-6273(01)00561-x. [DOI] [PubMed] [Google Scholar]
- Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S, Storm D, Duman RS. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci. 2000;20:4030–4036. doi: 10.1523/JNEUROSCI.20-11-04030.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmusk T, Metsis M. Regulation of BDNF promoters in the rat hippocampus. Neurochem Int. 1994;25:11–15. doi: 10.1016/0197-0186(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Palm K, Mesis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- Todd KG, McManus DJ, Baker GB. Chronic administration of the antidepressants phenelzine, desipramine, clomipramine, or maprotiline decrease binding to 5-hydroxytryptamine2A receptors without affecting benzodiazepine binding sites in rat brain. Cell Mol Neurobiol. 1995;15:361–370. doi: 10.1007/BF02089946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouvin JH, Gardier AM, Chanut E, Pages N, Jacquot C. Time course of brain serotonin metabolism after cessation of long-term fluoxetine treatment in the rat. Life Sci. 1993;52:187–192. doi: 10.1016/0024-3205(93)90116-k. [DOI] [PubMed] [Google Scholar]
- Ueyama T, Kawai Y, Nemoto K, Sekimoto M, Tone S, Senba E. Immobilization stress reduced the expression of neurotrophins and their receptors in the rat brain. Neurosci Res. 1997;28:103–110. doi: 10.1016/s0168-0102(97)00030-8. [DOI] [PubMed] [Google Scholar]
- Ulrichsen J, Partilla JS, Dax EM. Long-term administration of m-chlorophenylpiperazine (mCPP) to rats induces changes in serotonin receptor binding, dopamine levels and locomotor activity without altering prolactin and corticosterone secretion. Psychopharmacology. 1992;107:229–235. doi: 10.1007/BF02245142. [DOI] [PubMed] [Google Scholar]
- Unlap T, Jope RS. Dexamethasone attenuates kainate-induced AP-1 activation in rat brain. Brain Res Mol Brain Res. 1994;24:275–282. doi: 10.1016/0169-328x(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Xu H, Richardson JS, Li XM. Dose-related effects of chronic antidepressants on neuroprotective proteins BDNF, Bcl-2 and Cu/Zn-SOD in rat hippocampus. Neuropsychopharmacology. 2003;28:53–62. doi: 10.1038/sj.npp.1300009. [DOI] [PubMed] [Google Scholar]
- Young EA, Lopez JF, Murphy-Weinberg V, Watson SJ, Akil H. Mineralocorticoid receptor function in major depression. Arch Gen Psychiatry. 2003;60:24–28. doi: 10.1001/archpsyc.60.1.24. [DOI] [PubMed] [Google Scholar]