

Abstract
OBJECTIVE
Increases in brain cyclooxygenase-2 (COX2) are associated with the central inflammatory response and with delayed neuronal death, events that cause secondary insults after traumatic brain injury. A growing literature supports the benefit of COX2-specific inhibitors in treating brain injuries.
METHODS
DFU [5,5-dimethyl-3(3-fluorophenyl)-4(4-methylsulfonyl)phenyl-2(5H)-furanone] is a third-generation, highly specific COX2 enzyme inhibitor. DFU treatments (1 or 10 mg/kg intraperitoneally, twice daily for 3 d) were initiated either before or after traumatic brain injury in a lateral cortical contusion rat model.
RESULTS
DFU treatments initiated 10 minutes before injury or up to 6 hours after injury enhanced functional recovery at 3 days compared with vehicle-treated controls. Significant improvements in neurological reflexes and memory were observed. DFU initiated 10 minutes before injury improved histopathology and altered eicosanoid profiles in the brain. DFU 1 mg/kg reduced the rise in prostaglandin E2 in the brain at 24 hours after injury. DFU 10 mg/kg attenuated injury-induced COX2 immunoreactivity in the cortex (24 and 72 h) and hippocampus (6 and 72 h). This treatment also decreased the total number of activated caspase-3–immunoreactive cells in the injured cortex and hippocampus, significantly reducing the number of activated caspase-3–immunoreactive neurons at 72 hours after injury. DFU 1 mg/kg amplified potentially anti-inflammatory epoxyeicosatrienoic acid levels by more than fourfold in the injured brain. DFU 10 mg/kg protected the levels of 2-arachidonoyl glycerol, a neuro-protective endocannabinoid, in the injured brain.
CONCLUSION
These improvements, particularly when treatment began up to 6 hours after injury, suggest exciting neuroprotective potential for COX2 inhibitors in the treatment of traumatic brain injury and support the consideration of Phase I/II clinical trials.
Keywords: 2-Arachidonoyl glycerol, Caspase-3, COX2 inhibitor, Eicosanoids, Endocannabinoid, Neuroinflammation, Rat behavior
Traumatic brain injury (TBI) initiates a central inflammatory response that results in the increased production of prostaglandins and reactive oxygen species. These products may cause secondary insults to the brain (55, 90, 94). Cyclooxygenases catalyze the first step in the formation of prostaglandins from arachidonic acid, producing reactive oxygen species in the process. Cyclooxygenase-1 (COX1), present normally in most tissues, is thought to maintain essential physiological functions, such as gastric mucosal integrity, renal function, and platelet homeostasis (87, 101). COX2, the inducible isoform, is expressed in the normal brain (30, 87) but is rare or absent in most organs under normal conditions. As in the periphery (32, 79), COX2 in the brain can be regulated by inflammatory cytokines (87), and prostaglandin production increases in response to pathological conditions of the central nervous system (3, 70, 72, 89, 107). COX2 overexpression can also exacerbate neuroinflammatory responses (91).
Brain COX2 can also be induced by excitotoxicity in vivo (1, 16, 42, 52, 106) and in vitro (38, 95, 99). Recent findings demonstrate the prolonged overexpression of COX2 in the brain after TBI (96). In fact, COX2 has been associated with the delayed cell death that occurs after many types of brain injuries (9, 20, 47, 103). Thus, it is not surprising that cyclooxygenase inhibitors may benefit the injured central nervous system (11–13, 22, 47, 80, 85). However, the protective mechanisms of COX2-specific inhibitors have not been elucidated.
In the nervous system, arachidonic acid itself has electrophysiological and anticholinergic properties (44, 65). Its eicosanoid metabolites are numerous, possessing a variety of neurophysiological activities. Prostaglandins have been associated with many neural mechanisms (8, 29, 64), including long-term potentiation (65), which is implicated in memory consolidation. Some evidence exists for prostaglandin neurotoxicity (67, 68, 97, 110). In addition, there is a small literature on the neuroprotective and anti-inflammatory role of selected arachidonic acid metabolites. These include endocannabinoids (anandamide and 2-arachidonyl glycerol [24, 60]), energy modulation (hydroperoxyeicosatetraenoic acid [5-HPETE] [31]), anti-inflammatory effects (11,12-epoxyeicosatrienoic acid [EET] [66]), lipoxygenase-mediated analgesia (19, 61), and circadian modulation (100). The formation of these metabolites may be favored when the COX2 pathway is blocked. Our hypothesis proposes that the beneficial effects of COX2 inhibitors are mediated, at least in part, by increased formation of neuroprotective arachidonic acid metabolites.
5,5-Dimethyl-3(3-fluorophenyl)-4(4-methylsulfonyl)phenyl- 2(5H)-furanone (DFU) is a third-generation, highly COX2-specific enzyme inhibitor that is analogous to rofecoxib in structure and pharmacology (15, 81). DFU inhibits 100% of lipopolysaccharide-induced prostaglandin E2 (PGE2) synthesis in whole-blood assays (7) and shows efficacy in various in vivo models of acute and chronic systemic inflammation with an approximate ED50 value of 1 mg/kg given orally (14, 102). At this dose, the plasma concentration of DFU is below that required to inhibit platelet COX1. Thus, at least in animal models, inhibition of COX1 is not required to achieve anti-inflammatory, antipyretic, and analgesic effects.
No therapeutic strategy has proved effective for the treatment of human TBI. COX2 inhibitors can ameliorate neuro-toxicity in vitro (47, 95) and in vivo (10, 85). Their anti-inflammatory effects, relative safety profile, and hydrophobic chemical structure led us to investigate DFU therapy for experimental TBI. We have examined the effects of DFU on functional recovery, brain eicosanoid biochemistry, and histopathology in a rat model of cortical contusion TBI.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats, each weighing 300 to 400 g, were studied. Animals were cared for in accordance with the United States Public Health Service regulations. All protocols were approved by the Temple University Institutional Animal Care and Use Committee.
TBI Model
In the lateral cortical impact (LCI) rat model of TBI, moderate levels of injury result in a temporary loss of strength and coordination in the limbs contralateral to the injury, significant retrograde amnesia, learning deficits, induction of central inflammatory responses, and delayed loss of cortical and subcortical neurons. The relevance of the LCI model to clinical head trauma has been validated by several groups with respect to common histopathological findings and cognitive-behavioral outcome measures (26, 34, 56).
Surgical Preparation
The animals were preanesthetized with 2% isoflurane, then given oxygen with 0.75% isoflurane through a fixed face mask on a stereotactic platform. The skull was exposed, and a craniectomy was made laterally, midway between lambda and bregma, between the central suture and the left temporal ridge with a 6-mm trephine. The exposed dura was subjected to a 5-mm-diameter piston impact (bregma −2 to −7 mm, according to the coordinates of Paxinos and Watson [74]) of 3.0-mm depth, 4-m/s velocity, and 100-millisecond duration. The scalp was closed without replacement of the bone flap. Anesthesia was discontinued, and the animal was assessed for exclusion criteria (latency of pinna and corneal reflexes and righting response).
Drug Dosing
In the initial study, DFU (1 mg/kg) was injected intraperitoneally (i.p.) in a twice-daily dosage starting 10 minutes before and continuing for 3 days after LCI injury. In subsequent studies, DFU treatments (1 or 10 mg/kg i.p.) were initiated at the designated time (either 10 min before or 2 or 6 h after injury) and continued for 3 days after injury. Dosages corresponded to effective doses given in systemic inflammation models (15, 48) and were arrived at in consultation with Dr. Jilly Evans (Merck). The drug was suspended in 5% poly-ethoxylated vegetable oil (Emulphor; GAF Corp., New York, NY) in saline by mixing vigorously for 30 seconds at room temperature with a rotor-stator device.
Experimental Design
The initial study included two control groups and two pretreatment (10 min before) LCI injury groups: sham/vehicle (n = 15), surgery, no injury, and treated with vehicle (negative control, not included in statistical analyses); injured/vehicle (n = 12), treated with vehicle and injured (positive control); 1DFU/pre (n = 15), treated with DFU (1 mg/kg i.p.) and injured; and 10DFU/pre (n = 12), treated with DFU (10 mg/kg i.p.) and injured.
Subsequent studies included a positive control group and four posttreatment LCI injury groups: injured/vehicle (n = 43), injured, treated with vehicle 2 hours after injury (positive control); 1DFU/2 hours (n = 13), injured, treated with DFU (1 mg/kg i.p.) 2 hours after injury; 1DFU/6 hours (n = 13), injured, treated with DFU (1 mg/kg i.p.) 6 hours after injury; 10DFU/2 hours (n = 12), injured, treated with DFU (10 mg/kg i.p.) 2 hours after injury; and 10DFU/6 hours (n = 12), injured, treated with DFU (10 mg/kg i.p.) 6 hours after injury.
All subsequent twice-daily doses were the same as the initial dose. No differences were observed between the pretreatment and posttreatment positive controls; the groups were combined and analyzed together.
Functional Evaluations
Neurological and behavioral performances were assessed before and 3 days after injury. Treatment groups were assigned randomly by draw at the time of surgery, so that no order or group bias could be introduced by the surgeon/tester.
Neurological Reflexes
Neuroscores were based on three tests of limb reflexes (56): contraflexion (forelimb and head flexion in response to anticipation of falling), hindlimb extension (in response to repetitive raising and lowering by the tail), and lateral pulsion (attempting to roll the animal onto its back). Scores for strength and reflexive coordination (0–4, 4 = best) were assigned for left and right limbs separately, for a maximum of 24 on the three tasks.
Beam Walk Test
A balance-beam task with both cognitive and motor components was used to assess complex motor coordination. Animals were trained to escape an unpleasant pair of stimuli (150-W light and 90-decibel white noise) by running along a 2.5-cm × 1-m beam with four posts mounted on alternating sides, to enter a black box. The aversive stimuli were turned off once the animal entered the box. The time taken (best of three trials) for the animal to enter the box was the outcome measure (latency = 0–120 s).
Open-field Test
To test exploratory activity in a novel environment, animals were placed in an open field (30 × 61 × 20 cm deep) and observed for 2 minutes. The number of rearings and transits to each corner were summed for an activity score.
Morris Water Maze
The water maze, used to assess hippocampal-dependent spatial memory function (34), was conducted exactly as described previously (51). The best score from three probe trials was used for each animal.
Immunological Evaluations
Tissue Preparation
Sham (n = 2 per time point), injured/vehicle treated (n = 3–4 per time point), and injured DFU pretreated (n = 3–4 per time point) animals were killed at 6, 24, and 72 hours after injury via transcardial perfusion with phosphate-buffered saline (PBS) followed by PBS containing 4% formaldehyde (PBSF). Brains were dissected, fixed in PBSF for 1 more hour, cryoprotected in 20% sucrose in PBS for 3 days at 4°C, snap-frozen on powdered dry ice, and stored at −80°C. Histological sections (20 μm) were made with a cryostat microtome (International Equipment Co., Needham, MA; −20°C). Brain regions were evaluated in the anterior injured zone between bregma −2.8 and −4.3 mm (according to the coordinates of Paxinos and Watson [74]) using every 10th or 11th section (6 per animal).
Tissue Staining
Sections were stained with a COX2 monoclonal antibody (1:2000; Cayman Chemical, Ann Arbor, MI) as a marker of central inflammation as described previously (96). Caspase-3 gene induction (105) and enzymatic activation (17) have been associated with delayed cell death and neuropathological changes in the injured brain. Activated caspase-3 antisera (AC3, 1:250; Promega, Madison, WI) was used to visualize apoptotic cells. Fluorescently labeled secondary antibodies (1:200; Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) were used on brain sections, and the signal was visualized with a Nikon (Tokyo, Japan) fluorescence microscope. Control sections stained with secondary antibody only were performed with each experiment for use in background assessment. To confirm the specificity of these antibodies, immunoblots were performed on fresh-frozen tissue exactly as described previously (96). COX2 antibodies yielded a single band of 72 kD. AC3 antibodies gave no detectable signal to brain extracts but yielded two bands at approximately 20 kD by use of an extract of campothecin-treated Jurkat cells (kindly provided by Padmini Salgame, Temple University, Philadelphia, PA).
Immunoreactivity Assessment
Scoring for each stain was performed in a blinded manner by a single investigator. COX2 is expressed at low levels throughout the normal brain and is induced after injury (96), so we evaluated both the number of cells and the apparent amount expressed per cell. Cell numbers were assigned geometric values, as follows: 0 (none), 1 (1–2 cells), 2 (3–4 cells), 4 (5–8 cells), 8 (9–16 cells), 16 (17–32 cells), or 32 (>32). Signal intensity was graded on a scale of 1 to 4 and was integrated into the score by multiplication. A single area might have two or three scores summed together if cells of various intensities were observed. For activated caspase-3 staining, only the cell number was evaluated under the assumption that, once activated, cells were committed to apoptosis. In analysis of the results, only differences greater than twofold were considered noteworthy.
Colocalization Studies
Dual colocalization immunohistochemistry was performed on 20-μm sections of perfused (4% paraformaldehyde in PBS) and fixed, cryoprotected, frozen brains. Tissues were stained for the neuron-specific antigen neuN (1:2000; Chemicon International, Temecula, CA) and the activated caspase-3 antigen (AC3, 1:1000; Promega) in 4% Blotto (Carnation instant nonfat dry milk, Nestlé, Solon, OH) containing 0.0025% sodium azide for 3 days at 4°C. Appropriate immunoglobulin antibodies (1:250), labeled with fluorescein isothiocyanate and rhodamine, respectively, were used for detection. Stains were visualized by use of the Axioplan-2 fluorescence microscope and HRc digital camera (Carl Zeiss Co., Oberkochen, Germany) to record images at highest resolution.
Three animals were used in each group, and two fields from each area were averaged for these analyses. Autofluorescence is a confound in colocalization studies of injured tissues because of extensive inflammatory infiltrates. Thus, sets of three photographs were taken of each selected region, both ipsilateral and contralateral to injury. The rhodamine filter was used to identify the AC3-positive cells, the fluorescein isothiocyanate filter was used to identify neuN-positive cells, and the DAPI filter was used to reveal autofluorescence. Digital images (Tiff format) were processed by a blinded observer using Adobe Photoshop 7 (Adobe Systems, Mountain View, CA). Each triplet was layered into a single file. The layers were thresholded individually, moving the control tab to the right edge of the intensity histogram. The autofluorescence signal was subtracted from the other images. This approach eliminated false-positives but possibly eliminated a few real colocalized cells as well. Colocalized signal (>50 contiguous pixels) was circled and counted.
Eicosanoid Analyses
Tissue Preparation
Conscious animals were decapitated in a sharpened guillotine at 6, 24, 48, 72, and 144 hours after injury. Brains were rapidly dissected, frozen on powdered dry ice, and stored at −80°C. For prostaglandin assays, frozen brains were coronally cut into sections 300 μm thick with a cryostat microtome (International Equipment Co.; −8°C), briefly thaw-mounted, and stored at −80°C. Micropunches were dissected (1000-μm cannula) in a cold box (at −10 to −20°C) from the dorsal hippocampus and the parietal and perirhinal cortex, both ipsilateral and contralateral to the site of injury. Tissue was immediately sonicated in acidified ethanol (120 μl, 0°C), and an aliquot was set aside for protein determination (Bradford microassay; Biorad, Hercules, CA). A small amount (2 pg) of the radioactive tracer [3H]prostaglandin F1α (~200 Ci/mmol; NEN Life Science Products, Boston, MA) was added, the samples were clarified (10 min at 3000 × g, 0°C), and the supernatants were stored at −80°C.
Sep-Pak Vac 3-ml C18 cartridges (Waters, Milford, MA) were pretreated twice to improve recovery by rinsing with water (3 ml), acetonitrile (1.5 ml), chloroform (1.5 ml), chloroform (1.5 ml), acetonitrile (1.5 ml), and water (3 ml). The cartridges were activated with methanol (3 ml), ethanol (3 ml), and water (3 ml). Before loading, the samples were combined with water (1 ml) containing formic acid to a pH of 4.0. Samples were loaded onto the cartridges by gravity and rinsed with water (3 ml), followed by hexane (3 ml), and eluted slowly by gravity with a solution of ethyl acetate and 1% ethanol (1 ml). The eluted fraction was dried by vacuum centrifugation (Savant Instruments, Farmingdale, NY), redissolved in enzyme immunoassay buffer (Cayman Chemical Prostaglandin EIA Kit), and stored at −80°C. The recovery was determined by calculating the fraction of tritium recovered via scintillation counting.
Prostaglandin Quantification
Sample extracts were assayed for PGE2 by use of enzyme immunoassay kits (Cayman Chemical). Sample prostaglandin levels were corrected for recovery and normalized by the total protein in the specimen (BioRad micro-Bradford assay) as follows: [PGE2 (pg)/recovery/total protein (μg)].
Eicosanoid Measurements
The fluorescence-detected high-performance liquid chromatography (HPLC) method has been validated by use of a series of eicosanoid standards (Biomol Research Laboratories, Inc., Plymouth Meeting, PA) and brain tissue extracts (109). In brief, dried eicosanoid extracts were dissolved in anhydrous acetonitrile and reacted (30 min in a desiccator at 4°C) with freshly prepared fluorescent dye, 2-(2,3-naphthalimino)ethyl-trifluoromethane sulfonate (Molecular Probes, Inc., Eugene, OR) with N,N-diisopropylethylamine (Sigma Chemical Co., St. Louis, MO) as the catalyst. The reaction was dried under argon and stored at −80°C. Samples were redissolved in dry methanol and analyzed by HPLC (Jasco, Easton, MD) with a fluorescence detector (excitation wavelength, 260 nm; emission wavelength, 396 nm). The injection volume was 8 μl, and separations were performed on a Symmetry C18 column (Waters) at 30°C. Mobile Phase A consisted of 0.5% formic acid in water, and mobile Phase B consisted of 0.5% formic acid in acetonitrile. A flow rate of 1 ml/min was used to deliver the mobile Phase A and B gradient as follows: 50 to 65% B in 40 minutes; 65 to 100% B in 80 minutes; and 100% B for 20 minutes. Data acquisition used Borwin Version 1.50.08 software (Jasco).
The limit of detection for these eicosanoids ranged from 2 to 20 pg per sample (109). The separation of this method did not resolve all the individual eicosanoid regioisomer standards, but brain eicosanoid could be quantified by combining the peak areas from four eicosanoid classes. Eicosanoid classes used were prostaglandins (PGs), hydroxyeicosatetraenoic acids (HETEs), EETs, and their dihydroxy metabolites (Di-HETEs). Eicosanoid peak areas (corrected for recovery) were summed by class, corrected for tissue wet weight, and normalized to the contralateral side.
2-Arachidonoyl Glycerol Analysis
Animals (n = 5 per group) were decapitated as above, and the cerebral hemispheres were separated, snap-frozen on dry ice, and stored at −80°C. Extraction, purification, and gas chromatography–mass spectrometry were performed exactly as described (73). Values of 2-arachidonoyl glycerol (2-AG) were corrected for the original wet weight of the tissue.
Statistical Analyses
Results are represented as mean ± standard error of the mean. Statistical analyses were performed using Statview software. Analysis of variance was performed with post hoc Dunnett’s test or Scheffé’s test comparisons. A value of P < 0.05 was necessary to reject the null hypothesis that the group means were equivalent.
RESULTS
In these studies, two doses of DFU were used, either 1 or 10 mg/kg body weight. DFU (or vehicle) was initially administered to animals either 10 minutes before injury or at the designated time after injury (2 or 6 h). Animals were further treated with the same dose of DFU twice daily for 3 days. The biochemistry and histochemistry studies used animals in which treatment was initiated before injury.
COX2 Inhibitor Treatments Improve Functional Recovery at 3 Days after Injury
DFU improved neurological reflexes significantly in all posttreatment groups at 3 days after injury (Fig. 1A). Injured vehicle-treated animals had a significant decrease in neuroscore compared with sham-operated animals (19 ± 1 versus 23 ± 1, respectively; P < 0.01, Scheffé’s test). The injured DFU-treated animals had improved neuroscores compared with injured vehicle-treated animals (Fig. 1A; P < 0.01, Dunnett’s test). This improvement was statistically significant for both dosage groups (1 or 10 mg/kg) when the first dose of DFU was administered 2 or 6 hours after the injury.
FIGURE 1.
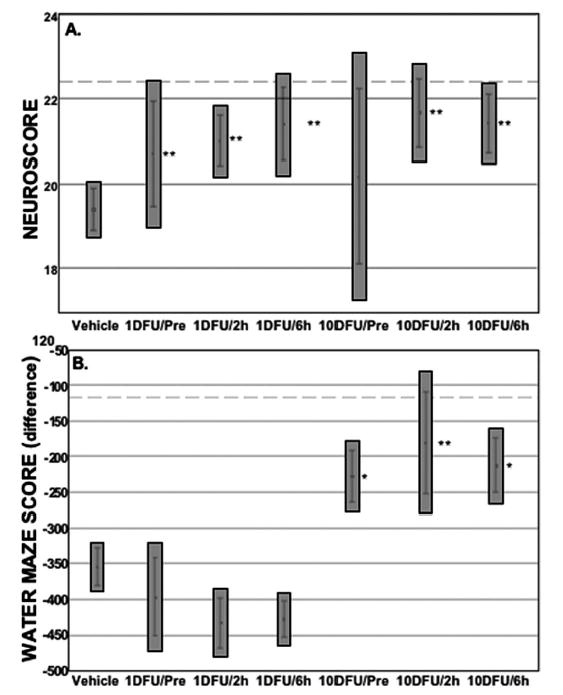
DFU treatment initiated before or after injury improved functional recovery after TBI. A, neuroscore: injured/vehicle-treated animals scored significantly lower compared with shams (19.3 ± 0.3 versus 22.4 ± 0.7 [dashed line]; P < 0.05, Scheffé’s test). Injured/DFU-treated animals improved their neuroscore compared with injured/vehicle-treated animals (P < 0.01, Dunnett’s test, except the 10 mg/kg pretreatment group). This improvement occurred even when the first dose of DFU was administered 2 to 6 hours after the injury. Neuroscore consisted of forelimb flexion, hindlimb extension, and lateral pulsion (see Materials and Methods). B, water maze difference scores were improved in the high-dose DFU groups, even when treatment was initiated 2 to 6 hours after injury. Sham: vehicle-treated and surgery, no injury (n = 15); vehicle: injured/vehicle-treated (n = 55); 1DFU/Pre: injured, 1 mg/kg DFU initiated 10 minutes before surgery (n = 15); 1DFU/2 hours: injured, 1 mg/kg DFU initiated 2 hours after injury (n = 13); 1DFU/6 hours: injured, 1 mg/kg DFU initiated 6 hours after injury (n = 13); 10DFU/Pre: injured, 10 mg/kg DFU initiated 10 minutes before surgery (n = 12); 10DFU/2 hours: injured, 10 mg/kg DFU initiated 2 hours after injury (n = 12); and 10DFU/6 hours: injured, 10 mg/kg DFU initiated 6 hours after injury (n = 12). Results are mean ± standard error of the mean; gray boxes represent the 99% confidence interval. *, P < 0.05, Dunnett’s test; **, P < 0.01, Dunnett’s test.
Cognitive function was improved by high-dose DFU treatments. Unmodified memory scores were higher in the high-dose group (Table 1) but reached statistical significance only for the group in which treatment was initiated 2 hours after injury. To minimize the high variances, a difference score was derived in which the memory score before injury was subtracted from the 3 days postinjury score for each animal (Fig. 1B). Memory performance was improved in all high-dose treatment groups (P < 0.05, Dunnett’s test) and nearly achieved sham levels of performance. No injury-related changes in swim speed or open-field behavior were observed among any of the groups (data not shown).
TABLE 1.
Effect of cyclooxygenase-2 inhibition on memory performance in the Morris water mazea
| Groupband time initiated | n | Water maze scorec | Dunnett t score |
|---|---|---|---|
| Injured/vehicle | |||
| Pre + Post | 55 | 81 ± 13 | |
| 1 mg/kg DFU | |||
| Pre | 13 | 132 ± 25 | 1.857 |
| 2 h post | 13 | 121 ± 12 | 1.454 |
| 6 h post | 13 | 115 ± 16 | 1.259 |
| 10 mg/kg DFU | |||
| Pre | 12 | 135 ± 16 | 1.912 |
| 2 h post | 12 | 162 ± 32d | 2.886 |
| 6 h post | 12 | 132 ± 17 | 1.796 |
DFU, 5,5-dimethyl-3(3-fluorophenyl)-4(4-methylsulfonyl)phenyl-2(3H)-furanone.
All animals were injured and treated as described in Materials and Methods.
The water maze scores increase with the time spent near the target zone; see Reference 51 for details. Analysis of variance (P = 0.0472), Dunnett critical value = 2.64.
P < 0.05 versus injured/vehicle.
DFU treatments did not significantly alter beam-walk performance. Injured beam-walk latencies were significantly impaired compared with the sham group (5.4 ± 1.1 s; P = 0.05, Dunnett’s test). However, a trend toward improvement was found for the high-dose 2 hours posttreatment group (15.9 ± 3.6 s) compared with the injured/vehicle control group (32.5 ± 5.8 s; P = 0.10, Dunnett’s test). Interestingly, the variance in this experimental group (171 s2) was an order of magnitude smaller than the mean variance among all experimental groups (1674 s2), indicating a possible drug effect.
Systemic DFU Treatment Inhibits Central PG Synthesis
PGE2 levels have been shown to rise precipitously after TBI (25, 27, 28, 89); thus, we measured PGE2 levels in the regions in which COX2 levels were known to increase (96). Systemically administered DFU (1 mg/kg) initiated 10 minutes before injury inhibited injury-induced PGE2 levels at 24 hours after injury in the dorsal hippocampus (Fig. 2) and cerebral cortex. Within 6 hours after LCI TBI, PGE2 levels were significantly elevated in the ipsilateral hippocampus compared with sham controls and remained significantly elevated at 24 hours (Fig. 2; P < 0.05, Dunnett’s test). At 72 hours and 6 days (144 h) after injury, the mean PGE2 content remained greater than sham levels but were not statistically significant. In contrast, PGE2 levels in the contralateral hippocampus (Fig. 2, thin lines) or ipsilateral and contralateral perirhinal cortex (not shown) were not significantly elevated above sham levels and did not change appreciably with time.
FIGURE 2.
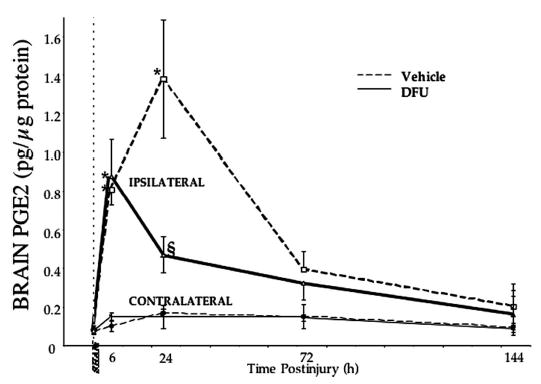
DFU treatment attenuated brain prostaglandin E2 levels after TBI. Hippocampal PGE2 production was maximal in the injured brain at approximately 24 hours after injury. DFU (1 mg/kg initiated 10 min before injury) decreased injury-induced PGE2 production at 24 hours after injury. Micropunches of fresh-frozen brain (bregma −3.1 to −3.6 according to the coordinates of Paxinos and Watson [74]) were dissected, and PGE2 levels were determined as described in Materials and Methods. Parietal cortex showed essentially the same profile (sham: 7.6 ± 0.5; injured/vehicle: 25.6 ± 8.5; injured/DFU: 12.8 ± 2.9 pg PGE2). Sham-operated animals from each time point (n = 2) showed no differences in PGE2 levels and were grouped together at time zero; otherwise, n = 3 to 4 per point. *, P < 0.05, Dunnett’s test; §, P < 0.05, Scheffé’s test.
At 6 hours after injury, there was no difference in the hippocampal PGE2 content between injured animals that were given vehicle or DFU. By 24 hours after injury, DFU blocked the injury-induced rise in PGE2 content in the ipsilateral hippocampus compared with vehicle-treated controls (Fig. 2; P < 0.05, Scheffé’s test). At 72 hours and 6 days, there was no significant difference in PGE2 levels in the ipsilateral hippocampus between DFU-treated and vehicle-treated groups.
COX2 Inhibition Attenuates COX2 Expression in the Injured Brain
DFU improved behavioral recovery, so we hypothesized that COX2 inhibition might attenuate central inflammation (namely, COX2 expression) and/or delayed cell death (namely, activation of caspase-3) in brain areas associated with functional deficits after TBI. Injury-induced COX2 immunoreactivity (COX2-ir) peaked between 24 and 72 hours in the ipsilateral cortex (Fig. 3A, left) and as early as 6 hours after injury in the ipsilateral hippocampus (Fig. 3B, left), consistent with previous findings (96).
FIGURE 3.
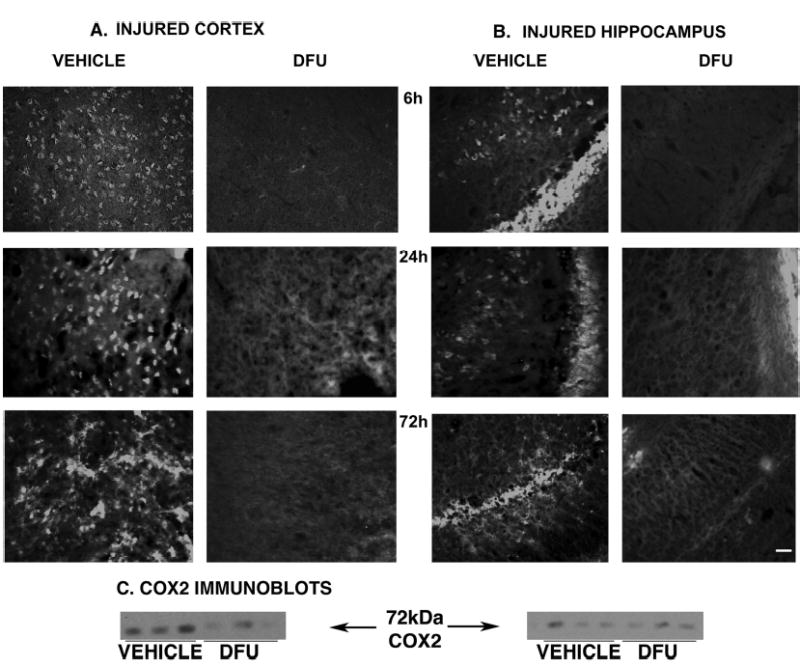
DFU treatment attenuated COX2 expression after TBI. DFU (10 mg/kg initiated 10 min before injury, right columns) reduced COX2 in both ipsilateral parietal cortex (A) and hippocampal CA2–CA3 regions (B) compared with vehicle-treated controls (left columns). Attenuation of COX2-ir was observed as early as 6 hours (top panels), at 24 hours (middle), and at 72 hours (bottom) after injury. COX2-ir was evaluated in 3 to 4 animals per group, according to both the cell number and the intensity of signal (see Materials and Methods). Scale bar = 50 μm. C, immunoblots of COX2 (72 kD) from the ipsilateral cortex (left) and hippocampus (right) at 72 hours after injury. DFU attenuated injury-induced COX2 in the ipsilateral cortex (P < 0.05, Dunnett’s test) but not in the hippocampus (right) by this method. Fresh-frozen micropunches of injured cortex and dorsal hippocampus (bregma − 3.6 to − 4.3 mm according to the coordinates of Paxinos and Watson [74]) were dissected and processed as described (96).
DFU (10 mg/kg) initiated 10 minutes before injury attenuated both the amount and intensity of COX2-ir in the cortex (Fig. 3A, right) after TBI compared with vehicle-treated controls. Specifically, there was less COX2-ir in the ipsilateral cortex, including cells of the cingulate, injured and adjacent parietal, perirhinal, and piriform regions. These decreases were observed at 6, 24, and 72 hours and were statistically significant by immunoblot at 72 hours in the ipsilateral cortex (Fig. 3C, left; P < 0.05, Dunnett’s test).
Similarly, DFU treatment reduced COX2-ir in the ipsilateral hippocampus at 6, 24, and 72 hours after injury (Fig. 3B, right). Attenuation of COX2-ir in the CA1 and CA2 regions was most pronounced at 6 hours (COX2 was no longer elevated in the CA1 and CA2 by 72 h). In the dentate gyrus, the greatest effect was seen at 24 hours, with pronounced decreases at 6 and 72 hours as well. In the CA3 region (most vulnerable to delayed neuronal death in this model), DFU attenuation of COX2-ir was greatest at 72 hours after injury, with pronounced decreases at 6 hours as well. COX2 immunoblots did not confirm these changes in the hippocampus, perhaps because of subregion-specific temporal changes after TBI (96).
DFU Attenuates Injury-induced Activated Caspase-3 Immunoreactivity
Immunohistochemical staining with the cell-death marker activated caspase-3 (AC3-ir) revealed a robust increase in the number of AC3-ir brain cells in the injured cortex and hippocampus (Fig. 4, B, D, and E, solid lines). These increases were more than twofold compared with shams and with respect to the corresponding contralateral regions (Fig. 4E). DFU treatment (10 mg/kg) initiated 10 minutes before injury substantially reduced the number of AC3-ir cells in brain regions around the injury site (Fig. 4, A, C, and E, dashed lines). Colocalization of AC3-ir with the neuronal marker neuN revealed that the benefit of DFU was, at least in part, because of neuroprotection (Fig. 4, A–D, Table 2).
FIGURE 4.
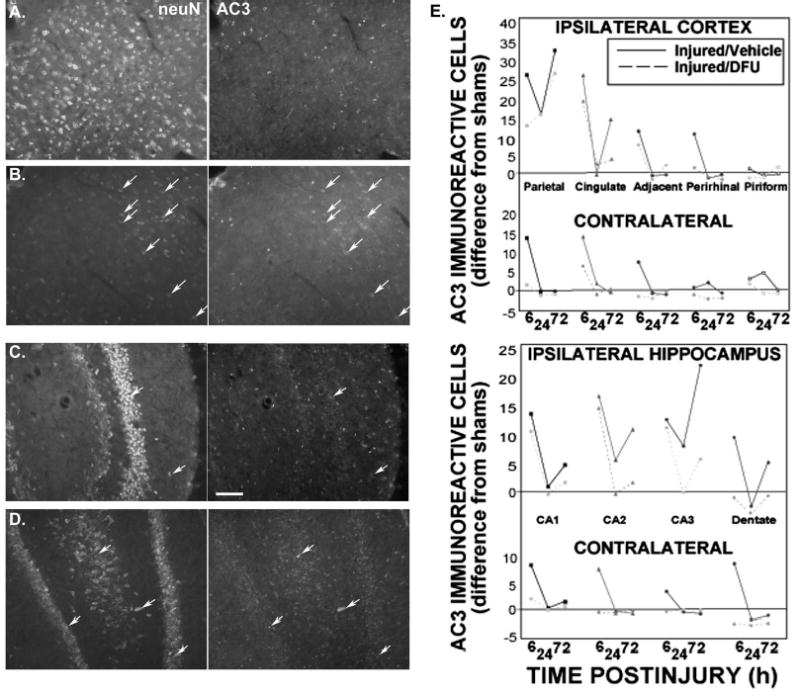
DFU treatment attenuated activated caspase-3 immunoreactivity after TBI. A–D, colocalization (arrows) of activated caspase-3 (AC3, right) with neurons (neuN, left) is an indicator of neuronal apoptosis at 72 hours after injury. A, DFU (10 mg/kg initiated 10 min before injury) abolishes injury-induced AC3/neuN colocalization in the ipsilateral parietal cortex; B, vehicle-treated injured brains. C, DFU attenuates AC3/neuN colocalization in hippocampal CA3 hilar pyramidal and dentate granule neurons; D, vehicle-treated injured brains. Scale bar = 50 μm. E, activated caspase-3 in all cells of selected brain regions, ipsilateral and contralateral to injury: injured/vehicle (solid lines) and injured/DFU (10 mg/kg, dashed lines) groups. Scores are the difference from the sham group (data averaged from all time points, range = 0–4) in the designated region. Notice the differences in scale in the contralateral regions. Fluorescence colocalization and cell counts performed as described in Materials and Methods.
TABLE 2.
COX2 inhibitor neuroprotection: DFU reduced colocalization of activated caspase-3 and neuN at 72 hours postinjurya
|
Cortexc |
Hippocampusc |
|||||
|---|---|---|---|---|---|---|
| Treatmentb | Cingulate | Parietal | CA1 | CA2 | CA3 | Dentate |
| Injured/vehicle | 14 ± 2 | 7.8 ± 2.3 | 7.0 ± 1.7 | 11 ± 3 | 7.0 ± 2.7 | 7.3 ± 0.9 |
| Injured/DFU | 3.8 ± 1.4 | 0 | 3.2 ± 1.5 | 14 ± 5 | 10 ± 3 | 4.0 ± 1.4 |
| Analysis of variance P value | 0.0071 | 0.0140 | 0.1208 | 0.6552 | 0.4035 | 0.1280 |
DFU, 5,5-Dimethyl-3(3-fluorophenyl)-4(4-methylsulfonyl)phenyl-2(3H)-furanone.
DFU treatment was 10 mg/kg initiated 10 minutes before injury. There was no colocalized staining in any of the sham animals at the 72-hour time point.
Average counts of colocalized cells from the reference space (bregma −2.8 to −4.3 mm).
At 6 hours after injury, the highest numbers of AC3-ir cells were found in the injured cortex and the ipsilateral CA1–CA2 hippocampal regions, where COX2 levels were maximal. By 24 hours, AC3-ir cell numbers in most brain regions decreased to sham levels, except around the ipsilateral parietal cortex and hippocampus (CA2, CA3). These areas demonstrated biphasic peaks with a minimum (still above sham levels) at 24 hours after injury. Surprisingly, contralateral cortex (perirhinal, piriform) showed some increase in AC3-ir cells by 24 hours (compared with ipsilateral counterparts), perhaps indicative of slight contra-coup injury.
The highest number of AC3-ir cells at any time point was observed in the injured parietal cortex at 72 hours after injury, as expected for a marker of delayed cell death. All other cortical regions returned to sham levels except the ipsilateral cingulate, probably because of its proximity to the injured cortex. Ipsilateral hippocampal regions also demonstrated increases in AC3-ir at 72 hours. The cell number was highest in the CA3 region, coincident with previously reported profiles of cell death (21) and maximal COX2 expression (96). In contrast, the number of AC3-ir cells in all contralateral regions of the hippocampus was at or below sham levels.
DFU treatment attenuated the number of AC3-ir cells at 6 hours after injury in all ipsilateral and contralateral cortical regions in which an increase was observed (Fig. 4E, dashed lines). In the hippocampus, DFU did not reduce the number of AC3-ir cells in the ipsilateral Ammon’s horn until later time points. At 24 and 72 hours, DFU effectively diminished the number of AC3-ir cells in the entire ipsilateral hippocampus.
DFU-treated animals had an 83% reduction in AC3-ir neurons in injured cortex compared with vehicle-treated animals (analysis of combined cingulate and parietal regions adjacent to injury, 2.5 ± 1.2 versus 14.3 ± 3.2; P < 0.01, Scheffé’s test). The injured parietal cortex itself contained excessive autofluorescent cells, barring the fluorescence colocalization analyses in this area. Although DFU treatment halved the number of AC3-ir neurons in the ipsilateral CA1 and dentate gyrus, these differences were not statistically significant (Table 2). Again, recurrent autofluorescence in the CA2–CA3 regions at 72 hours in this model confounded the analysis. No significant differences in AC3-ir neurons were observed among contralateral brain regions.
COX2 Inhibition Alters Eicosanoid Metabolism in the Injured Brain
Assessment of relative brain eicosanoid levels at 24 hours after injury confirmed increases in PGs and revealed increases in HETEs and EETs, cytochrome P450 epoxygenase metabolites of arachidonic acid (Fig. 5). No change in DiHETEs, the metabolites of the EETs, was observed at this time.
FIGURE 5.
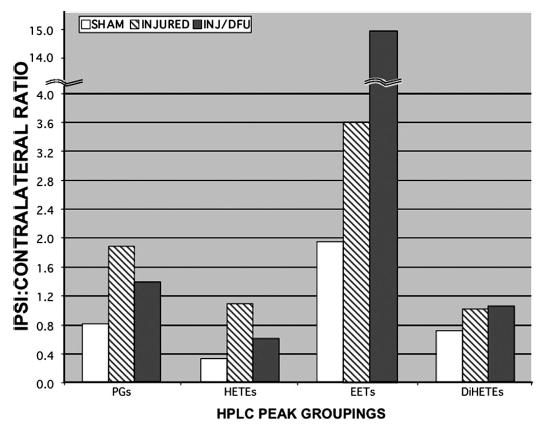
DFU treatment altered relative eicosanoid levels in the injured cortex. Eicosanoid levels increased in the ipsilateral cortex by 24 hours after injury. DFU (1 mg/kg) initiated 10 minutes before injury decreased PGs and HETEs and resulted in an amplification of EETs, possibly because of arachidonic acid shunting. Fresh-frozen parietal cortex was microdissected from 300-μm sections, and lipids were extracted and derivatized as described in Materials and Methods. PGs, HETEs, EETs, and their dihydroxy metabolites (DiHETEs) were quantified by fluorescence-detected HPLC (109). Individual peak areas (corrected for recovery) were grouped by class and summed, corrected for tissue wet weight, and normalized to the contralateral side. Averages (n = 3 animals per group) shown for sham/vehicle (SHAM, light bars), injured/vehicle (INJURED, striped bars), and injured/DFU (INJ/DFU, 1 mg/kg, dark bars). Note the discontinuity in the y axis, showing the extent of EET increase.
DFU treatment (1 mg/kg) initiated 10 minutes before injury not only attenuated injury-induced prostaglandin synthesis at 24 hours but also unexpectedly attenuated the increase in HETEs (Fig. 5). Significantly, DFU amplified the production of the EETs at 24 hours after injury by more than fourfold (Fig. 5; note the discontinuity in the y axis).
COX2 Inhibition Preserves 2-AG Levels in the Injured Brain
The brain levels of 2-AG, a potentially neuroprotective endocannabinoid (73), were determined in the ipsilateral hemispheres of the sham/vehicle, injured/vehicle, and injured/DFU (10 mg/kg initiated 10 min before injury) groups at 4 hours and 24 hours after injury. TBI decreased 2-AG levels (21.1 ± 4.3 nmol/g wet weight) compared with sham (56 ± 13 nmol/g wet weight; P < 0.05, Dunnett’s test). DFU treatment protected 2-AG levels in the injured brain (43.2 ± 7.6 nmol/g wet weight; P < 0.05 versus injured/vehicle, Scheffé’s test). There was no significant difference between sham/vehicle and injured/DFU-treated brains.
DISCUSSION
The acute treatment of brain trauma is still limited to ventilatory and fluid resuscitation, removal of extracerebral hematomas or contused brain matter, and efforts to control rising intracranial pressure (18). To date, no clinical treatment has been demonstrated to improve outcome from TBI (49, 63). Brain injuries trigger increases in COX2 expression, which has been implicated in the cause of neuronal cell death (16, 43). In agreement with these findings, cyclooxygenase inhibitors have been shown to improve neuronal survival in vitro as well as neuropathology and recovery in vivo (10, 12, 22, 38, 62, 85, 95). The present studies demonstrate that DFU improves functional recovery and reduces cell death and inflammation when administered systemically before or after TBI. In addition, we provide evidence that this COX2 inhibitor attenuates injury-induced prostaglandin production in the brain, shifting arachidonic acid metabolism toward potentially neuroprotective eicosanoids.
DFU (1 or 10 mg/kg i.p., twice daily, 3 d), initiated as late as 6 hours after injury, improved functional recovery 3 days after TBI. DFU inhibited injury-induced prostaglandin production by 24 hours after injury, apparently crossing the blood-brain barrier. DFU treatment diminished COX2 expression and caspase-3 activation at 72 hours after injury in the injured cortex and hippocampus. This benefit occurred in brain areas most vulnerable to delayed cell death at 72 hours after TBI (21, 82). Activated caspase-3, a marker of cellular apoptosis, has been observed in neurons, astrocytes, and oligodendrocytes after TBI (5). The effect of DFU was, at least in part, neuroprotection. Although neuronal colocalization with activated caspase-3 may not in itself confirm neuronal apoptosis, it is consistent with the clinical, in vivo, and in vitro appearance of apoptosis after trauma (5, 36, 53, 75–77, 84, 105, 108). The limited neuroprotection observed in the CA3 hippocampal region might be because some dying neurons were missed. AC3-ir cells most likely include the damaged neurons that had lost their neuN staining, a phenomenon readily observed in pyknotic neurons of injured cortex (5). A decrease in caspase-3 activation in hippocampal neurons and inflammatory cells is consistent with recent reports on the activity of COX2 inhibitors in vivo (10, 85).
DFU also reduced the appearance of COX2 protein. Rofecoxib has been shown to reduce the inflammation and excitotoxic cell death resulting from intraparenchymal injection of quisqualic acid (85). There is mounting evidence that COX2-specific enzyme inhibitors diminish lipopolysaccharide-induced COX2 messenger RNA in vitro (4, 23, 39, 41, 50, 98, 104). We provide the first in vivo evidence of this phenomenon in the brain. These data support a “vicious cycle” of neuroinflammation in which COX2 activity results in prolonged COX2 gene expression (Fig. 6).
FIGURE 6.
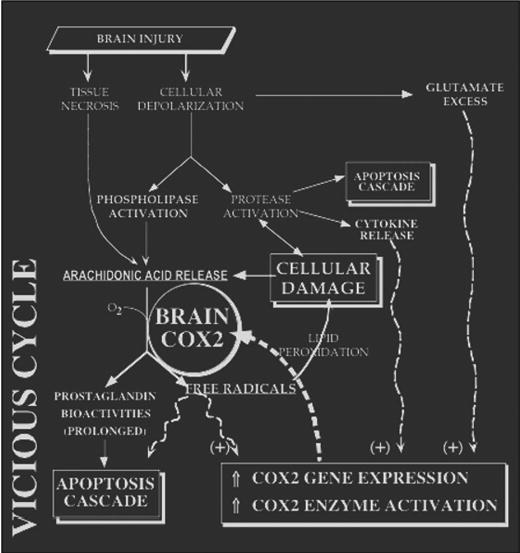
Vicious cycle of neuroinflammation. COX2 enzyme activity and gene expression increase both acutely and chronically after brain injuries. This may establish a vicious cycle that worsens functional recovery via overproduction of prostaglandins and free radicals. COX2 inhibition benefits the injured brain and attenuates the injury-induced rise in COX2 activity (acutely) and gene expression (by 72 h after injury).
Central inflammation is often accompanied by the migration of peripheral inflammatory cells into the brain (57, 58). After hypoxic injury, up-regulation of the adhesion molecules vascular cell adhesion molecule (VCAM), intercellular adhesion molecule (ICAM), and E-selectin on cerebrovascular endothelial cells can be inhibited by indomethacin (92, 93). These adhesion molecules happen to be down-regulated by arachidonic acid-derived 11,12-EET in cardiac endothelial cells (66). Thus, COX2 inhibitors may also protect the injured brain by shunting arachidonic acid down alternative metabolic pathways.
Shunting has been alluded to by Christie et al. (19) in a model of opioid-nonsteroidal anti-inflammatory drug (NSAID) synergy. They speculated that “blockade of cyclooxygenase and/or 5-lipoxygenase might lead to shunting of arachidonic acid metabolism … [and] enhanced formation of 12-LOX metabolites, thereby enhancing the efficacy of opioids” (19, pp 2–3) in the periaqueductal gray matter. Arachidonic acid shunting may be most significant when its levels increase (i.e., after phospholipase activation because of tissue damage or inflammation). Arachidonic acid can be oxidized to many biologically and chemically active derivatives, the most prevalent being prostaglandins. When COX2 activity increases, proportionately more arachidonic acid is converted to prostanoids and less to other metabolites. However, with the inhibition of COX2 activity, arachidonic acid may accumulate or be converted to other eicosanoid metabolites.
As expected, DFU diminishes PGE2 levels (and prostaglandins in general) in the injured brain. At 6 hours after injury, there was no difference in the PGE2 content between injured/vehicle and injured/DFU animals. This may be because of the route of central nervous system penetration and its time-dependent binding to COX2 (81). Surprisingly, DFU also attenuated the increase in HETEs observed 24 hours after injury. There is precedent for the synthesis of HETEs (specifically) by COX2 after the enzyme is covalently modified by aspirin (59, 71). DFU reversibly inhibits COX2 catalytic activity (81), so the reduction in HETEs leads us to question whether naturally occurring COX2 might produce these eicosanoids or whether brain injury initiates a cyclooxygenase-dependent HETE pathway.
Panikashvili et al. (73) clearly demonstrated that 2-AG, systemically administered to mice, was neuroprotective and improved functional outcomes after closed-head injury. In our studies, DFU protected the injured rat brain from a decrease in this endogenous cannabinoid. Interestingly, Kozak et al. (46) have provided evidence that COX2 metabolizes 2-AG, in which case COX2 inhibition would protect the brain from 2-AG catabolism, particularly in the environment of increasing COX2 levels seen after TBI. Most recently, Hampson and Grimaldi (35) demonstrated that 12- and 15-HETE provided neurons with protection against glutamate-mediated excitotoxicity. Surprisingly, certain acetylated (e.g., by aspirin) forms of COX2 may preferentially produce 15-HETE (45). This points to a potentially protective role for covalent “inhibition” of cyclooxygenases. Thus, COX2 inhibitors may lead to neuroprotective arachidonic acid metabolites via several mechanisms. Further study using sensitive chromatography techniques (109) will be necessary to determine which eicosanoids arise in the injured brain treated with COX2 inhibitors.
TBI sets into motion destructive/pathological responses as well as protective/adaptive responses. However, the distinction between these two types of processes is blurred, and indeed, responses that are initially protective can become destructive or vice versa (86, 94). In animal models of TBI, brain prostaglandin levels rise rapidly after injury (25, 28, 89). The release of such factors may have some physiologically adaptive purpose. For example, the early rise in thromboxane may limit hemorrhage after TBI. However, the chronic inappropriate expression of COX2 produces excessive prostaglandins and free radicals, probably promoting the central inflammatory response, alterations in the blood-brain barrier, cerebral autoregulation, and neuronal death (33, 40, 96, 97).
Two classes of anti-inflammatory drugs have been used to reduce injury-induced central prostaglandin production. Glucocorticoids block the induction of COX2 without affecting the expression of COX1 (54, 78). Although these anti-inflammatory steroids lack the gastric side effects of the NSAIDs, they produce even more serious side effects than the NSAIDs (37), and the usefulness of glucocorticoids in alleviating injury-induced central inflammation may be limited (69). Moreover, glucocorticoids did not improve outcomes in pre-clinical (88) or clinical studies (2, 82a).
First- and second-generation NSAIDs, conversely, act by inhibiting both COX1 and COX2 enzyme activity and thus boost gastric acid production as they alleviate inflammation (59). In rat brain injury models, cyclooxygenase inhibitors limit prostaglandin production (88), suppress hypermetabolism (83), and decrease brain damage after temporary focal ischemia (10, 62). Clinically, indomethacin has been suggested to improve intracranial pressure and cerebral perfusion pressure after brain injury in humans (6). Recent studies focusing on the role of COX2 in neuropathology, excitotoxicity, and control of cerebral microcirculation have used the third-generation, highly specific COX2 inhibitors in both basic and clinical investigations. These NSAIDs (e.g., rofecoxib, valdecoxib, celecoxib) should have fewer side effects than indomethacin, ibuprofen, or aspirin. The primary focus has been on the specificity of these drugs for COX2 over COX1 and the reduced production of prostaglandins, prostacyclins, thromboxanes, and free radicals.
COX2 inhibition benefits the brain-injured rat by improving both functional recovery and neuropathology. Although decreased prostaglandin and free radical production is one likely mechanism of action, the potential shunting of arachidonic acid toward anti-inflammatory and neuroprotective eicosanoids and the inhibition of 2-AG catabolism may also benefit the injured brain. Given the safety profile of the third-generation COX2-specific inhibitors, Phase I/II clinical trials of these agents in neuroprotection after brain injuries seem to be warranted.
Acknowledgments
We thank Dr. Jilly Evans, Merck and Co., Inc., for the generous gift of DFU and Dr. Rafael Mechoulam, Hebrew University, Jerusalem, for assistance with the 2-AG assays. This work was supported in part by National Institute of Neurological Disorders and Stroke Grant R01-NS38654 (KIS, RKN), the Irving and Felicia Rubin Family Brain Injury Research Grant Fund (KIS), and the American Association of Neurological Surgeons/Congress of Neurological Surgeons Joint Section on Neurotrauma and Critical Care Fellowship Award (ASM).
Contributor Information
Jonas J. Gopez, Department of Neurosurgery, Temple University, School of Medicine, Philadelphia, Pennsylvania
Hongfei Yue, Department of Chemistry, Temple University, Philadelphia, Pennsylvania
Ram Vasudevan, Department of Neurosurgery, Temple University, School of Medicine, Philadelphia, Pennsylvania
Amir S. Malik, Department of Neurosurgery, University of Texas, Houston Medical Center, Houston, Texas
Lester N. Fogelsanger, Department of Neurosurgery, Temple University, School of Medicine, Philadelphia, Pennsylvania
Shawn Lewis, Department of Neurosurgery, University of Cincinnati College of Medicine, Cincinnati, Ohio.
David Panikashvili, Department of Pharmacology, Hebrew University, Jerusalem, Israel
Esther Shohami, Department of Pharmacology, Hebrew University, Jerusalem, Israel
Susan A. Jansen, Department of Chemistry, Temple University, Philadelphia, Pennsylvania
Raj K. Narayan, Department of Neurosurgery, University of Cincinnati College of Medicine, Cincinnati, Ohio
Kenneth I. Strauss, Department of Neurosurgery, University of Cincinnati College of Medicine, Cincinnati, Ohio.
References
- 1.Adams J, Collaco-Moraes Y, de Belleroche J. Cyclooxygenase-2 induction in cerebral cortex: An intracellular response to synaptic excitation. J Neurochem. 1996;66:6–13. doi: 10.1046/j.1471-4159.1996.66010006.x. [DOI] [PubMed] [Google Scholar]
- 2.Alderson P, Roberts I. Corticosteroids in acute traumatic brain injury: Systematic review of randomised controlled trials. BMJ. 1997;314:1855–1859. doi: 10.1136/bmj.314.7098.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almer G, Teismann P, Stevic Z, Halaschek-Wiener J, Deecke L, Kostic V, Przedborski S. Increased levels of the pro-inflammatory prostaglandin PGE2 in CSF from ALS patients. Neurology. 2002;58:1277–1279. doi: 10.1212/wnl.58.8.1277. [DOI] [PubMed] [Google Scholar]
- 4.Beasley D. COX-2 and cytosolic PLA2 mediate IL-1β-induced cAMP production in human vascular smooth muscle cells. Am J Physiol. 1999;276(Suppl 4):H1369–H1378. doi: 10.1152/ajpheart.1999.276.4.H1369. [DOI] [PubMed] [Google Scholar]
- 5.Beer R, Franz G, Srinivasan A, Hayes RL, Pike BR, Newcomb JK, Zhao X, Schmutzhard E, Poewe W, Kampfl A. Temporal profile and cell subtype distribution of activated caspase-3 following experimental traumatic brain injury. J Neurochem. 2000;75:1264–1273. doi: 10.1046/j.1471-4159.2000.0751264.x. [DOI] [PubMed] [Google Scholar]
- 6.Biestro AA, Alberti RA, Soca AE, Cancela M, Puppo CB, Borovich B. Use of indomethacin in brain-injured patients with cerebral perfusion pressure impairment: Preliminary report. J Neurosurg. 1995;83:627–630. doi: 10.3171/jns.1995.83.4.0627. [DOI] [PubMed] [Google Scholar]
- 7.Brideau C, Kragman S. A human whole blood assay for clinical evaluation of biochemical efficacy of cyclooxygenase inhibitors. Inflamm Res. 1996;45:68–74. doi: 10.1007/BF02265118. [DOI] [PubMed] [Google Scholar]
- 8.Busija D, Thore C, Beasley T, Bari F. Induction of cyclooxygenase-2 following anoxic stress in piglet cerebral arteries. Microcirculation. 1996;3:379–386. doi: 10.3109/10739689609148310. [DOI] [PubMed] [Google Scholar]
- 9.Caggiano AO, Breder CD, Kraig RP. Long-term elevation of cyclooxygenase-2, but not lipoxygenase, in regions synaptically distant from spreading depression. J Comp Neurol. 1996;376:447–462. doi: 10.1002/(SICI)1096-9861(19961216)376:3<447::AID-CNE7>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Candelario-Jalil E, Alvarez D, Castaneda JM, Al-Dalain SM, Martinez-Sanchez G, Merino N, Leon OS. The highly selective cyclooxygenase-2 inhibitor DFU is neuroprotective when given several hours after transient cerebral ischemia in gerbils. Brain Res. 2002;927:212–215. doi: 10.1016/s0006-8993(01)03358-3. [DOI] [PubMed] [Google Scholar]
- 11.Candelario-Jalil E, Alvarez D, Gonzalez-Falcon A, Garcia-Cabrera M, Martinez-Sanchez G, Merino N, Giuliani A, Leon OS. Neuroprotective efficacy of nimesulide against hippocampal neuronal damage following transient forebrain ischemia. Eur J Pharmacol. 2002;453:189–195. doi: 10.1016/s0014-2999(02)02422-6. [DOI] [PubMed] [Google Scholar]
- 12.Candelario-Jalil E, González-Falcón A, García-Cabrera M, León OS, Fiebich BL. Wide therapeutic time window for nimesulide neuroprotection in a model of transient focal cerebral ischemia in the rat. Brain Res. 2004;1007:98–108. doi: 10.1016/j.brainres.2004.01.078. [DOI] [PubMed] [Google Scholar]
- 13.Cernak I, O’Connor C, Vink R. Inhibition of cyclooxygenase 2 by nimesulide improves cognitive outcome more than motor outcome following diffuse traumatic brain injury in rats. Exp Brain Res. 2002;147:193–199. doi: 10.1007/s00221-002-1245-z. [DOI] [PubMed] [Google Scholar]
- 14.Chan C, Panneton M. A selective inhibitor of COX-2 reverses endotoxin-induced pyretic responses in non-human primates. Eur J of Pharmacol. 1997;327:221–225. doi: 10.1016/s0014-2999(97)89664-1. [DOI] [PubMed] [Google Scholar]
- 15.Chan CC, Boyce S, Brideau C, Charleson S, Cromlish W, Ethier D, Evans J, Ford-Hutchinson AW, Forrest MJ, Gauthier JY, Gordon R, Gresser M, Guay J, Kargman S, Kennedy B, Leblanc Y, Leger S, Mancini J, O’Neill GP, Ouellet M, Patrick D, Percival MD, Perrier H, Prasit P, Rodger I. Rofecoxib [Vioxx, MK-0966; 4-(4′-methylsulfonylphenyl)-3-phenyl-2-(5H)-furanone]: A potent and orally active cyclooxygenase-2 inhibitor—Pharmacological and biochemical profiles. J Pharmacol Exp Ther. 1999;290:551–560. [PubMed] [Google Scholar]
- 16.Chen J, Marsh T, Zhang JS, Graham SH. Expression of cyclo-oxygenase 2 in rat brain following kainate treatment. Neuroreport. 1995;6:245–248. [PubMed] [Google Scholar]
- 17.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chestnut R: Treating raised intracranial pressure in head injury, in Narayan R, Wilberger JR, Povlishock JT (eds): Neurotrauma New York, McGraw-Hill, 1996, pp 445–470.
- 19.Christie MJ, Vaughan CW, Ingram SL. Opioids, NSAIDs and 5-lipoxygenase inhibitors act synergistically in brain via arachidonic acid metabolism. Inflamm Res. 1999;48:1–4. doi: 10.1007/s000110050367. [DOI] [PubMed] [Google Scholar]
- 20.Collaco-Moraes Y, Aspey B, Harrison M, de Belleroche J. Cyclo-oxygenase-2 messenger RNA induction in focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:1366–1372. doi: 10.1097/00004647-199611000-00035. [DOI] [PubMed] [Google Scholar]
- 21.Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dash P, Mach S, Moore A. Regional expression and role of cyclooxygenase-2 following experimental traumatic brain injury. J Neurotrauma. 2000;17:69–81. doi: 10.1089/neu.2000.17.69. [DOI] [PubMed] [Google Scholar]
- 23.Debey S, Meyer-Kirchrath J, Schror K. Regulation of cyclooxygenase-2 expression by iloprost in human vascular smooth muscle cells: Role of transcription factors CREB and ICER. Biochem Pharmacol. 2003;65:979–988. doi: 10.1016/s0006-2952(02)01661-1. [DOI] [PubMed] [Google Scholar]
- 24.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 25.Dewitt D, Kong D, Lyeth B, Jenkins L, Hayes R, Wooten E, Prough D. Experimental traumatic brain injury elevates brain prostaglandin E2 and thromboxane B2 levels in rats. J Neurotrauma. 1988;5:303–313. doi: 10.1089/neu.1988.5.303. [DOI] [PubMed] [Google Scholar]
- 26.Dixon C, Clifton G, Lighthall J, Yaghmai A, Hayes R. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- 27.Ellis EF, Police RJ, Rice LY, Grabeel M, Holt S. Increased plasma PGE2, 6-keto-PGF1α, and 12-HETE levels following experimental concussive brain injury. J Neurotrauma. 1989;6:31–37. doi: 10.1089/neu.1989.6.31. [DOI] [PubMed] [Google Scholar]
- 28.Ellis EF, Wright KF, Wei EP, Kontos HA. Cyclooxygenase products of arachidonic acid metabolism in cat cerebral cortex after experimental concussive brain injury. J Neurochem. 1981;37:892–896. doi: 10.1111/j.1471-4159.1981.tb04476.x. [DOI] [PubMed] [Google Scholar]
- 29.Ericsson A, Arias C, Sawchenko P. Evidence for an intramedullary prostaglandin-dependent mechanism in the activation of stress-related neuroendocrine circuitry by intravenous interleukin-1. J Neurosci Res. 1997;17:7166–7179. doi: 10.1523/JNEUROSCI.17-18-07166.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng L, Xia Y, Garcia G, Hwang D, Wilson C. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-α, and lipopolysaccharide. J Clin Invest. 1995;95:1669–1675. doi: 10.1172/JCI117842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foley TD. 5-HPETE is a potent inhibitor of neuronal Na+,K+-ATPase activity. Biochem Biophys Res Commun. 1997;235:374–376. doi: 10.1006/bbrc.1997.6790. [DOI] [PubMed] [Google Scholar]
- 32.Fu J, Masferrer J, Seibert K, Raz A, Needleman P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J Biol Chem. 1990;265:16737–16740. [PubMed] [Google Scholar]
- 33.Graham D: Neuropathology of head injury, in Narayan R, Wilberger JR, Povlishock JT (eds): Neurotrauma New York, McGraw-Hill, 1996, pp 43–60.
- 34.Hamm RJ, Dixon CE, Gbadebo DM, Singha AK, Jenkins LW, Lyeth BG, Hayes RL. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J Neurotrauma. 1992;9:11–20. doi: 10.1089/neu.1992.9.11. [DOI] [PubMed] [Google Scholar]
- 35.Hampson AJ, Grimaldi M. 12-hydroxyeicosatetrenoate (12-HETE) attenuates AMPA receptor-mediated neurotoxicity: Evidence for a G-protein-coupled HETE receptor. J Neurosci. 2002;22:257–264. doi: 10.1523/JNEUROSCI.22-01-00257.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harter L, Keel M, Hentze H, Leist M, Ertel W. Caspase-3 activity is present in cerebrospinal fluid from patients with traumatic brain injury. J Neuroimmunol. 2001;121:76–78. doi: 10.1016/s0165-5728(01)00409-x. [DOI] [PubMed] [Google Scholar]
- 37.Haynes R: Adrenocorticotrophic hormone: Adrenocortical steroids and their synthetic analogs: Inhibitors of the synthesis and actions of adrenocortical hormones, in Gilman A, Rall T, Nies A, Taylor P (eds): The Pharmacological Basis of Therapeutics New York, Pergamon Press, 1990.
- 38.Hewett S, Uliasz T, Vidwans A, Hewett J. Cyclooxygenase-2 contributes to N-methyl-d-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharm Exp Ther. 2000;293:417–425. [PubMed] [Google Scholar]
- 39.Hinz B, Brune K, Pahl A. Prostaglandin E2 upregulates cyclooxygenase-2 expression in lipopolysaccharide-stimulated RAW 264.7 macrophages. Biochem Biophys Res Commun. 2000;272:744–748. doi: 10.1006/bbrc.2000.2859. [DOI] [PubMed] [Google Scholar]
- 40.Hurley SD, Olschowka JA, O’Banion MK. Cyclooxygenase inhibition as a strategy to ameliorate brain injury. J Neurotrauma. 2002;19:1–15. doi: 10.1089/089771502753460196. [DOI] [PubMed] [Google Scholar]
- 41.Karim S, Berrou E, Levy-Toledano S, Bryckaert M, MacLouf J. Regulatory role of prostaglandin E2 in induction of cyclo-oxygenase-2 by a thromboxane A2 analogue (U46619) and basic fibroblast growth factor in porcine aortic smooth-muscle cells. Biochem J. 1997;326:593–599. doi: 10.1042/bj3260593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufmann W, Worley P, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:2317–2321. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kelley K, Ho L, Winger D, Freire-Moar J, Borelli C, Aisen P, Pasinetti G. Potentiation of excitotoxicity in transgenic mice overexpressing neuronal cyclooxygenase-2. Am J Pathol. 1999;155:995–1004. doi: 10.1016/S0002-9440(10)65199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kjome JR, Swenson KA, Johnson MN, Bordayo EZ, Anderson LE, Klevan LC, Fraticelli AI, Aldrich SL, Fawcett JR, Venters HD, Jr, Ala TA, Frey WH., II Inhibition of antagonist and agonist binding to the human brain muscarinic receptor by arachidonic acid. J Mol Neurosci. 1998;10:209–217. doi: 10.1007/BF02761775. [DOI] [PubMed] [Google Scholar]
- 45.Kozak KR, Prusakiewicz JJ, Rowlinson SW, Schneider C, Marnett LJ. Amino acid determinants in cyclooxygenase-2 oxygenation of the endocannabinoid 2-arachidonylglycerol. J Biol Chem. 2001;276:30072–30077. doi: 10.1074/jbc.M104467200. [DOI] [PubMed] [Google Scholar]
- 46.Kozak KR, Rowlinson SW, Marnett LJ. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J Biol Chem. 2000;275:33744–33749. doi: 10.1074/jbc.M007088200. [DOI] [PubMed] [Google Scholar]
- 47.Kunz T, Oliw E. The selective cyclooxygenase-2 inhibitor rofecoxib reduces kainate-induced cell death in the rat hippocampus. Eur J Neurosci. 2001;13:569–575. doi: 10.1046/j.1460-9568.2001.01420.x. [DOI] [PubMed] [Google Scholar]
- 48.Li CS, Black WC, Brideau C, Chan CC, Charleson S, Cromlish WA, Claveau D, Gauthier JY, Gordon R, Greig G, Grimm E, Guay J, Lau CK, Riendeau D, Therien M, Visco DM, Wong E, Xu L, Prasit P. A new structural variation on the methanesulfonylphenyl class of selective cyclooxygenase-2 inhibitors. Bioorg Med Chem Lett. 1999;9:3181–3186. doi: 10.1016/s0960-894x(99)00559-4. [DOI] [PubMed] [Google Scholar]
- 49.Maas AIR, Steyerberg EW, Murray GD, Bullock R, Baethmann A, Marshall LF, Teasdale GM. Why have trials of neuroprotective agents in head injury failed to show convincing efficacy? A pragmatic analysis and theoretical considerations. Neurosurgery. 1999;44:1286–1298. [PubMed] [Google Scholar]
- 50.Maldve RE, Kim Y, Muga SJ, Fischer SM. Prostaglandin E2 regulation of cyclooxygenase expression in keratinocytes is mediated via cyclic nucleotide-linked prostaglandin receptors. J Lipid Res. 2000;41:873–881. [PubMed] [Google Scholar]
- 51.Malik AS, Narayan RK, Wendling WW, Cole RW, Pashko LL, Schwartz AG, Strauss KI. A novel dehydroepiandrosterone analog improves functional recovery in a rat traumatic brain injury model. J Neurotrauma. 2003;20:463–475. doi: 10.1089/089771503765355531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marcheselli VL, Bazan NG. Sustained induction of prostaglandin endoperoxide synthase-2 by seizures in hippocampus: Inhibition by a platelet-activating factor antagonist. J Biol Chem. 1996;271:24794–24799. doi: 10.1074/jbc.271.40.24794. [DOI] [PubMed] [Google Scholar]
- 53.Martin LJ, Kaiser A, Yu JW, Natale JE, Al-Abdulla NA. Injury-induced apoptosis of neurons in adult brain is mediated by p53-dependent and p53-independent pathways and requires Bax. J Comp Neurol. 2001;433:299–311. doi: 10.1002/cne.1141. [DOI] [PubMed] [Google Scholar]
- 54.Masferrer JL, Seibert K, Zweifel B, Needleman P. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc Natl Acad Sci U S A. 1992;89:3917–3921. doi: 10.1073/pnas.89.9.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McIntosh TK. Neurochemical sequelae of traumatic brain injury: Therapeutic implications. Cerebrovasc Brain Metab Rev. 1994;6:109–162. [PubMed] [Google Scholar]
- 56.McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Soares H, Faden AL. Traumatic brain injury in the rat: Characterization of a lateral fluid-percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- 57.McKeating EG, Andrews PJ, Mascia L. The relationship of soluble adhesion molecule concentrations in systemic and jugular venous serum to injury severity and outcome after traumatic brain injury. Anesth Analg. 1998;86:759–765. doi: 10.1097/00000539-199804000-00016. [DOI] [PubMed] [Google Scholar]
- 58.McKeating EG, Andrews PJ, Signorini DF, Mascia L. Transcranial cytokine gradients in patients requiring intensive care after acute brain injury. Br J Anaesth. 1997;78:520–523. doi: 10.1093/bja/78.5.520. [DOI] [PubMed] [Google Scholar]
- 59.Meade E, Smith W, DeWitt D. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other nonsteroidal antiinflammatory drugs. J Biol Chem. 1993;268:6610–6614. [PubMed] [Google Scholar]
- 60.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 61.Mechoulam R, Fride E, Di Marzo V. Endocannabinoids. Eur J Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- 62.Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, Chen J, Graham SH. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci U S A. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Narayan R, Michel M, Ansell B, Baethmann A, Biegon A, Bracken M, Bullock M, Choi S, Clifton G, Contant C, Coplin W, Dietrich W, Ghajar J, Grady S, Grossman R, Hall E, Heetderks W, Hovda D, Jallo J, Katz R, Knoller N, Kochanek P, Maas A, Majde J, Marion D, Marmarou A, Marshall L, McIntosh T, Miller E, Mohberg N, Muizelaar JP, Pitts L, Quinn P, Riesenfeld G, Robertson C, Strauss K, Teasdale G, Temkin N, Tuma R, Wade C, Walker M, Weinrich M, Whyte J, Wilberger J, Young A, Yurkewicz L. Clinical trials in head injury. J Neurotrauma. 2002;19:503–557. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nicol G, Vasko M, Evans A. Prostaglandins suppress an outward potassium current in embryonic rat sensory neurons. J Neurophysiol. 1997;77:167–176. doi: 10.1152/jn.1997.77.1.167. [DOI] [PubMed] [Google Scholar]
- 65.Nishizaki T, Nomura T, Matsuoka T, Tsujishita Y. Arachidonic acid as a messenger for the expression of long-term potentiation. Biochem Biophys Res Commun. 1999;254:446–449. doi: 10.1006/bbrc.1998.9961. [DOI] [PubMed] [Google Scholar]
- 66.Node K, Huo YQ, Ruan XL, Yang BC, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Banion MK. Cyclooxygenase-2: Molecular biology, pharmacology, and neurobiology. Crit Rev Neurobiol. 1999;13:45–82. doi: 10.1615/critrevneurobiol.v13.i1.30. [DOI] [PubMed] [Google Scholar]
- 68.O’Banion MK, Olschowka JA. Localization and distribution of cyclooxygenase-2 in brain tissue by immunohistochemistry. Methods Mol Biol. 1999;120:55–66. doi: 10.1385/1-59259-263-5:55. [DOI] [PubMed] [Google Scholar]
- 69.O’Callaghan JP, Brinton RE, McEwen BS. Glucocorticoids regulate the synthesis of glial fibrillary acidic protein in intact and adrenalectomized rats but do not affect its expression following brain injury. J Neurochem. 1991;57:860–869. doi: 10.1111/j.1471-4159.1991.tb08230.x. [DOI] [PubMed] [Google Scholar]
- 70.Ogorochi T, Narumiya S, Mizuno N, Yamashita K, Miyazaki H, Hayaishi O. Regional distribution of prostaglandins D2, E2, and F2α and related enzymes in postmortem human brain. J Neurochem. 1984;43:71–82. doi: 10.1111/j.1471-4159.1984.tb06680.x. [DOI] [PubMed] [Google Scholar]
- 71.O’Neill GP, Mancini JA, Kargman S, Yergey J, Kwan MY, Falgueyret J-P, Abramovitz M, Kennedy BP, Ouellet M, Cromlish W, Culp S, Evans JF, Ford-Hutchinson AW, Vickers PJ. Overexpression of human prostaglandin G/H synthase-1 and -2 by recombinant vaccinia virus: Inhibition by nonsteroidal anti-inflammatory drugs and biosynthesis of 15-hydroxyeicosatetraenoic acid. Mol Pharmacol. 1994;45:245–254. [PubMed] [Google Scholar]
- 72.O’Neill P, Walton S, Foy PM, Shaw MD. Role of prostaglandins in delayed cerebral ischemia after subarachnoid hemorrhage. Neurosurgery. 1992;30:17–22. doi: 10.1227/00006123-199201000-00004. [DOI] [PubMed] [Google Scholar]
- 73.Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- 74.Paxinos G, Watson C: The Rat Brain in Stereotaxic Coordinates San Diego, Academic Press, 1986.
- 75.Pike BR, Flint J, Dutta S, Johnson E, Wang KK, Hayes RL. Accumulation of non-erythroid αII-spectrin and calpain-cleaved αII-spectrin breakdown products in cerebrospinal fluid after traumatic brain injury in rats. J Neurochem. 2001;78:1297–1306. doi: 10.1046/j.1471-4159.2001.00510.x. [DOI] [PubMed] [Google Scholar]
- 76.Pike BR, Zhao X, Newcomb JK, Glenn CC, Anderson DK, Hayes RL. Stretch injury causes calpain and caspase-3 activation and necrotic and apoptotic cell death in septo-hippocampal cell cultures. J Neurotrauma. 2000;17:283–298. doi: 10.1089/neu.2000.17.283. [DOI] [PubMed] [Google Scholar]
- 77.Qiu J, Whalen MJ, Lowenstein P, Fiskum G, Fahy B, Darwish R, Aarabi B, Yuan J, Moskowitz MA. Upregulation of the Fas receptor death-inducing signaling complex after traumatic brain injury in mice and humans. J Neurosci. 2002;22:3504–3511. doi: 10.1523/JNEUROSCI.22-09-03504.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raz A, Wyche A, Needleman P. Temporal and pharmacological division of fibroblast cyclooxygenase expression into transcriptional and translational phases. Proc Natl Acad Sci U S A. 1989;86:1657–1661. doi: 10.1073/pnas.86.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raz A, Wyche A, Siegel N, Needleman P. Regulation of fibroblast cyclo-oxygenase synthesis by interleukin-1. J Biol Chem. 1988;263:3022–3028. [PubMed] [Google Scholar]
- 80.Resnick D, Graham S, Dixon C, Marion D. Role of cyclooxygenase 2 in acute spinal cord injury. J Neurotrauma. 1998;15:1005–1013. doi: 10.1089/neu.1998.15.1005. [DOI] [PubMed] [Google Scholar]
- 81.Riendeau D, Percival MD, Boyce S, Brideau C, Charleson S, Cromlish W, Ethier D, Evans J, Falgueyret JP, Ford-Hutchinson AW, Gordon R, Greig G, Gresser M, Guay J, Kargman S, Leger S, Mancini JA, O’Neill G, Ouellet M, Rodger IW, Therien M, Wang Z, Webb JK, Wong E, Chan CC. Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX-2 inhibitor. Br J Pharmacol. 1997;121:105–117. doi: 10.1038/sj.bjp.0701076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- 82a.Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, Cottingham R, Svoboda P, Brayley N, Mazairac G, Laloe V, Munoz-Sanchez A, Arango M, Hartzenberg B, Khamis H, Yutthakasemsunt S, Komolafe E, Olldashi F, Yadav Y, Murillo-Cabezas F, Shakur H, Edwards P CRASH trial collaborators. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH Trial): Randomised placebo-controlled trial. Lancet. 2004;364:1321–1328. doi: 10.1016/S0140-6736(04)17188-2. [DOI] [PubMed] [Google Scholar]
- 83.Roe SY, Rothwell NJ. Whole body metabolic responses to brain trauma in the rat. J Neurotrauma. 1997;14:399–408. doi: 10.1089/neu.1997.14.399. [DOI] [PubMed] [Google Scholar]
- 84.Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1393–1401. doi: 10.1097/00006123-200106000-00051. [DOI] [PubMed] [Google Scholar]
- 85.Scali C, Giovannini MG, Prosperi C, Bellucci A, Pepeu G, Casamenti F. The selective cyclooxygenase-2 inhibitor rofecoxib suppresses brain inflammation and protects cholinergic neurons from excitotoxic degeneration in vivo. Neuroscience. 2003;117:909–919. doi: 10.1016/s0306-4522(02)00839-4. [DOI] [PubMed] [Google Scholar]
- 86.Scherbel U, Raghupathi R, Nakamura M, Saatman KE, Trojanowski JQ, Neugebauer E, Marino MW, McIntosh TK. Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc Natl Acad Sci U S A. 1999;96:8721–8726. doi: 10.1073/pnas.96.15.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci U S A. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shapira Y, Davidson E, Weidenfeld Y, Cotev S, Shohami E. Dexamethasone and indomethacin do not affect brain edema following head injury in rats. J Cereb Blood Flow Metab. 1988;8:395–402. doi: 10.1038/jcbfm.1988.75. [DOI] [PubMed] [Google Scholar]
- 89.Shohami E, Shapira Y, Sidi A, Cotev S. Head injury induces increased prostaglandin synthesis in rat brain. J Cereb Blood Flow Metab. 1987;7:58–63. doi: 10.1038/jcbfm.1987.8. [DOI] [PubMed] [Google Scholar]
- 90.Siesjö BK, Katsura K. Ischemic brain damage: Focus on lipids and lipid mediators. Adv Exp Med Biol. 1992;318:41–56. doi: 10.1007/978-1-4615-3426-6_5. [DOI] [PubMed] [Google Scholar]
- 91.Spielman L, Winger D, Ho L, Aisen PS, Shohami E, Pasinetti GM. Induction of the complement component C1qB in brain of transgenic mice with neuronal overexpression of human cyclooxygenase-2. Acta Neuropathol (Berl) 2002;103:157–162. doi: 10.1007/s004010100447. [DOI] [PubMed] [Google Scholar]
- 92.Stanimirovic D, Shapiro A, Wong J, Hutchison J, Durkin J. The induction of ICAM-1 in human cerebromicrovascular endothelial cells (HCEC) by ischemia-like conditions promotes enhanced neutrophil/HCEC adhesion. J Neuroimmunol. 1997;76:193–205. doi: 10.1016/s0165-5728(97)00057-x. [DOI] [PubMed] [Google Scholar]
- 93.Stanimirovic DB, Wong J, Shapiro A, Durkin JP. Increase in surface expression of ICAM-1, VCAM-1 and E-selectin in human cerebromicrovascular endothelial cells subjected to ischemia-like insults. Acta Neurochir Suppl (Wien) 1997;70:12–16. doi: 10.1007/978-3-7091-6837-0_4. [DOI] [PubMed] [Google Scholar]
- 94.Strauss KI. Recent advances in understanding the pathophysiology of neurotrauma. Trauma Q. 1998;13:353–372. [Google Scholar]
- 95.Strauss KI, Marini AM. Cyclooxygenase-2 inhibition protects cultured cerebellar granule neurons from glutamate-mediated excitotoxic cell death. J Neurotrauma. 2002;19:627–638. doi: 10.1089/089771502753754091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Strauss KI, Barbe M, Marshall R, Raghupathi R, Mehta S, Narayan R. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takadera T, Yumoto H, Tozuka Y, Ohyashiki T. Prostaglandin E2 induces caspase-dependent apoptosis in rat cortical cells. Neurosci Lett. 2002;317:61–64. doi: 10.1016/s0304-3940(01)02449-1. [DOI] [PubMed] [Google Scholar]
- 98.Takahashi Y, Taketani Y, Endo T, Yamamoto S, Kumegawa M. Studies on the induction of cyclooxygenase isozymes by various prostaglandins in mouse osteoblastic cell line with reference to signal transduction pathways. Biochim Biophys Acta. 1994;1212:217–224. doi: 10.1016/0005-2760(94)90256-9. [DOI] [PubMed] [Google Scholar]
- 99.Tocco G, Freire-Moar J, Schreiber SS, Sakhi SH, Aisen PS, Pasinetti GM. Maturational regulation and regional induction of cyclooxygenase-2 in rat brain: Implications for Alzheimer’s disease. Exp Neurol. 1997;144:339–349. doi: 10.1006/exnr.1997.6429. [DOI] [PubMed] [Google Scholar]
- 100.Uz T, Manev H. Circadian expression of pineal 5-lipoxygenase mRNA. Neuroreport. 1998;9:783–786. doi: 10.1097/00001756-199803300-00003. [DOI] [PubMed] [Google Scholar]
- 101.Vane JR. Towards a better aspirin. Nature. 1994;367:215–216. doi: 10.1038/367215a0. [DOI] [PubMed] [Google Scholar]
- 102.Visco D, Widmer R, Shen F, Orevillo C, Christen A, Chan CC, Leger S, Prasit P, Therien M, Wang Z, Rodger I, MacIntyre E: The effects of selective COX-2 inhibitor on adjuvant-induced arthritis in rat. Presented at The 10th International Conference on Prostaglandins and Related Compounds, Vienna, Austria, September 22–27, 1996; in Sinzinger H, Samuelsson B, Vane JR, Paoletti R, Wong PYK (eds): Advances In Experimental Medicine And Biology New York, Kluwer Academic/Plenum Publishers, 1998, vol 433, p 202.
- 103.Wallace C, Lyford G, Worley P, Steward O. Differential intracellular sorting of immediate early gene mRNAs depends on signals in the mRNA sequence. J Neurosci Res. 1998;18:26–35. doi: 10.1523/JNEUROSCI.18-01-00026.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu XM, Sansores-Garcia L, Chen XM, Matijevic-Aleksic N, Du M, Wu KK. Suppression of inducible cyclooxygenase 2 gene transcription by aspirin and sodium salicylate. Proc Natl Acad Sci U S A. 1999;96:5292–5297. doi: 10.1073/pnas.96.9.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: Regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- 107.Yang SY, Gao ZX. Determination and clinical significance of plasma levels of prostaglandins in patients with acute brain injury. Surg Neurol. 1999;52:238–245. doi: 10.1016/s0090-3019(99)00083-x. [DOI] [PubMed] [Google Scholar]
- 108.Yang X, Yang S, Zhang J, Xue L, Hu Z. Role of Caspase 3 in neuronal apoptosis after acute brain injury. Chin J Traumatol. 2002;5:250–253. [PubMed] [Google Scholar]
- 109.Yue H, Strauss KI, Borenstein MR, Barbe MF, Rossi LJ, Jansen SA. Determination of bioactive eicosanoids in rodent brain tissue by a sensitive reversed-phase liquid chromatography method with fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;803:267–277. doi: 10.1016/j.jchromb.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 110.Zhang J, Rivest S. Anti-inflammatory effects of prostaglandin E2 in the central nervous system in response to brain injury and circulating lipopolysaccharide. J Neurochem. 2001;76:855–864. doi: 10.1046/j.1471-4159.2001.00080.x. [DOI] [PubMed] [Google Scholar]