

Abstract
Disorders of sodium and water balance are common in critically ill adult neurologic patients. Normal aspects of sodium and water regulation are reviewed. The etiology of possible causes of sodium disturbance is discussed in both the general inpatient and neurologic populations. Areas of importance are highlighted with regard to the differential diagnosis of sodium disturbance in neurologic patients and management strategies are discussed. Specific discussions of the etiology, diagnosis and management of cerebral salt wasting syndrome, the syndrome of inappropriate antidiuretic hormone secretion and central diabetes insipidus are presented, as well as the problems of over-treatment. The importance of diagnosis at an early stage of these diseases is stressed, with a recommendation for conservative management of milder cases.
Keywords: hypernatremia, hyponatremia, cerebral salt wasting, SIADH, diabetes insipidus
Normal physiology of salt and water regulation
Sodium is the major extracellular cation in the body and is therefore one of the most important osmotically active solutes. The extracellular to intracellular sodium concentration gradient is maintained by the cell-membrane Na-K ATPase pump and total body sodium is controlled via renal excretion. Sodium re-absorption occurs predominantly at the proximal convoluted tubule and is affected by sympathetic innervation, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP). ANP is released from the cardiac atrium whereas BNP is found in the brain and cardiac ventricle.1 ANP and BNP cause natriuresis via a direct effect on the inner medullary collecting duct as well as inhibiting renin and aldosterone release.2 C-type natriuretic peptide (CNP) and dendroaspis natriuretic peptide (DNP) may also be implicated in sodium regulation.3
Total body water volume is predominantly controlled by renal manipulation of body sodium, with resulting water volume adjustment to maintain tonicity. Water makes up 60% of the mass of the human body and moves freely between intracellular and extracellular fluid spaces as dictated by the movements of osmotically active particles. Water balance is monitored by osmoreceptors in the hypothalamus, low pressure baroreceptors located in the right atrium and great veins, and by high pressure baroreceptors in the carotid sinus. The two main mechanisms for controlling water balance are antidiuretic hormone (ADH) secretion and thirst. Increases in extracellular fluid (ECF) tonicity cause secretion of ADH from the posterior pituitary promoting free water reabsorption in the kidney leading to concentrated urine. Hypovolemia is also a potent stimulus for ADH release via the renin-angiotensin-aldosterone system. For example, the hypo-osmolar state of sodium depletion will result in low plasma volume as a homeostatic response to maintain osmolality and the hypo-osmolar state of water retention will promote sodium loss for water offloading. Sodium loss from the kidneys or gastrointestinal tract may therefore promote a diuresis, but the resulting low plasma volume causes activation of the renin-angiotensin-aldosterone system promoting net re-absorption of sodium, and water retention
Changes in sodium and water balance have a profound effect on the brain and central nervous system (CNS) and the behaviour of the brain cell in response to changing plasma osmolality has previously been described.4 In acute hyperosmolar states there is loss of intracellular water with cell shrinkage followed by gradual restoration of brain volume via the generation of non-electrolyte osmotically active intracellular solute. In the hypo-osmolar state there is cellular expansion which is corrected over time by the loss of intracellular solute. Total brain volume is therefore preserved by alterations in the intracellular milieu.
Dysnatremia
Disturbance of sodium balance, referred to as dysnatremia in this review, is a frequent finding in adults in the hospital in-patient setting,5 and accounts for the bulk of electrolyte disturbances in this patient population.6 The symptoms and signs of sodium disturbance are related to the balance between sodium and water balance and are shown in table 1. This review will focus on dysnatremia in the adult neurologic patient, but within the context of sodium disturbance in the in-patient general hospital population.
Table 1.
Symptoms and signs of hypo- and hypernatremia
| Moderate | Severe | |
|---|---|---|
| Hyponatremia | Lethargy | Drowsiness/Confusion |
| Nausea/Vomiting | Depressed Reflexes | |
| Irritability | Seizures | |
| Headache | Coma | |
| Muscle Weakness/Cramps | Death | |
| Hypernatremia | Lethargy | Hyperreflexia |
| Thirst | Ataxia | |
| Irritability | Seizures | |
| Coma |
Hyponatremia
Epidemiology
The reported incidence of hyponatremia depends on its threshold for diagnosis, but is usually defined as serum sodium <135 mmol/L. Hyponatremia has been reported in 1–15% of hospital inpatients7,8,9 and is associated with a mortality increase of 7 to 60%.8 Furthermore, acute hyponatremia leads to greater mortality than chronic states.10 Hyponatremia is more common in neurologic patients than in the general hospital population11 and is particularly associated with aneurismal subarachnoid haemorrhage (SAH),12 traumatic brain injury (TBI)13 and basilar meningitis.14
Causes
The causes of hyponatremia are numerous (see table 2), but in the adult neurologic patient population, hyponatremia most commonly occurs because of the syndrome of inappropriate antidiuretic hormone secretion (SIADH) or the cerebral salt wasting syndrome (CSWS). In addition to pathologic causes, administration of hypotonic fluid is a common iatrogenic cause of hyponatremia in hospitalized patients.5,7 Hyponatremia has also been noted in patients receiving normal saline as maintenance fluid,15 and this may occur because of perioperative ADH secretion, as a stress response. It is important to remember that hyponatremia can occur in the setting of hypo-, eu- or hypervolemia and the causes of hyponatremia may be distinguished by the associated volume disturbance.
Table 2.
Common Causes of Hyponatremia
Decreased ECF Volume
|
Normal ECF Volume
|
Increased ECF Volume
|
Syndrome of inappropriate antidiuretic hormone secretion
SIADH was first described by Schwartz in 1957 in patients with bronchogenic carcinoma.16 The key findings were urinary sodium loss without corresponding loss of water, leading to a decrease in plasma osmolality in the presence of hypertonic urine.
Treatment by fluid restriction led to resolution of these abnormalities.
The pathophysiology of SIADH is not fully understood, although ADH release correlates with a change in the threshold for the thirst response, with a lower threshold for thirst in patients with SIADH.17 However, there is also loss of control of ADH release, with plasma ADH levels being unchanged by drinking17 or by osmotic stimulus.18
SIADH is associated with many conditions and these are best classified into four major categories: neoplasia, non-malignant lung disease, drugs and neurologic diseases. The most common causes in the neurologic group include meningitis/encephalitis, brain tumour, SAH and TBI. SIADH has also been reported following spinal surgery.19 Drug related hyponatremia secondary to the antiepileptic drugs carbamazepine and oxcarbazepine is of particular relevance in the care of neurologic patients. The etiology of hyponatremia has recently been reviewed in detail elsewhere.9
Cerebral salt wasting syndrome
CSWS, a syndrome characterized by polyuria and natriuresis, was first described by Peters in 195020 and was subsequently ascribed to disruption of hypothalamic-renal pathways.21 A current working definition of CSWS is renal loss of sodium due to intracranial disease leading to hyponatremia and hypovolemia. Whilst the pathophysiology of CSWS is not fully understood, the evidence suggests that raised levels of circulating ANP and BNP mediate, at least in part, increased natriuresis and hyponatremia in acute brain injury, especially after SAH.22 There is an initial elevation of ANP following SAH, but ANP levels subsequently fall and the initial rise is not correlated with hyponatremia.23 An increased ANP level has also been reported as a normal response to trauma and this cannot be suppressed with correction of volume status.24 Elevated BNP levels are also associated with hyponatremia and plasma levels have been correlated with urinary sodium excretion and intracranial pressure.25
CSWS is predominantly associated with SAH but has also been described in conjunction with TBI, glioma, tuberculous or carcinomatous meningitis.22
Investigation of the hyponatremic neurologic patient
Except in life-threatening disturbances of sodium homeostasis, the initial finding of an abnormal sodium level should always prompt specific investigation into the underlying cause before management is initiated. The speed of onset of the hyponatremia, as well as the presence of symptoms, is most important because patients with the most rapid onset are more likely to become symptomatic.26 A differentiation must be made between hypervolemia with normal total body sodium (suggesting SIADH) and hypovolemia with disproportionately low total body sodium (suggesting CSWS). This differentiation is crucial because the managements of these two conditions are diametrically opposed.27,28
The diagnostic criteria of SIADH were summarised by Harrigan in 199629 and are shown in table 3. Although these criteria are adequate in the presence of an appropriate level of clinical suspicion, other diagnostic tests for SIADH have been described, including measurement of elevated levels of plasma and/or urinary ADH levels.30,31 Attempts have been made to identify derived parameters of sodium and water homeostasis for the differentiation between SIADH and CSWS in order to avoid the need to measure total body water and circulating blood volume.32 In SIADH there is high free water absorption, low fractional water and sodium excretion, low or normal urine output and increased ADH, whereas in CSWS there is normal free water absorption, increased fractional water and sodium excretion, high urine output and an increase in ADH (secondary and hence appropriate). Urine biochemical analysis will demonstrate dilute or concentrated urine and should be reviewed with simultaneous plasma osmolality assessment. ADH and ANP levels are high in both SIADH and CSWS, either as a primary or secondary event.
Table 3.
Diagnostic criteria for the syndrome of inappropriate antidiuretic hormone secretion
|
Biochemical criteria, however, may fail to distinguish between the two pathologies and the key difference is the presence of volume depletion in CSWS.22,3 Particular attention should be paid to skin turgor and mucous membranes. Intravascular volume can be assessed by examining jugular venous distension and by observation of orthostatic variations in blood pressure and pulse. Daily weight may provide helpful additional information because water accounts for a large proportion of body mass. Extracellular volume status can be difficult to assess clinically and may rarely be a confounding factor. In addition, some studies assessing volume status have highlighted the presence of hypovolemia in certain patients fulfilling the diagnostic criteria for SIADH.33 This is due to the volume depletion of CSWS causing a secondary rise in ADH. Under such conditions, patients should correctly be diagnosed with CSWS rather than SIADH.33
The pathophysiologic changes and biochemical findings of SIADH and CSWS are summarised in table 4 and a practical algorithm and differential for assessment of the hyponatremic patient are shown in figure 1 and table 2.
Table 4.
Biochemical and water changes in the syndrome of inappropriate ADH secretion (SIADH), cerebral salt wasting syndrome (CSWS) and diabetes insipidus (DI)
| Finding | SIADH | CSWS | DI |
|---|---|---|---|
| Plasma volume | Raised | Lowered | Lowered |
| Sodium balance | Positive/equal | Negative | Equal |
| Water balance | Positive | Negative | Negative |
| Serum sodium | Low | Low | High |
| Serum osmolality | Lowered | High/normal | High |
| Urine sodium | High | High | Normal |
| Urine osmolality | High | Normal/high | Low |
(Normal values: Plasma osmolality 278–305 mmol/kg, Plasma sodium 135–145 mmol/L, urine osmolality 350–1000 mmol/kg, urine sodium 20–60 mmol/L : 100–250 mmol/24hr)
Figure 1.
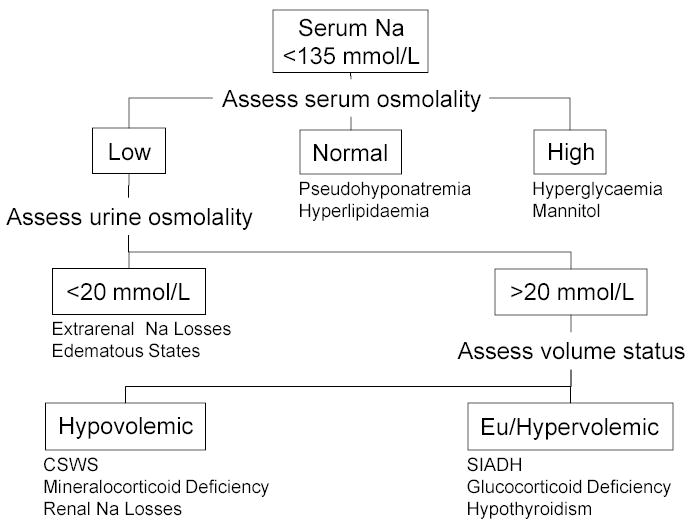
Algorithm for Assessment of the Hyponatremic Patient
Treatment of hyponatremia
An expectant and supportive management strategy is best adopted in asymptomatic patients as the physiological disturbance is often transient. Treatment is however indicated in the presence of acute symptomatic hyponatremia which can be a prelude to life-threatening complications (see Table 1).
The correction of hyponatremia, especially in the chronic setting, can lead to neurologic sequelae.9 This risk can be minimized by gradual correction of sodium deficits (see below). It is especially important to note that treatment should be targeted to the point of alleviation of symptoms rather than to the achievement of biochemical normality.34
Use of volume status to guide therapy in hyponatremia has produced varied results. Measurement of CVP and daily weight has been described to manage hyponatremic patients following SAH35 but this strategy is prone to errors and the simple response to saline infusion may be more reliable.36 CVP readings alone have been used to classify neurosurgical patients with hyponatremia and natriuresis as hypovolemic, normovolemic or hypervolemic in order to guide therapy.37 Hypovolemic patients received salt and additional amounts of fluid whilst normovolemic patients received salt with standard maintenance fluid volumes. 75% of patients achieved normal serum sodium within 72 hours with the non-responding group requiring more aggressive therapy. The effect of intravenous fluid administration on serum sodium level can be estimated using simple formulae (table 5).
Table 5.
Formulae for Estimation of Effect of Intravenous Fluid Administration on Serum Sodium64
| ΔSerum[Na+]= Infusate Volume{(Infusate[Na+]+Infusate[K+])−Serum[Na+]} /(Total Body Water+1) |
| Required Na+ Load = Total Body Water (desired [Na+] − current [Na+]) |
| Total Body Water can be estimated as a fraction of body weight.65 |
Specific treatment of SIADH
The appropriate management of SIADH is fluid restriction, initially to 1L per day. This usually results in a slow rise in sodium of 1.5mmol/l/day.7 If supplemental intravenous fluid is required, 0.9% saline is the usual choice. More recent studies support this management strategy, although some authors recommend maintenance fluid replacement therapy with intravenous 5% dextrose.38,39
Pharmacologic treatment is a further option when the diagnosis is certain. SIADH treatment using furosemide is described,40 with saline or salt supplementation to counteract the sodium loss which accompanies the free water loss. Lithium may be beneficial in patients with SIADH after brain trauma with dosage adjusted to maintain a plasma lithium concentration of 1 mmol/L.41 Lithium acts as a blocker of 3,5-adenosine monophosphatase and inhibits the action of ADH on the renal tubule. The predictability and safety of this treatment has been questioned7. Demeclocycline is an ADH antagonist which can also be used in the treatment of SIADH.
Initially democlocycline hydrochloride 900–1200 mg daily is given orally in divided doses, reduced after therapeutic effect is achieved to a daily oral maintenance dose of 600–900 mg. It is less toxic than lithium42 but may take up to three weeks to achieve maximum effect.9
However, it should be remembered that many patients have self-limiting disease. In one study, a third of consecutive neurosurgical patients fulfilled criteria for SIADH but only half of these required treatment with fluid restriction with the remainder achieving spontaneous resolution of their dysnatremias.43
Specific treatment of CSWS
Best management of CSWS is treatment with fluid and sodium resuscitation, although the saline solution used in the treatment of CSWS is more controversial. 0.9% saline solution should generally be used in the first instance,3 although in acute symptomatic hyponatremia, hypertonic (3%) saline has also been recommended,7 with the administration of furosemide in the event of hypervolemia. Administration of 3% saline requires central venous access and the use of 1.5% saline, which can be administered peripherally, has been advocated as an equally effective alternative.3 It should be noted that administration of sodium in CSWS may, in some patients, drive further urinary sodium loss with accompanying loss of water. In cases refractory to salt and fluid therapy, prophylactic fludrocortisone may limit the negative sodium balance after SAH44 by increasing sodium reabsorption from the renal tubule. Oral fludrocortisone doses of between 0.1 and 0.4 mg daily are recommended.45,44 Care must be taken to ensure that hypokalemia does not then occur as a secondary effect.46
Neurologic complications of treatment of hyponatremia
The dangers of rapid correction of hyponatremia are serious and well described7,47 and remind us that the best treatments of sodium disturbance are often supportive with management of the underlying condition. Myelinolysis is a neurologic disorder affecting pontine and extrapontine structures that can occur after rapid elevation of serum sodium levels. Risk factors for its development include alcoholism, malnutrition and liver disease. The symptoms and signs include mutism, dysarthria and lethargy followed by spastic quadraparesis and pseudobulbar palsy. The risk of myelinolysis can be minimised by gradual correction of sodium deficit at a rate of less than 10 mmol/L/24 hrs, although lower correction rates are also reported.47 Close monitoring of the hyponatremic patient undergoing treatment is mandatory and, if over-rapid correction is suspected, reversal using desmopressin and water may be appropriate.48
Hypernatremia
Epidemiology
Hypernatremia is defined as serum sodium >145 mmol/L. It is less common than hyponatremia with an incidence of around 1% across the spectrum of all hospital patients.49,50 Hypernatremia is relatively more common in neurologic and critically ill patients than in the general inpatient population and water depletion is especially common in the intensive care setting, where a 9% incidence of Na >150mmol/L has been reported.51 Hypernatremia is often a paraphenomenon, being an indicator of the severity of the underlying disease process.
Causes
Hypernatremia is usually related to water deficiency, such as inadequate water supplementation or water loss, e.g. associated with fever,51 and only rarely does it represent salt excess, such as ingestion of salt or infusion of saline or hypertonic fluids.52,53 Except in cases of uncontrollable diabetes insipidus, a hypernatremic state can only be maintained when access to water or thirst is impaired and patients with altered mental state or decreased level of consciousness are therefore particularly susceptible.54 The elderly are also at high risk, possibly due to impaired urinary concentrating power, reduced thirst reflexes and frailty resulting in the inability to access water.
Causes of hypernatremia are listed in table 6 and include central and nephrogenic diabetes insipidus, dehydration, fever and osmotic diuresis. Accounts of nephrogenic diabetes can be found elsewhere55,56 and this review will focus on central diabetes insipidus (CDI).
Table 6.
Common Causes of Hypernatremia
Decreased ECF Volume
|
Increased ECF Volume
|
Central diabetes insipidus
CDI is a failure of homeostatic release of ADH from the hypothalamo-pituitary axis. It is characterized by the inability to concentrate urine and the passage of a large volume of inappropriately dilute urine with a consequent rise is plasma osmolality due to disproportionate loss of water over sodium, and progressive dehydration. In neurologic practise it is particularly associated with pituitary surgery,57 TBI58 and anterior communicating artery aneurismal SAH.59 Patients who become brain dead often develop severe CDI and this is particularly relevant in the management of potential organ donors.60
The anatomic and pathologic relationships of CDI have been reviewed previously.61 Compromise of the hypothalamic centres or the supraoptic tract above the median eminence may lead to permanent CDI, whilst damage below the median eminence or removal of the posterior lobe of the pituitary leads to transient CDI because ADH is subsequently released from fibres ending in the median eminence.
The incidence of CDI in a neurosurgical unit has been reported as 3.7%,59 with one third following SAH, one third following TBI, one sixth after pituitary surgery and one sixth following intracerebral haemorrhage. Overall mortality in this group was high (72.4%), although this may be due in part to the fact that the bulk of the patients had suffered severe acute brain injury. The incidence of CDI after severe TBI is around 3% and is strongly associated with basal skull fracture.39 Development of CDI in non-pituitary surgery patients is often associated with severe cerebral edema and impending death.59 Pituitary stalk hematoma is a rare complicating factor after severe brain injury and MRI has been recommended for patients in whom the CDI is out of proportion to the severity of the injury.62
Investigation of the hypernatremic neurologic patient
Clinical assessment of volume status allows differentiation between hypernatremic hypovolemia, with disproportionate water loss over sodium, and hypernatremic eu- or hypervolemia resulting from hypertonic sodium gain. The majority of hypernatremic neurologic patients will be hypovolemic and in this context CDI must be distinguished from simple dehydration by urine volume assessment and biochemical analysis. Although conscious patients will become thirsty, neurologic patients may be cognitively impaired, either as a result of their primary pathology or the subsequent electrolyte disturbance, so thirst is often is an unreliable sign.
In the context of neurologic insult the diagnosis of CDI can be made in the light of an abnormally high serum osmolality (>305 mmol/kg) and serum sodium (>145 mmol/L), in combination with an abnormally low urine osmolality (<350 mmol/kg), reflecting the inability of the kidney to concentrate urine in an appropriate manner. These laboratory tests may take some time and measurement of urine specific gravity (SG) is a useful adjunct to diagnosis when urgent treatment is required to prevent significant hypovolemia because of excessive urine volumes. A urine SG of less than 1.005, in the light of raised serum sodium, points strongly to a diagnosis of CDI. It is important to realise however that this criterion may provide a false negative when the serum sodium is grossly raised and the diagnosis should therefore always be confirmed with laboratory measurement of urine and serum osmolality.
Neurologic patients may have other causes of water diuresis, including pre-hospital and intra-operative volume resuscitation. Diuresis of solute may also be caused by osmotic diuresis secondary to the use of mannitol or hypertonic saline, for control of intracranial pressure, or hyperglycemia. An algorithm for assessment of the hypernatremic patient is shown in figure 2.
Figure 2.
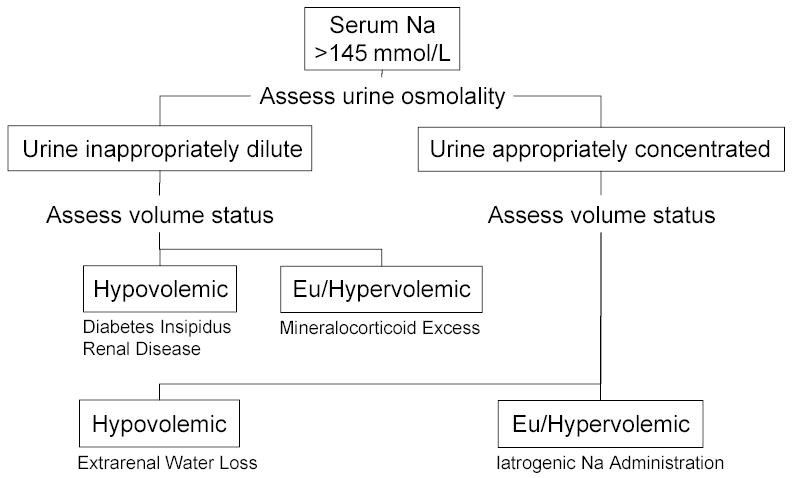
Algorithm for Assessment of the Hypernatremic Patient
Management of hypernatremia
The management of the hypernatremic patient is invariably water replacement and retention. Rarely are patients hypernatremic as a result of excessive sodium administration, but such patients will respond to a normalisation of intake. Conscious patients with CDI are able to increase oral intake and if this fails, or if urine output is greater than 250ml/hr, parenteral or intranasal vasopressin may be administered. Small doses, such as DDAVP 0.4 μg intravenously or 100–200 μg intranasally, minimize the risk of an over-prolonged action and can be repeated as required, dependent on clinical effect. In the unconscious patient, fluid replacement is best achieved with either intravenous 5% dextrose or water via a nasogastric tube with concomitant DDAVP administration. However, excessive intravenous fluid administration, given to replace high urinary output, may exacerbate the problem and accurate clinical assessment of volume status is required to guide treatment. Saline solutions may aggravate the renal water loss since urine concentration cannot be achieved in the absence of ADH. Over-rapid correction of hypernatremia may have serious consequences, most notably pulmonary and cerebral edema.59 A reduction in serum sodium concentration of 10 mmol/L per day has therefore been suggested, with a more rapid reduction only indicated in those who have developed hypernatremia over of a period of hours.54
Summary
Disturbances of sodium balance are common in the general in-patient hospital population but are more common in neurologic patients. A high index of suspicion should therefore be maintained in this patient group. In particular, the hyponatremic neurosurgical patient is at risk not only from the dysnatremia, but also from the consequences of over treatment. Asymptomatic patients can be managed successfully with a logical approach to diagnosis and a period of close monitoring. Treatment should be restricted to acute symptomatic patients in whom the associated mortality is significantly higher than the normal population.63
References
- 1.Levin ER, Gardner DG, Samson WK. Natriuretic peptides. N Engl J Med. 1998;339:321–328. doi: 10.1056/NEJM199807303390507. [DOI] [PubMed] [Google Scholar]
- 2.Palmer BF. Hyponatremia in patients with central nervous system disease: SIADH versus CSW. Trends Endocrinol Metab. 2003;14:182–187. doi: 10.1016/s1043-2760(03)00048-1. [DOI] [PubMed] [Google Scholar]
- 3.Rabinstein AA, Wijdicks EF. Hyponatremia in critically ill neurological patients. Neurologist. 2003;9:290–300. doi: 10.1097/01.nrl.0000095258.07720.89. [DOI] [PubMed] [Google Scholar]
- 4.Pollock AS, Arieff AI. Abnormalities of cell volume regulation and their functional consequences. Am J Physiol. 1980;239:F195–F205. doi: 10.1152/ajprenal.1980.239.3.F195. [DOI] [PubMed] [Google Scholar]
- 5.Arieff AI, Llach F, Massry SG. Neurological manifestations and morbidity of hyponatremia: correlation with brain water and electrolytes. Medicine (Baltimore) 1976;55:121–129. doi: 10.1097/00005792-197603000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Anderson RJ. Hospital-associated hyponatremia. Kidney Int. 1986;29:1237–1247. doi: 10.1038/ki.1986.134. [DOI] [PubMed] [Google Scholar]
- 7.Arieff AI. Management of hyponatraemia. BMJ. 1993;307:305–308. doi: 10.1136/bmj.307.6899.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fried LF, Palevsky PM. Hyponatremia and hypernatremia. Med Clin North Am. 1997;81:585–609. doi: 10.1016/s0025-7125(05)70535-6. [DOI] [PubMed] [Google Scholar]
- 9.Baylis PH. The syndrome of inappropriate antidiuretic hormone secretion. Int J Biochem Cell Biol. 2003;35:1495–1499. doi: 10.1016/s1357-2725(03)00139-0. [DOI] [PubMed] [Google Scholar]
- 10.Soupart A, Decaux G. Therapeutic recommendations for management of severe hyponatremia: current concepts on pathogenesis and prevention of neurologic complications. Clin Nephrol. 1996;46:149–169. [PubMed] [Google Scholar]
- 11.Reeder RF, Harbaugh RE. Administration of intravenous urea and normal saline for the treatment of hyponatremia in neurosurgical patients. J Neurosurg. 1989;70:201–206. doi: 10.3171/jns.1989.70.2.0201. [DOI] [PubMed] [Google Scholar]
- 12.Hasan D, Wijdicks EF, Vermeulen M. Hyponatremia is associated with cerebral ischemia in patients with aneurysmal subarachnoid hemorrhage. Ann Neurol. 1990;27:106–108. doi: 10.1002/ana.410270118. [DOI] [PubMed] [Google Scholar]
- 13.Atchison JW, Wachendorf J, Haddock D, et al. Hyponatremia-associated cognitive impairment in traumatic brain injury. Brain Inj. 1993;7:347–352. doi: 10.3109/02699059309034961. [DOI] [PubMed] [Google Scholar]
- 14.Hohenegger M. Problems of electrolyte metabolism in meningitis and encephalitis. Hyponatremias and the cerebral salt-losing syndrome. Wien Med Wochenschr. 1967;117:882–884. [PubMed] [Google Scholar]
- 15.Steele A, Gowrishankar M, Abrahamson S, et al. Postoperative hyponatremia despite near-isotonic saline infusion: a phenomenon of desalination. Ann Intern Med. 1997;126:20–25. doi: 10.7326/0003-4819-126-1-199701010-00003. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz WB, Bennett W, Curelop S, et al. A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone. 1957. J Am Soc Nephrol. 2001;12:2860–2870. doi: 10.1681/ASN.V12122860. [DOI] [PubMed] [Google Scholar]
- 17.Smith D, Moore K, Tormey W, et al. Downward resetting of the osmotic threshold for thirst in patients with SIADH. Am J Physiol Endocrinol Metab. 2004;287:E1019–E1023. doi: 10.1152/ajpendo.00033.2004. [DOI] [PubMed] [Google Scholar]
- 18.Kamoi K, Toyama M, Takagi M, et al. Osmoregulation of vasopressin secretion in patients with the syndrome of inappropriate antidiuresis associated with central nervous system disorders. Endocr J. 1999;46:269–277. doi: 10.1507/endocrj.46.269. [DOI] [PubMed] [Google Scholar]
- 19.Amini A, Schmidt MH. Syndrome of inappropriate secretion of antidiuretic hormone and hyponatremia after spinal surgery. Neurosurg Focus. 2004;16:E10. doi: 10.3171/foc.2004.16.4.11. [DOI] [PubMed] [Google Scholar]
- 20.Peters J, Welt L, Sims E, et al. A salt-wasting syndrome associated with cerebral disease. Trans Assoc Am Physicians. 1950;63:57–64. [PubMed] [Google Scholar]
- 21.Cort JH. Cerebral salt wasting. Lancet. 1954;266:752–754. doi: 10.1016/s0140-6736(54)92715-4. [DOI] [PubMed] [Google Scholar]
- 22.Betjes MG. Hyponatremia in acute brain disease: the cerebral salt wasting syndrome. Eur J Intern Med. 2002;13:9–14. doi: 10.1016/s0953-6205(01)00192-3. [DOI] [PubMed] [Google Scholar]
- 23.Diringer M, Ladenson PW, Stern BJ, et al. Plasma atrial natriuretic factor and subarachnoid hemorrhage. Stroke. 1988;19:1119–1124. doi: 10.1161/01.str.19.9.1119. [DOI] [PubMed] [Google Scholar]
- 24.Putensen C, Mutz N, Pomaroli A, et al. Atrial natriuretic factor release during hypovolemia and after volume replacement. Crit Care Med. 1992;20:984–989. doi: 10.1097/00003246-199207000-00014. [DOI] [PubMed] [Google Scholar]
- 25.Berendes E, Walter M, Cullen P, et al. Secretion of brain natriuretic peptide in patients with aneurysmal subarachnoid haemorrhage. Lancet. 1997;349:245–249. doi: 10.1016/s0140-6736(96)08093-2. [DOI] [PubMed] [Google Scholar]
- 26.Oh MS, Carroll HJ. Disorders of sodium metabolism: hypernatremia and hyponatremia. Crit Care Med. 1992;20:94–103. doi: 10.1097/00003246-199201000-00021. [DOI] [PubMed] [Google Scholar]
- 27.Zafonte RD, Mann NR. Cerebral salt wasting syndrome in brain injury patients: a potential cause of hyponatremia. Arch Phys Med Rehabil. 1997;78:540–542. doi: 10.1016/s0003-9993(97)90173-8. [DOI] [PubMed] [Google Scholar]
- 28.Maesaka JK, Gupta S, Fishbane S. Cerebral salt-wasting syndrome: does it exist? Nephron. 1999;82:100–109. doi: 10.1159/000045384. [DOI] [PubMed] [Google Scholar]
- 29.Harrigan MR. Cerebral salt wasting syndrome: a review. Neurosurgery. 1996;38:152–160. doi: 10.1097/00006123-199601000-00035. [DOI] [PubMed] [Google Scholar]
- 30.Isotani E, Suzuki R, Tomita K, et al. Alterations in plasma concentrations of natriuretic peptides and antidiuretic hormone after subarachnoid hemorrhage. Stroke. 1994;25:2198–2203. doi: 10.1161/01.str.25.11.2198. [DOI] [PubMed] [Google Scholar]
- 31.Weinand ME, O'Boynick PL, Goetz KL. A study of serum antidiuretic hormone and atrial natriuretic peptide levels in a series of patients with intracranial disease and hyponatremia. Neurosurgery. 1989;25:781–785. doi: 10.1097/00006123-198911000-00014. [DOI] [PubMed] [Google Scholar]
- 32.Lolin Y, Jackowski A. Hyponatraemia in neurosurgical patients: diagnosis using derived parameters of sodium and water homeostasis. Br J Neurosurg. 1992;6:457–466. doi: 10.3109/02688699208995035. [DOI] [PubMed] [Google Scholar]
- 33.Nelson PB, Seif SM, Maroon JC, et al. Hyponatremia in intracranial disease: perhaps not the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) J Neurosurg. 1981;55:938–941. doi: 10.3171/jns.1981.55.6.0938. [DOI] [PubMed] [Google Scholar]
- 34.Ayus JC, Krothapalli RK, Arieff AI. Changing concepts in treatment of severe symptomatic hyponatremia. Rapid correction and possible relation to central pontine myelinolysis. Am J Med. 1985;78:897–902. doi: 10.1016/0002-9343(85)90209-8. [DOI] [PubMed] [Google Scholar]
- 35.Ogawasara K, Kinouchi H, Nagamine Y, et al. Differential diagnosis of hyponatremia following subarachnoid hemorrhage. No Shinkei Geka. 1998;26:501–505. [PubMed] [Google Scholar]
- 36.Chung HM, Kluge R, Schrier RW, et al. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;83:905–908. doi: 10.1016/0002-9343(87)90649-8. [DOI] [PubMed] [Google Scholar]
- 37.Damaraju SC, Rajshekhar V, Chandy MJ. Validation study of a central venous pressure-based protocol for the management of neurosurgical patients with hyponatremia and natriuresis. Neurosurgery. 1997;40:312–316. doi: 10.1097/00006123-199702000-00015. [DOI] [PubMed] [Google Scholar]
- 38.Doczi T, Tarjanyi J, Huszka E, et al. Syndrome of inappropriate secretion of antidiuretic hormone (SIADH) after head injury. Neurosurgery. 1982;10:685–688. doi: 10.1227/00006123-198206010-00001. [DOI] [PubMed] [Google Scholar]
- 39.Born JD, Hans P, Smitz S, et al. Syndrome of inappropriate secretion of antidiuretic hormone after severe head injury. Surg Neurol. 1985;23:383–387. doi: 10.1016/0090-3019(85)90212-5. [DOI] [PubMed] [Google Scholar]
- 40.Iwasa H, Yamada T, Nakahara N, et al. Marked effect of furosemide and hypertonic saline in the treatment of SIAD after head injury. No Shinkei Geka. 1984;12:651–655. [PubMed] [Google Scholar]
- 41.Finsterer U, Beyer A, Jensen U, et al. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH)--treatment with lithium. Intensive Care Med. 1982;8:223–229. doi: 10.1007/BF01694525. [DOI] [PubMed] [Google Scholar]
- 42.Forrest JN, Jr, Cox M, Hong C, et al. Superiority of demeclocycline over lithium in the treatment of chronic syndrome of inappropriate secretion of antidiuretic hormone. N Engl J Med. 1978;298:173–177. doi: 10.1056/NEJM197801262980401. [DOI] [PubMed] [Google Scholar]
- 43.Fox JL, Falik JL, Shalhoub RJ. Neurosurgical hyponatremia: the role of inappropriate antidiuresis. J Neurosurg. 1971;34:506–514. doi: 10.3171/jns.1971.34.4.0506. [DOI] [PubMed] [Google Scholar]
- 44.Hasan D, Lindsay KW, Wijdicks EF, et al. Effect of fludrocortisone acetate in patients with subarachnoid hemorrhage. Stroke. 1989;20:1156–1161. doi: 10.1161/01.str.20.9.1156. [DOI] [PubMed] [Google Scholar]
- 45.Ishikawa SE, Saito T, Kaneko K, et al. Hyponatremia responsive to fludrocortisone acetate in elderly patients after head injury. Ann Intern Med. 1987;106:187–191. doi: 10.7326/0003-4819-106-2-187. [DOI] [PubMed] [Google Scholar]
- 46.Morinaga K, Hayashi S, Matsumoto Y, et al. Therapeutic effect of a mineralocorticoid in patients with hyponatremia of central origin. No To Shinkei. 1995;47:671–674. [PubMed] [Google Scholar]
- 47.Karp BI, Laureno R. Pontine and extrapontine myelinolysis: a neurologic disorder following rapid correction of hyponatremia. Medicine (Baltimore) 1993;72:359–373. [PubMed] [Google Scholar]
- 48.Soupart A, Ngassa M, Decaux G. Therapeutic relowering of the serum sodium in a patient after excessive correction of hyponatremia. Clin Nephrol. 1999;51:383–386. [PubMed] [Google Scholar]
- 49.Snyder NA, Feigal DW, Arieff AI. Hypernatremia in elderly patients. A heterogeneous, morbid, and iatrogenic entity. Ann Intern Med. 1987;107:309–319. doi: 10.7326/0003-4819-107-2-309. [DOI] [PubMed] [Google Scholar]
- 50.Palevsky PM, Bhagrath R, Greenberg A. Hypernatremia in hospitalized patients. Ann Intern Med. 1996;124:197–203. doi: 10.7326/0003-4819-124-2-199601150-00002. [DOI] [PubMed] [Google Scholar]
- 51.Polderman KH, Schreuder WO, Strack van Schijndel RJ, et al. Hypernatremia in the intensive care unit: an indicator of quality of care? Crit Care Med. 1999;27:1105–1108. doi: 10.1097/00003246-199906000-00029. [DOI] [PubMed] [Google Scholar]
- 52.Moder KG, Hurley DL. Fatal hypernatremia from exogenous salt intake: report of a case and review of the literature. Mayo Clin Proc. 1990;65:1587–1594. doi: 10.1016/s0025-6196(12)62194-6. [DOI] [PubMed] [Google Scholar]
- 53.Herfel R, Stone CK, Koury SI, et al. Iatrogenic acute hyponatraemia in a college athlete. Br J Sports Med. 1998;32:257–258. doi: 10.1136/bjsm.32.3.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adrogue HJ, Madias NE. Hypernatremia. N Engl J Med. 2000;342:1493–1499. doi: 10.1056/NEJM200005183422006. [DOI] [PubMed] [Google Scholar]
- 55.Morello JP, Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol. 2001;63:607–630. doi: 10.1146/annurev.physiol.63.1.607. [DOI] [PubMed] [Google Scholar]
- 56.Sasaki S. Nephrogenic diabetes insipidus: update of genetic and clinical aspects. Nephrol Dial Transplant. 2004;19:1351–1353. doi: 10.1093/ndt/gfh172. [DOI] [PubMed] [Google Scholar]
- 57.Poon WS, Lolin YI, Yeung TF, et al. Water and sodium disorders following surgical excision of pituitary region tumours. Acta Neurochir (Wien ) 1996;138:921–927. doi: 10.1007/BF01411280. [DOI] [PubMed] [Google Scholar]
- 58.Agha A, Thornton E, O'Kelly P, et al. Posterior pituitary dysfunction after traumatic brain injury. J Clin Endocrinol Metab. 2004;89:5987–5992. doi: 10.1210/jc.2004-1058. [DOI] [PubMed] [Google Scholar]
- 59.Wong MF, Chin NM, Lew TW. Diabetes insipidus in neurosurgical patients. Ann Acad Med Singapore. 1998;27:340–343. [PubMed] [Google Scholar]
- 60.Smith M. Physiological changes during brain stem death - lessons for management of the organ donor. J Heart Lung Transplantation. 2004;23:S217–22. doi: 10.1016/j.healun.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 61.Shucart WA, Jackson I. Management of diabetes insipidus in neurosurgical patients. J Neurosurg. 1976;44:65–71. doi: 10.3171/jns.1976.44.1.0065. [DOI] [PubMed] [Google Scholar]
- 62.Kawai K, Aoki M, Nakayama H, et al. Posterior pituitary hematoma in a case of posttraumatic diabetes insipidus. Case report J Neurosurg. 1995;83:368–371. doi: 10.3171/jns.1995.83.2.0368. [DOI] [PubMed] [Google Scholar]
- 63.Roca-Ribas F, Ninno JE, Gasperin A, et al. Cerebral salt wasting syndrome as a postoperative complication after surgical resection of acoustic neuroma. Otol Neurotol. 2002;23:992–995. doi: 10.1097/00129492-200211000-00030. [DOI] [PubMed] [Google Scholar]
- 64.Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342:1581–1589. doi: 10.1056/NEJM200005253422107. [DOI] [PubMed] [Google Scholar]
- 65.Oh MS and Carroll HJ.Regulation of intracellular and extracellular volume. In: Arieff, A. I. and DeFronzo, R. A., eds. Fluid, electrolyte and acid-base disorders 2nd ed.New York: Churchill Livingstone, 1995:1–28.