

Abstract
Wild emmer wheat, Triticum dicoccoides, is the progenitor of modern tetraploid and hexaploid cultivated wheats. Our objective was to map domestication-related quantitative trait loci (QTL) in T. dicoccoides. The studied traits include brittle rachis, heading date, plant height, grain size, yield, and yield components. Our mapping population was derived from a cross between T. dicoccoides and Triticum durum. Approximately 70 domestication QTL effects were detected, nonrandomly distributed among and along chromosomes. Seven domestication syndrome factors were proposed, each affecting 5–11 traits. We showed: (i) clustering and strong effects of some QTLs; (ii) remarkable genomic association of strong domestication-related QTLs with gene-rich regions; and (iii) unexpected predominance of QTL effects in the A genome. The A genome of wheat may have played a more important role than the B genome during domestication evolution. The cryptic beneficial alleles at specific QTLs derived from T. dicoccoides may contribute to wheat and cereal improvement.
From agriculture origins in the Near East 10,000 years ago, emmer was the principal wheat of the newly established farming settlements, subsequently spreading 7,000 years ago to Egypt, India, and Europe (1). Wild emmer wheat, Triticum dicoccoides, genome AABB, is a tetraploid, a predominantly self-pollinating wild progenitor of modern tetraploid and hexaploid cultivated wheats (2, 3). T. dicoccoides proved important in wheat breeding in the past, and it deserves in-depth study as a potential genetic resource for cereal improvement (1).
Analysis of quantitative trait (QT) loci (QTL) of domestication-related traits was studied in rice (4), maize (5), sorghum (6, 7), and millet (8). In recent years, QTLs were mapped in hexaploid wheat for agronomic traits (9, 10). To date, QTLs have been reported for tetraploid wheat mainly for quality traits (11, 12) but not for domestication traits. Grass domestication evolution is the key to exploiting genome diversity for future cereal improvement (13). Therefore, the objective of this study was to map genetic loci underlying domestication-related traits, including brittle rachis (Br), heading date (HD), plant height (HT), grain size, yield, yield components, and the genomic distribution pattern of domestication QTLs in T. dicoccoides.
Materials and Methods
Mapping Population.
A cross was made between a highly stripe-rust-resistant T. dicoccoides accession, Hermon H52 (H52) (1), with typical wild traits, from Mt. Hermon, Israel, and a Triticum durum cultivar, Langdon (Ldn). The F2 mapping population consisted of 150 genotypes. Leaf samples were collected in the greenhouse at the elongation stage, frozen in liquid nitrogen, and stored at –80°C until DNA isolation (14). The F2 individuals were bagged to ensure selfing for producing F3 families.
Phenotyping of Domestication-Related Traits.
A randomized block design field experiment was conducted in Neve Yaar, Israel, from 1997 to 1998. The trial totaled 162 single-row plots, 150 for the F3 families, and 6 for each of the parents. QTs were measured on F3 progenies of F2 plants genotyped for molecular markers (15). Eleven traits were scored for 10 individual F3 plants from each plot per genotype: HT (centimeters); HD; spike number/plant; spike weight/plant (grams), including grains, hulls, and rachis; single spike weight (grams); kernel number/plant; kernel number/spike; kernel number/spikelet; 100-grain weight (GWH, grams); grain yield/plant (YLD, grams); and spikelet number/spike. Br was qualitatively and individually scored.
Molecular Framework Map.
Genotyping was conducted by using standard molecular markers, i.e., microsatellite, or short sequence repeat, amplified fragment length polymorphism, random amplified polymorphic DNA, and restriction fragment length polymorphism (15). Two versions of genetic maps were constructed for each chromosome by mapmaker 3.0b (16) due to the paucity of linkage information from repulsively linked dominant markers (17) and verified by our own algorithm (18). These two maps consist mostly of dominant markers linked in the coupling phase and of codominant markers. The H version map consists mainly of dominant markers with the dominant allele from T. dicoccoides H52. The L version map consists mainly of dominant markers with the dominant allele from Ldn. The codominant markers appear in both maps (15). The map distances in the framework maps were computed by using the haldane mapping function (16). To reduce the effect of missing information caused by the abundance of dominant markers (about 2/3 in our mapping population) on mapping efficiency, marker “virtual restoration” procedure of the multiqtl software (http://esti.haifa.ac.il/∼poptheor/) was used. We calculated the probabilities of heterozygote and dominant homozygote for each dominant marker phenotype, based on available scores of nearest-neighbor markers, and these probabilities served in calculating the likelihood function.
QTL Detection.
To detect QTLs, the entire genome was scanned for each of the QTs by using the general interval mapping approach (19, 20). The analysis was conducted by using maximum likelihood algorithms of multiqtl (19–21). Before conducting the QTL analysis, the initial trait distributions were transformed to reduce the effect of strong deviation from normality on the mapping results. The detected QTLs were graphically presented by mapchart software (22).
Single-QTL Model.
In this case, the model can be presented as
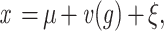 |
where g = (QQ, qq, or Qq) and v(QQ) = d, v(qq) = −d, v(Qq) = h, and ξ is a random value with distribution N(0, σ2). Therefore, mean values for the QTL groups can be represented as ExQQ = μ + d, Exqq = μ − d, ExQq = μ + h, where d is the additive effect (hence D = 2d = XQQ − Xqq is the allele substitution effect at a QTL Q/q for trait X), and h is the dominance effect. On the basis of the relative magnitude of additive and dominance effects, a suitable simplified genetic model [“pure additive” (h = 0), “full dominance” (h = d) of T. dicoccoides QTL alleles, or recessive (h = −d) effects, or “general model” (h deviates significantly from 0, d and −d)] was chosen for each of the putative QTLs. A standard permutation test (23) was used to obtain chromosome-wise statistical significance of the putative QTL when the single-QTL model was fitted. Based on 10,000 runs per chromosome, the thresholds of the test statistics were obtained for bootstrap analysis aimed at evaluating interval estimates of the main parameters (QTL effect, its chromosomal position, and the proportion of explained phenotypic variation).
Linked-QTL Model.
The single-QTL logarithm of odds (LOD) graphs for several chromosomes indicated the possibility of two QTLs per chromosome. This pattern was similar for different traits and paralleled in the two versions of the QTL maps, H and L. Therefore, a model with linked QTLs was fitted for each trait-chromosome combination (24). The hypothesis of two-linked QTLs (H2) could be compared with two alternatives, H1 (only one QTL in the chromosome affecting the trait) and H0 (the chromosome has no effect on the trait). Most difficult is comparing H2 vs. H1. For this reason, two approaches conditioned on the selected model can be used with multiqtl tools: (i) Monte Carlo simulations and (ii) bootstrap analysis. For the first approach, in addition to the linked-QTL model, a single-QTL model is also fitted. Then, the obtained maximum likelihood estimates of the single-QTL model are used to generate a large number (1,000) of Monte Carlo data sets. For each set, the program fits both the linked- and single-QTL models. The LOD difference of these models is compared with that obtained for the real data. If it exceeds the LOD difference of real data, the “overcomes” number increases by one. Significance is calculated as the ratio of the “overcomes” number to the total number of runs. One thousand bootstrap samples were analyzed by using the model fitted to the initial data. The two-linked-QTL model was used if the effect of each QTL (either additive or heterozygous) was at least 2-fold compared with its standard deviation calculated across the bootstrap runs. Computer simulations indicate that these two tests give similar results, albeit the second test seems to be more conservative for relatively small sample sizes and QTL effects (not shown).
Genome-Wise Statistical Significance.
For effects to be declared significant on the chromosome-trait basis, a correction for multiple comparisons should be done, recalling that the experiment included 14 × 11 = 154 chromosome-trait combinations. The approach based on controlling the false discovery rate (FDR) (25) was used to set significance levels. Critical chromosome-trait error rates (P values), estimated by the permutation tests for FDR = 0.05 and FDR = 0.1, were P < 0.011 and P < 0.032 for map version H, and P < 0.0085 and P < 0.026 for version L, respectively (see Tables 3 and 4, which are published as supporting information on the PNAS web site, www.pnas.org.).
Results
Phenotypic Distribution of Domestication-Related Traits.
The wild parent, T. dicoccoides H52, demonstrated traits typical of a wild marginal population: short stature (HT = 64 cm), late flowering date (HD = 148 days), small seeds (GWH = 1.0 g), poor yield (YLD = 0.48 g), and yield components. By contrast, the domesticated parent, T. durum cv. Ldn, possessed taller stature (HT = 117 cm), early flowering date (HD = 143 days), big seed (GWH = 3.0 g), good yield (YLD = 8.2 g), and yield components. Mean offspring values for most observed traits (10/11) were between the wild and domesticated parents. Segregation of the F3 family means was observed for all 11 traits in the F2 mapping population with the coefficient of variation (CV) ranging from 2.7% to 50.9%. All traits, except HD (CV = 2.7%), showed high variability. In particular, spike number/plant, spike weight/plant, single spike weight, kernel number/spike, YLD, and spikelet number/spike were highly variable, with CV >20%. Transgressive segregation was observed for all traits except GWH.
Significance at the Chromosome-Trait and Experimental-Wise Levels.
Single- and linked-QTL analyses and the permutation test revealed 75, 50, and 39 QTL effects at P = 5%, 1%, and 0.1% by using the H version map; and 76, 52, and 42 QTLs by using the L version map (of total 14 × 11 = 154 chromosome-trait combinations). The number of chromosomes with QTL effects for the specific traits ranged from two (for HT) to eight (for GWH). The number of QTL effects for the specific chromosomes ranged from zero (for chromosomes 1A, 3B, and 6A) to 19 (for chromosome 5A), and more than half of the 75 and 76 significant effects were located on chromosomes 1B, 2A, and 5A (Table 1). However, the above test of the significance of individual chromosome-trait combinations ignores multiple comparisons caused by multiple traits and chromosomes. Thus, the FDR correction was applied (25). This procedure resulted in the reduction of the numbers of detected QTLs to 22 and 24 for the H and L version maps, respectively. Namely, these QTLs were not significant at FDR = 5% (for FDR = 10% the reduction was for five and six effects, respectively). However, most of these nonsignificant QTLs (using FDR = 5% or 10%) proved reproducible between the H and L version maps. Moreover, a part of the weak QTL effects displayed overlapping location with strong QTL effects for other traits rather than random distribution across the genome. Therefore, only the QTL effects that were not significant in the FDR test and were not reproducible between the H and L version maps were excluded.
Table 1.
Distribution of detected domestication QTLs among genomes and chromosomes
| Chromosome | Single
|
Linked
|
S
+ L
|
|||
|---|---|---|---|---|---|---|
| H | L | H | L | H | L | |
| 1A | 0 | 0 | 0 | 0 | 0 | 0 |
| 2A | 5 (6) | 5 (6) | 8 | 8 | 13 | 13 |
| 3A | 4 (6) | 3 (6) | 0 | 0 | 4 | 3 |
| 4A | 1 | 1 | 0 | 0 | 1 | 1 |
| 5A | 0 (3) | 0 (3) | 16 | 16 | 16 | 16 |
| 6A | 0 | 0 | 0 | 0 | 0 | 0 |
| 7A | 1 (3) | 0 (1) | 2 (4) | 2 (4) | 3 | 2 |
| SubT | 11 | 9 | 26 | 26 | 37 | 35 |
| SubT (adj) | 19 | 17 | 28 | 28 | ||
| X2-c | 17.2** | 18.2** | 56.5** | 56.5** | 49.9** | 52.1** |
| X2-c (adj) | 18.2** | 19.4** | 55.4** | 55.4** | ||
| 1B | 2 (4) | 5 | 8 | 8 | 10 | 13 |
| 2B | 0 (1) | 0 (1) | 0 | 0 | 0 | 0 |
| 3B | 0 | 0 | 0 | 0 | 0 | 0 |
| 4B | 0 (1) | 0 (1) | 0 | 0 | 0 | 0 |
| 5B | 1 (4) | 1 (4) | 2 | 2 | 3 | 3 |
| 6B | 1 (2) | 0 (2) | 2 (4) | 0 (4) | 3 | 0 |
| 7B | 0 (2) | 1 (2) | 0 | 0 (2) | 0 | 1 |
| SubT | 4 | 7 | 12 | 10 | 16 | 17 |
| SubT (adj) | 14 | 15 | 14 | 16 | ||
| X2-c | 7.2 | 16.1* | 25.9** | 28.9** | 32.8** | 43.17** |
| X2-c (adj) | 8.3 | 9.9 | 27.7** | 23.5** | ||
| Total | 15 | 16 | 38 | 36 | 53 | 52 |
| Total (adj) | 33 | 32 | 42 | 44 | ||
| X2-g | 3.4† | 0.3 | 5.28* | 7.37** | 8.55** | 6.36* |
Single, number of QTLs detected by single-QTL analysis; Linked, number of QTLs detected by linked-QTL analysis; S + L, summation of Single and Linked QTLs. The numbers in brackets are the adjusted QTL numbers. X2-g, χ2 value for the between-genome comparison; X2-c, χ2 value for between-chromosome comparisons within each genome.
,
,
,
, significance at the levels of 0.10, 0.05, 0.01, and 0.001, respectively. adj, adjusted; the data in the corresponding columns or rows were obtained by using the adjusted numbers of QTLs by taking into account the weak (insignificant by FDR test) but reproducible QTLs between the two versions of maps and coinciding with other (significant) QTLs.
Modes of QTL Action for Domestication-Related Traits.
As shown in Table 1, for the H version map, 21 and 33 of the 54 trait-chromosome effects fitted the linked- and single-QTL models, respectively; similarly, for the L version, this proportion was 22 and 32, respectively. Of the 76 detected effects in the L version map, 15, 35, 11, and 12 followed the additive, dominance, recessive, and heterotic modes, respectively (relative to the T. dicoccoides allele) (Table 3). Corresponding proportions for the 75 detected effects in the H version map were 19, 29, 11, and 14, respectively (not shown). Therefore, nearly 80% of the detected QTL effects fitted the additive, dominant, or recessive modes of action of the T. dicoccoides QTL allele. Using arbitrary thresholds of 10% and 25% of percentage explained variance (PEV) to delineate “minor,” “intermediate,” and “major” QTLs (26), 35 major QTL effects with PEV >25%, LOD >3.0, and FDR = 5.0% were detected by the L version map (Table 3).
Genomic Distribution of QTLs for Domestication Traits.
The map positions of the QTLs for various traits on the same chromosome were highly overlapped (Table 3). Fig. 1 and Table 4 show that most of the significant QTLs, referred to as domestication syndrome factors (DSFs), are clustered in limited intervals of chromosomes 1B, 2A, 3A, and 5A (see also Table 3). Table 1 indicates that the map-version differences for the number of QTL effects uncovered on the levels of chromosome, genome, and the entire genome were all nonsignificant. For almost all domestication traits, the number of QTL effects mapped on the A genome exceeded or equaled that on the B genome (not shown). Despite the high number of significant QTLs, they are highly clustered on the maps. Consequently, the total number of intervals of the QTL locations was 16 in each version of the map; most of these clusters appear in pairs or triplets within the corresponding chromosomes (1B, 2A, 5A, 5B, 6B, 7A).
Figure 1.

Map locations of DSFs and their involved QTLs in L version maps of wild
emmer wheat, T. dicoccoides. (Upper)
Short arms of chromosomes. (Right) DSFs and
corresponding QTLs:
 , DSF;
█, KNS;
, DSF;
█, KNS;
 , kernel
number/spikelet (KNL); ▥, YLD;
, kernel
number/spikelet (KNL); ▥, YLD;
 , HT;
⊞, spikelet number/spike (SLS);
, HT;
⊞, spikelet number/spike (SLS);
 , single
spike weight (SSW);
, single
spike weight (SSW);
 , spike
weight/plant (SWP);
, spike
weight/plant (SWP);
 , kernel
weight/plant (KNP);
, HD; ▧, GWH; ▨, spike
number/plant (SNP). The regular trait name represents a single QTL;
the italic trait name represents a single QTL (Q2) detected by
linked-QTL analysis; the regular trait name tailed with Q1 means the
first QTL and tailed with Q2, the second QTL in a pair of linked QTLs.
A tailed trait name (5) means that the QTL effect is not significant at
the level of 5% of FDR but is significant at FDR 10%; (10) means that
the effect is not significant at FDR 10%.
, kernel
weight/plant (KNP);
, HD; ▧, GWH; ▨, spike
number/plant (SNP). The regular trait name represents a single QTL;
the italic trait name represents a single QTL (Q2) detected by
linked-QTL analysis; the regular trait name tailed with Q1 means the
first QTL and tailed with Q2, the second QTL in a pair of linked QTLs.
A tailed trait name (5) means that the QTL effect is not significant at
the level of 5% of FDR but is significant at FDR 10%; (10) means that
the effect is not significant at FDR 10%.
Discussion
Our discussion covers (i) clustering of domestication QTLs of wild emmer wheat and a comparison with other cereals; (ii) cryptic alleles; (iii) DSFs in genomic gene rich regions; and (iv) unexpected predominance of the major QTL effects in the A genome.
Domestication QTLs in Wild Emmer: Comparison with Other Cereals.
Br-related factors.
Br is a typical characteristic of wild cereals including T. dicoccoides. The breakage of rachis sheds seeds at maturity. This trait is agriculturally deleterious; hence, transformation of Br to non-Br is the first symbol of domestication. In this study, the Br of T. dicoccoides was controlled by a dominant gene (Br) and mapped on the long arm of chromosome 2A (Fig. 1). Remarkably, in an interval of 32.2 cM between this gene and a microsatellite marker, Xgwm294, a cluster of eight major linked domestication QTLs (with LOD of 3.7–9.3) was also mapped. Possibly, the domestication syndrome factor involving these eight QTL effects is tightly linked to the Br gene and/or is the result of a pleiotropic effect of Br. Br in T. dicoccoides functions as an abscission layer in millet (8), seed dispersal in sorghum and maize, and seed shedding in rice (6).
Seed size.
Seed size was strongly selected in all domesticated cereals: wheat, barley, oats, and rye in the Near East; maize in America; rice in Asia; and sorghum and millet in Africa (6, 13). We mapped eight QTLs for GWH on chromosomes 1B, 2A, 4A, 5A, 5B, 6B, 7A, and 7B. Major GWH QTLs were located on chromosomes 2A, 4A, and 5B with LOD > 3.7 and P ≤ 0.001. Two of the seed-size QTLs are involved in the proposed DSFs, but two major GWH QTLs (LOD = 4.7–6.1, P ≤ 0.001) on chromosomes 4A seem to be independent from the DSFs (Table 4). Three major seed-size QTLs correspond closely in sorghum, rice, and maize, and another five QTLs correspond between two of these genera when the taxa are compared in a pairwise fashion (6). Eight rice seed-size QTLs were mapped to chromosomes 2, 3, 4, 5, 8, 9, 11, and 12 (4). Parallel synteny existing between wheat and rice chromosomes (27) indicates that all detected seed-size QTLs in T. dicoccoides correspond to their rice counterparts.
Developmental timing.
Flowering time was also selected in the major cereals. Short-day flowering wild grasses were transformed into domesticates in which flowering time was unaffected by day length (13). HD/flowering time is an important criterion for regional adaptation and yield in all cereals (28). In our study, the wild parent, T. dicoccoides H52, was sensitive to day length and HD, flowering later than the cultivar Ldn. Four HD QTLs were mapped on chromosomes 2A, 4B, 5A, and 6B (Table 3). On the basis of the map positions, we postulated that HD QTL on 5A is involved in DSF7, and HD QTL on 2A is involved in DSF2 (Fig. 1). The wild allele for the QTL on 5A will increase the value of HD and so is responsible for the late flowering of T. dicoccoides, whereas the wild HD alleles on chromosomes 2A, 4B, and 6B can accelerate the flowering date (Table 3). These “earliness” alleles, plus the early genes from the T. durum cultivar, might explain the significant transgressive segregation (the majority of the individuals were earlier than the early parent Ldn) for HD in the mapping population.
Yield.
Primary domestication targets were likely the genes that facilitated harvesting and enabled colonization of new environments. Yield must have soon assumed priority, minimizing labor input and land needs (13). The wild parent in our mapping population had a very poor yield (YLD = 0.5 g vs. YLD = 8.2 g in the domesticated parent). Five yield QTLs were mapped on chromosomes 1B, 2A, 3A, 5A, and 5B. Three yield QTLs on chromosomes 1B, 2A, and 5A are highly significant (LOD = 5.5–10.1, P < 0.001). The eight yield QTLs overlapped with QTLs for other traits on chromosomes 1B, 2A, 3A, and 5A (Fig. 1), incorporated in various DSFs on different chromosomes or chromosomal intervals.
Cryptic Alleles.
Soller and Beckmann (29) pointed out that QTL mapping could uncover “cryptic” genetic variation (beneficial alleles) that is otherwise hidden in a sea of deleterious alleles. Here, the wild H52, T. dicoccoides parent, possesses stripe-rust resistance (14), short stature, and high tillering capacity, with agriculturally deleterious features such as slow growth (late heading); small grains; few, small, and light spikes; and low yield. Nevertheless, among the 75 domestication QTL effects for 11 traits, wild QTL alleles of T. dicoccoides for 18 (24%) effects were agriculturally beneficial, e.g., contributing to short plant, early HD, more spike number/plant, higher spike weight/plant, more kernel number per spikelet, higher GWH, and higher yield (Table 3). Thus, these cryptic alleles together with genes for resistance or tolerance to biotic and abiotic stresses and high protein content (1) could advance utilization of T. dicoccoides for wheat improvement. Such favorable alleles for yield components were also found in wild relatives of other crops, like rice (4), pearl millet (8), and others.
DSFs.
DSFs in wheat and other cereals.
A general transition from small-seeded plants with natural seed dispersal to larger-seeded nonshattering plants until harvest applies to all seed crops. These parallels transcend the deepest divisions within the angiosperms, with both monocot and dicot crops developing a similar adaptive domestication syndrome to human cultivation over the last 10,000 years (30). Here, 4 of 14 chromosomes of tetraploid wheat harbored 80.4% of the 56 strong-to-moderate QTLs underlying the differences between wild T. dicoccoides and cultivated T. durum for 11 traits, clustered in seven intervals (Fig. 1). We propose seven DSFs, each involving a pleiotropic QTL or cluster of QTLs affecting 5–11 traits (see Table 3). Substantial clustering of domestication QTLs was revealed in sunflowers, and genomic regions influencing multiple traits harbored QTL with antagonistic effects (26). Morphological differences between maize and teosinte result from QTLs clustered in only five genomic regions (31). Independent domestication of sorghum, rice, and maize involved convergent selection for large seeds, nonshattering spikes, and day-length insensitive flowering. These similar phenotypes are largely determined by a small number of QTLs that closely resemble each other in the three taxa (6). Loci underlying morphological differences between pear millet and its wild ancestor are mainly concentrated in four linkage groups (8). Thus, our findings of a limited number of DSFs corroborate the results in other cereal crops showing that the domestication syndrome is under a relatively simple and rapidly evolving genetic control (6).
Linkage vs. pleiotropy.
We have previously demonstrated nonrandom distribution of molecular markers along chromosomes (15). This feature is prevalent in most mapping studies of wheat (32, 33) and in many other organisms (34). It has been attributed to the nonrandomness of crossover distribution along chromosomes, i.e., reduced recombination in the proximal regions and hotspots of recombination in the subtelomeric regions of chromosomes (34).
The mechanism for QTL clustering may be the same as for molecular markers, i.e., tight linkage. If so, the coincidental mapping of several QTLs derives both from the small sample size effect on mapping resolution and scant recombination within blocks of QTL genes. Pleiotropy of the underlying genes could provide alternative interpretation. Coupling tight linkage and pleiotropy could better explain the data, especially due to the high correlation between some traits. Factor analysis reveals that variation of the 11 traits can be represented by three “independent factors” accounting for 84% of the multiple trait variation (not shown). Thus, DSFs simultaneously affecting the traits that depend on more than one “independent factor” can indeed include tightly linked loci with pleiotropic effects on the considered traits. It is noteworthy that the signs of the effects of the DSFs on different traits corroborated quite well with the correlations between the traits (Table 4). Due to the small number (seven) of the revealed strong DSFs, it would be highly desirable to attempt further dissection of these factors by using special mapping designs based on accumulation of recombinants for the flanking markers and saturation of target intervals by additional markers (35).
DSFs and gene-rich regions.
Gene distribution in Triticeae (wheat and barley) chromosomes is highly nonrandom, with a few gene-rich regions alternating with gene-poor regions, as in other eukaryotes (36–41). Gene-rich regions correspond to hot spots of recombination (36–42). Remarkably, the map positions of all (!) seven major wheat DSFs appeared to overlap with gene-rich regions (Table 2). Therefore, the high pleiotropy and/or tight linkage of most wheat domestication QTLs suggest an important role of recombination in either consolidation of positive mutations within the DSF clusters (43) and/or in reducing the antagonism between artificial and background (purifying) selection (44). The presumed coincidence between DSFs and gene-rich regions could facilitate component dissection of these factors, their further fine mapping, and finally, map-based cloning.
Table 2.
Putative DSFs in wild emmer wheat, T. dicoccoides
| DSF | Chr. | Involved QTLs | Map location interval | Position, cM | Chr. reference* |
|---|---|---|---|---|---|
| DSF1 | 1B | KNP, YLD, GWH, SNP, SWP | Xgwm273a-Xgwm403a | 68.6 ± 8.8 (H) | 1BS (38) |
| 75.3 ± 10.6 (L) | |||||
| DSF2 | 1B | KNP, KNS, KNL, YLD, SNP, SLS, SWP, SSW | Xgwm124-P57M52u | 133.9 ± 22.8 (H) | 1BL (38) |
| 132.3 ± 19.4 (L) | |||||
| DSF3 | 2A | KNP, KNS, YLD, HD, SNP, SWP | Xgwm630c-Xgwm294 | 150.8 ± 31.4 (H) | 2AI (39) |
| 200.0 ± 28.2 (L) | |||||
| DSF4 | 2A | KNP, KNS, KNL, YLD, GWH, SLS, SSW, SWP | Xgwm294-Br | 216.8 ± 11.8 (H) | 2AL (39) |
| 252.1 ± 11.7 (L) | |||||
| DSF5 | 3A | KNP, KNS, KNL, YLD, SSW, SWP | Xgwm218-Xgwm638 | 148.1 ± 28.4 (H) | 3AS (40) |
| 99.7 ± 27.8 (L) | |||||
| DSF6 | 5A | KNP, KNS, KNL, YLD, HT, SLS, SSW, SWP | Xgwm154-P56M50m | 72.5 ± 12.6 (H) | 5AL (37, 41) |
| 78.8 ± 26.7 (L) | |||||
| DSF7 | 5A | All traits | Xgwm186-P56M53c | 144.2 ± 29.3 (H) | 5AL (41) |
| 160.2 ± 31.0 (L) |
DSF, domestication syndrome factor. Chr, chromosome. QTL is represented by the trait names, and italics indicate the QTLs revealed by linked-QTL analysis, otherwise by single-QTL analysis. The first figure indicates the distance from the top of the corresponding chromosome, and the second indicates the interval length.
Corresponding gene-rich region in breadwheat.
Relative Importance of A and B Genomes in Domestication.
For 10 of the 11 domestication QTs, the adjusted number of QTLs on the A genome equaled or exceeded that on the B genome. For H and L version maps, the numbers of QTL effects revealed on the A/B genome at the FDR = 5% level were 37/16 and 35/17, respectively, and the genome differences were significant at the level of P = 0.004 and 0.012, respectively. Overall, the number of QTL effects on the A genome exceeded that on the B genome (Table 1). Interparental PstI-based amplified fragment length polymorphisms (AFLP) (15) also showed that markers are not randomly distributed among A and B genomes of tetraploid wheat: ≈60% of polymorphic AFLP loci were mapped to the B genome. PstI restriction enzyme preferentially targets low-copy regions of the genome (probably because of the high G+C content of its recognition sites), and in maize genome, PstI-based AFLPs are preferentially located in the hypomethylated noncentromeric regions associated with genes (45). Likewise, higher polymorphism in the B than in the A genome applies to microsatellites (46) and restriction fragment length polymorphism markers (47) in common hexaploid wheat as well as in T. dicoccoides in Israel (48). These nonrandom patterns may mirror the genetic differentiation of structure and function among genomes and chromosomes between T. dicoccoides and T. durum during domestication. It is noteworthy that by screening the graingenes web site (http://wheat.pw.usda.gov/), we found that of 161 mapped wheat disease resistance genes, 75 were from B genomes (P < 0.008) (unpublished results).
A few other manifestations of asymmetry between the A and B genomes in polyploid wheat are worth mentioning. Previous molecular cytogenetic analysis conducted at the Institute of Evolution (University of Haifa) by using genomic in situ hybridization revealed a pattern that was interpreted as evidence of a process of enrichment of the A genome with repetitive sequences of the B genome type (49). Such genomic changes may provide the physical basis for several phenomena described in polyploid wheats, such as A genome gene silencing (50) or nonrandom (clustered) distribution of markers (putatively caused by peculiarities of recombination) that was characteristic of B, but not A, chromosomes (15). Experiments with synthetic allohexaploids of wheat show that interactions between the components of a polyploid genome leading to diploidization of meiotic chromosome behavior may also include very fast nonrandom sequence elimination (51). The pattern of elimination in synthetic allohexaploids was similar to that of the natural hexaploid wheat.
Supplementary Material
Acknowledgments
We are greatly indebted to T. Krugman at the Institute of Evolution, University of Haifa, for assistance in the experiments; to K. Wendehake at the Institute of Plant Genetics and Crop Plant Research, Gatersleben, Germany, for technical assistance in microsatellite genotyping; and to Dr. L. R. Joppa, Northern Crop Science Laboratory, Fargo, ND, for the F1 seeds of our mapping population. We express our deep gratitude to Barbara Schaal, Piet Stam, Olga Raskin, and Alex Belyayev for constructive comments that improved the paper. This work was supported by the European Molecular Biology Organization (Grant No. ASTF9195 to J.P.); the Israel Discount Bank Chair of Evolutionary Biology; the Ancell–Teicher Research Foundation for Genetics and Molecular Evolution; the Israeli Ministry of Science (Grant No. 5757-1-95); the Ministry of Absorption; the Israel Science Foundation (Grant No. 9048/99); the Graduate School of the University of Haifa; and the German–Israeli Project Cooperation [funded by the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie (BMBF) and supported by the BMBF's International Bureau at the DRL], DIP Project No. DIP-B 4.3.
Abbreviations
- QT
quantitative trait
- QTL
QT loci
- HT
plant height
- HD
heading date
- GWH
100-grain weight
- YLD
grain yield/plant
- Br
brittle rachis
- FDR
false discovery rate
- LOD
logarithm of odds
- DSF
domestication syndrome factor
- H52
Hermon H52
- Ldn
Langdon
References
- 1.Nevo E, Korol A B, Beiles A, Fahima T. Evolution of Wild Emmer and Wheat Improvement—Population Genetics, Genetic Resources, and Genome Organization of Wheat's Progenitor, Triticum dicoccoides. Berlin: Springer; 2002. [Google Scholar]
- 2.Zohary D. In: Genetic Resouces in Plants–Their Exploitation and Conservation. Frankel O H, Bennett E, editors. Edinburgh: Blackwell; 1970. pp. 239–247. [Google Scholar]
- 3.Feldman M, Sears E R. Sci Am. 1981;244:102–112. [Google Scholar]
- 4.Xiao J, Li J, Grandillo S, Sang N A, Yuan L, Tanksley S D, McCouch S R. Genetics. 1998;150:899–909. doi: 10.1093/genetics/150.2.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doebley J, Stec A, Hubbard L. Nature. 1997;386:485–488. doi: 10.1038/386485a0. [DOI] [PubMed] [Google Scholar]
- 6.Paterson A H, Lin Y R, Li Z, Schertz K F, Doebley J F, Pinsom S R M, Liu S C, Stansel J W, Irvine J E. Science. 1995;269:1714–1718. doi: 10.1126/science.269.5231.1714. [DOI] [PubMed] [Google Scholar]
- 7.Lin Y R, Schertz K F, Paterson A H. Genetics. 1995;141:391–411. doi: 10.1093/genetics/141.1.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poncet V, Lamy F, Devos K M, Gale M D, Sarr A, Robert T. Theor Appl Genet. 2000;100:147–159. [Google Scholar]
- 9.Anderson J A, Sorrells M E, Tanksley S D. Crop Sci. 1993;33:453–459. [Google Scholar]
- 10.Messmer M M, Seyfarth R, Keller M, Schachermayr G, Winzeler M. Theor Appl Genet. 2000;100:419–431. [Google Scholar]
- 11.Blanco A, De Giovanni C, Laddomada B, Sciancalepore A, Simeone R, Devos K M, Gale M D. Plant Breeding. 1996;115:310–316. [Google Scholar]
- 12.Joppa L R, Du C, Hart G E, Hareland G A. Crop Sci. 1997;37:1586–1589. [Google Scholar]
- 13.Bucker E S, IV, Thornsberry J M, Kresovich S. Genet Res. 2001;77:213–218. doi: 10.1017/s0016672301005158. [DOI] [PubMed] [Google Scholar]
- 14.Peng J H, Fahima T, Röder M S, Li Y C, Dahan A, Grama A, Ronin Y I, Korol A B, Nevo E. Theor Appl Genet. 1999;98:862–872. [Google Scholar]
- 15.Peng J H, Korol A B, Fahima T, Röder M S, Ronin Y I, Li Y C, Nevo E. Genome Res. 2000;10:1509–1531. doi: 10.1101/gr.150300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lincoln S, Daly M, Lander E. Constructing Genetic Maps with MAPMAKER/EXP3.0, Whitehead Institute Technical Report. 3rd Ed. Cambridge, MA: Whitehead Institute; 1992. [Google Scholar]
- 17.Knapp S J, Holloway J L, Bridges W C, Liu B H. Theor Appl Genet. 1995;91:74–81. doi: 10.1007/BF00220861. [DOI] [PubMed] [Google Scholar]
- 18. Mester, D. I., Ronin, Y. I., Hu, Y., Peng, J., Nevo, E. & Korol, A. B. (2003) Theor. Appl. Genet., in press. [DOI] [PubMed]
- 19.Lander E S, Botstein D. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korol A B, Ronin Y I, Kirzhner V. Genetics. 1995;140:1137–1147. doi: 10.1093/genetics/140.3.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korol A, Ronin Y, Itzcovich A, Nevo E. Genetics. 2001;157:1789–1803. doi: 10.1093/genetics/157.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voorrips R E. J Hered. 2002;93:77–78. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]
- 23.Churchill G A, Doerge R W. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korol A B, Ronin Y I, Nevo E, Hayes P. Heredity. 1998;80:273–284. [Google Scholar]
- 25.Benjamini Y, Hochberg Y. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 26.Burke J M, Tang S, Knapp S J, Reiseberg L H. Genetics. 2002;161:1257–1267. doi: 10.1093/genetics/161.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bennetzen J L, Freeling M. Genome Res. 1997;7:301–306. doi: 10.1101/gr.7.4.301. [DOI] [PubMed] [Google Scholar]
- 28.Sarma R N, Gill B S, Sasaki T, Galiba G, Sutka J. Theor Appl Genet. 1998;97:103–109. [Google Scholar]
- 29.Soller M, Beckmann J S. In: Proc. Second Intern. Conf. Quant. Genet. Weir B S, Eisen E J, Goodman M M, Namkoog G, editors. Sinauer, MA: Sunderland; 1988. pp. 161–188. [Google Scholar]
- 30.Harlan J R. Crops and Man. Madison, WI: American Society of Agronomy; 1992. [Google Scholar]
- 31.Doebley J, Stec A. Genetics. 1993;134:559–570. doi: 10.1093/genetics/134.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gale M D, Atkinson M D, Chinoy C N, Harcourt R L, Jia J, Li Q Y, Devos K M. In: Proc. 8th Intern. Wheat Genet. Li Z S, Xin Z Y, editors. Beijing: China Agricultural Scientech Press; 1995. pp. 79–87. [Google Scholar]
- 33.Blanco A, Bellomo M P, Cenci A, De Giovanni C, D'Ovidio R, Iacono E, Laddomada B, Pagnotta M A, Porceddu E, Sciancalepore A, et al. Theor Appl Genet. 1998;97:721–728. [Google Scholar]
- 34.Korol A B, Preygel I, Preygel S. Recombination Variability and Evolution. London: Chapman & Hall; 1994. [Google Scholar]
- 35. Ronin, Y., Korol, A., Shtemberg, M., Nevo, E. & Soller, M. Genetics, in press. [DOI] [PMC free article] [PubMed]
- 36.Gill K S, Gill B S, Endo T R, Boyko E V. Genetics. 1996;143:1001–1012. doi: 10.1093/genetics/143.2.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gill K S, Gill B S, Endo T R, Taylor T. Genetics. 1996;144:1883–1891. doi: 10.1093/genetics/144.4.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delaney D E, Nasuda S, Endo T R, Gill B S, Hulbert S H. Theor Appl Genet. 1995;91:568–573. doi: 10.1007/BF00223281. [DOI] [PubMed] [Google Scholar]
- 39.Delaney D E, Nasuda S, Endo T R, Gill B S, Hulbert S H. Theor Appl Genet. 1995b;91:780–782. doi: 10.1007/BF00220959. [DOI] [PubMed] [Google Scholar]
- 40.Faris J D, Haen K M, Gill B S. Genetics. 2000;154:823–835. doi: 10.1093/genetics/154.2.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kunzel G, Korzun L, Meister A. Genetics. 2000;154:397–412. doi: 10.1093/genetics/154.1.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lichten M, Goldman A S. Annu Rev Genet. 1995;29:423–444. doi: 10.1146/annurev.ge.29.120195.002231. [DOI] [PubMed] [Google Scholar]
- 43.Otto S P, Barton N H. Genetics. 1997;147:879–906. doi: 10.1093/genetics/147.2.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rice W R. Nat Rev Genet. 2002;3:241–250. doi: 10.1038/nrg760. [DOI] [PubMed] [Google Scholar]
- 45.Castiglioni P, Ajmone-Marsan P, van Wijk R, Motto M. Theor Appl Genet. 1999;99:425–431. doi: 10.1007/s001220051253. [DOI] [PubMed] [Google Scholar]
- 46.Röder M S, Korzun V, Wendehake K, Plaschke J, Tixier M H, Leroy P, Ganal M W. Genetics. 1998;149:2007–2023. doi: 10.1093/genetics/149.4.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Y G, Tsunewaki K. Jap J Genet. 1991;66:617–633. doi: 10.1266/jjg.66.617. [DOI] [PubMed] [Google Scholar]
- 48.Li Y C, Fahima T, Korol A B, Peng J H, Röder M S, Kirzhner V, Beiles A, Nevo E. Mol Biol Evol. 2000;17:851–862. doi: 10.1093/oxfordjournals.molbev.a026365. [DOI] [PubMed] [Google Scholar]
- 49.Belyayev A, Raskina O, Korol A, Nevo E. Genome. 2000;43:1021–1026. [PubMed] [Google Scholar]
- 50.Harberd N P, Flavell R B, Thompson R D. Mol Gen Genet. 1987;209:326–332. doi: 10.1007/BF00329661. [DOI] [PubMed] [Google Scholar]
- 51.Ozkan H, Levy A A, Feldman M. Plant Cell. 2001;13:1735–1747. doi: 10.1105/TPC.010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.