

Abstract
We examined the effects of daily repetitive brief (15 min) myocardial ischemia and reperfusion (I/R) in WT C57/BL6 and extracellular superoxide dismutase (EC-SOD)-overexpressing mice. In the absence of myocardial necrosis, I/R resulted in persistent fibrosis in ischemic areas of C57/BL6 mice associated with persistent global and segmental anterior wall dysfunction. The I/R protocol induced chemokines (peak 3 days) followed sequentially by infiltration of macrophages and myofibroblasts (5 days). Fibrosis peaked at 7 days and was stable at 28 days despite regression of the chemokine and cellular response. Discontinuation of I/R at 7 or 28 days led to regression of fibrosis and ventricular dysfunction. In contrast, the EC-SOD mice developed markedly less chemokine induction, cell response, and fibrosis, with no ventricular dysfunction. Reversible fibrosis and ventricular dysfunction are features of human hibernating myocardium. The reduction of the cellular and functional response in EC-SOD mice suggests a role for reactive O2 in the pathogenesis of ischemic cardiomyopathy.
In the presence of ischemic heart disease, hibernating myocardium refers to a state of persistently impaired left ventricular function at rest, which may be reversed by revascularization (1). In the clinical context it may be the result of repetitive ischemic episodes precipitated by increased demand in the setting of limited flow reserve or chronically low resting myocardial blood flow (2). Numerous studies have examined the morphological alterations in hibernating human myocardial segments (3, 4) demonstrating cardiomyocyte changes, interstitial fibrosis (4), matrix remodeling (5), and evidence of inflammatory activity (6). However, investigations examining the mechanisms responsible for reversible ischemic myocardial dysfunction have been hampered by the difficulty in developing reproducible animal models of hibernating myocardium (7).
We have established a murine model of closed chest myocardial ischemia and reperfusion (I/R) (8). A single 15-min ischemia followed by reperfusion in murine heart is associated with a reactive oxygen intermediate-dependent induction of chemokines (9). Monocyte chemoattractant protein (MCP)-1 is induced after brief I/R in canine myocardium and also depends on free radicals (10). In both animal models a single brief ischemic insult does not lead to inflammatory cell infiltration or tissue injury. We hypothesized that repetitive brief myocardial I/R may induce a persistent oxidative stress-mediated inflammatory response, leading to structural myocardial changes and dysfunction.
We found that repetitive murine I/R induces sequential and reversible chemokine induction, macrophage infiltration, fibrosis, and cardiac dysfunction in the absence of myocardial infarction. The reaction to repeated occlusion is largely prevented by overexpression of extracellular superoxide dismutase (EC-SOD). These findings suggest a role for a reactive oxygen-driven inflammatory response in the pathogenesis of ischemic cardiomyopathy and myocardial hibernation.
Methods
Surgical and Daily I/R Procedures.
All animals received humane care in compliance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. WT C57/BL6 mice were obtained commercially (Harlan Sprague–Dawley), and EC-SOD mice were developed as described (11). EC-SOD mice breeding was performed in a C57/BL6 background and screened with PCR using tail DNA. Heterozygous EC-SOD and WT C57/BL6 mice underwent initial surgery at 8–10 weeks of age, 18- to 22-g body weight by using the closed-chest model of I/R, developed at our institution (8). Briefly, an 8-0 Prolene suture (Ethicon, Somerville, NJ) was placed around the left anterior descending artery, both ends of it threaded through a piece of PE-10 plastic tube (Becton Dickinson), exteriorized through the thorax wall, and stored s.c. After 7–9 days of recovery, the skin was reopened, and the ends of the 8-0 suture were attached to heavy metal picks. The picks were pulled apart until ST elevation occurred in electrocardiogram. The 15-min I/R (resolution of the ST elevation) was performed daily for 3, 5, 7, 14, 21, and 28 days in WT and 3, 5, and 7 days in EC-SOD mice. For the regression studies mice underwent 7- and 28-day I/R protocol, the 8-0 suture was removed, and animals stayed in the vivarium for 30- and 60-day recovery periods. Eight mice were included in each I/R and respective sham group.
The daily I/R procedure was performed with 1.5% isoflurane anesthesia (Isoflo; Abbott), and buprenorphine was given i.p. for analgesia and cefazolin as topical antibiotic (Bristol-Myers Squibb). Mice with persistent electrocardiogram changes after daily reperfusion were excluded from the study. For heart excision, mice were anesthetized with an overdose of sodium pentobarbital (Nembutal; Abbott) 5 h after the last ischemic episode.
Histology and Quantitative Analysis.
Hearts were fixed in zinc-formalin (Z-fix; Anatech, Battle Creek, MI) and embedded in paraffin. Sections were made at 200-μm intervals from base to apex and stained with hematoxylin and eosin for initial evaluation. The area below the suture was detected, and then serial sections were stained with picrosirius red to identify collagen fibers (12). The picrosirius red-stained slides were scanned by using a microscope equipped with a digital camera (Zeiss), and quantitative evaluation was performed with Zeiss image analysis software. The collagen-stained area was calculated as a percentage of the total myocardial area.
Immunohistochemistry.
Serial sections were stained immunohistochemically with the following antibodies: α-smooth muscle actin (Sigma), macrophage-specific rat anti-mouse antibody F4/80 (Serotec) (13), and anti-chicken tenascin C antibody (Chemicon). Staining was performed by using a peroxidase-based technique (Vectastain ELITE kit; Vector Laboratories), developed with diaminobenzidine and nickel (Vector Laboratories), and counterstained with eosin.
Echocardiography.
Echocardiographic measurements were performed 3–4 h after the last ischemic episode by using an 8-MHz probe (Sequoia C256; Acuson, Mountain View, CA). Short axis M-mode was used for measurement of diastolic and systolic ventricular and anterior wall (AW) diameters. The calculations were performed on three different images of each mouse. Fractional shortening was calculated by using a standard formula, and regional function was assessed by using the following formula for AW thickening, where AWs and AWd indicate systolic and diastolic AW thickness, respectively:
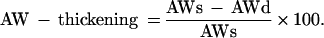 |
RNA Studies.
Total RNA was isolated from whole heart according to the acid guanidinium thiocyanate-phenol-chloroform method (14). Quantitation and quality of RNA were assessed by A260/A280 UV absorption, and samples with ratio above 1.9 were further used.
The mRNA expression level of MCP-1, macrophage inflammatory protein (MIP)-1α, MIP-1β, MIP-2, tumor necrosis factor α, IL-1β, osteopontin, transforming growth factor (TGF)-β1, TGF-β2, and TGF-β3, vascular endothelial growth factor, fibroblast growth factor (FGF)-1, FGF-2, and flt-1 was determined by using a ribonuclease protection assay (RiboQuant; PharMingen) according to the manufacturer's protocol as described (9). PhosphorImaging of the gels was performed (Storm 860; Molecular Dynamics), and signals were quantified by using image quant (Molecular Dynamics) software and normalized to housekeeping gene L32. To make a valid comparison, samples from WT and EC-SOD hearts were examined simultaneously on each gel.
Statistical Analysis.
Data are presented as mean ± SEM. ANOVA with a Student's–Newman-Keuls corrected post hoc analysis was done to compare differences between the groups. Differences with P < 0.05 were considered significant.
Results
Repetitive I/R in Murine Myocardium.
We performed initial surgical procedure for the placement of the left anterior descending artery suture loop in a total of 345 C57/BL6 mice. Seven to 9 days later 266 mice were alive and underwent the I/R protocol. Four I/R mice, but no sham animals, died during the protocol. Reperfusion could not be achieved for technical reasons in 22 animals, which showed histological evidence of myocardial infarction and were excluded from the study. No animals were discarded or died in the recovery groups.
Repetitive Brief I/R Induces Early Transient Inflammatory Reaction and Interstitial Remodeling.
After 7 days of I/R the heart sections from ischemic left ventricular AW showed increased cellularity and widened extracellular space when compared with sham animals (Fig. 1 A and B). The cellular infiltration peaked at 7 days and largely abated by 28 days of I/R, when the extracellular matrix appeared compact (Fig. 1C).
Figure 1.

Histopathological findings in C57/BL6 mice undergoing repetitive I/R protocol (daily 15-min I/R). Hematoxylin and eosin staining of the left ventricular AW in sham (A), 5-day I/R (B), and 28-day I/R (C) mouse (×400) demonstrates marked cellular infiltration after 5 days of brief repetitive I/R. Collagen deposition stained with picrosirius red of the same area in sham (D), 7-day I/R (E), and 28-day I/R (F) mouse (×100). Note that fibrotic changes are interstitial and there is no myocyte loss. In contrast, (G) myocardial infarction (1 h of ischemia and 7 days of reperfusion) is associated with replacement fibrosis (H) (×400 and 200). (I) Quantitative analysis of the collagen deposition in the left ventricular AW revealed significant increase in fibrosis after 7 days of brief repetitive I/R (*, P < 0.05 vs. respective shams). (J) Staining for α-smooth muscle actin shows only positive arteriolar wall in sham animals, but (K) after 5 days of brief repetitive I/R identifies phenotypically modulated myofibroblasts infiltrating the interstitial space (×200). (L) Myofibroblasts diminish after 28 days of I/R. (M) Tenascin expression was not found in sham mice (×200). Tenascin staining reached a maximum after 5 days (N) and diminished thereafter until 28 days (O) of I/R. (P) Sham hearts contain few macrophages stained with F4/80, in contrast to (Q) extensive macrophage infiltration after 5 days of I/R (×200). (R) Macrophages diminish thereafter until 28 days.
Control and sham hearts sections stained for collagen showed predominantly perivascular, thin perimysial, and endomysial strands (Fig. 1D). In mice subjected to I/R, collagen deposition in the ischemic area peaked at 7 days, with no evidence of replacement fibrosis (Fig. 1E). The extent of fibrosis remained comparable with repeated I/R up to 28 days (Fig. 1F). There was no fibrosis in the septal portion of the myocardium (not shown). The findings were clearly different from those in mice undergoing myocardial infarction protocol, which after 7 days of reperfusion show diminished cellularity (Fig. 1G), and an extensive area of replacement fibrosis (Fig. 1H). Quantitative planimetric analysis of collagen showed significant increase after 5 days of I/R, which reached a steady state after 7 days in the left ventricular AW (Fig. 1I). Interstitial myofibroblasts (stained with an α-smooth muscle actin antibody) infiltrated the I/R area, peaking at 5 days (Fig. 1K), but were not seen in shams (Fig. 1J). After 14 days myofibroblasts were reduced and appeared only sporadically by 21–28 days (Fig. 1L).
To characterize matrix components related to the remodeling, we examined expression of tenascin, a matricellular protein induced in actively remodeling tissues. Tenascin was not present in sham hearts (Fig. 1M), but peaked at 5 days of I/R (Fig. 1N) and disappeared by 14–21 days (Fig. 1O). Macrophage staining with F4/80 antibody revealed few positive cells in sham animals (Fig. 1P). Extensive macrophage infiltration was noted after 5 days of repetitive I/R (Fig. 1Q) and also disappeared over the next 2 weeks (Fig. 1R).
Effect of Brief Repetitive I/R on Ventricular Function.
M-mode echocardiography (Fig. 2A) in control and sham animals showed comparable values for fractional shortening (46.8 ± 2.7%) and left ventricular AW thickening (63.1 ± 5.9%). The fractional shortening suggested significant global ventricular dysfunction after 7 days of I/R (Fig. 2B). The AW thickening gradually decreased and showed persistent segmental AW dysfunction during the rest of the I/R protocol (Fig. 2C).
Figure 2.

Ventricular function during the repetitive I/R protocol. (A) Short axis M-mode echocardiography picture of sham animal (Upper) shows normokinetic left ventricular AW in contrast to 7 days of I/R mouse with hypokinetic AW (Lower). (B) Fractional shortening indicates normal global function in sham mice and significant dysfunction in I/R animals. (C) Quantitation of AW thickening identifies significant persistent regional ventricular dysfunction. *, P < 0.05 vs. respective shams.
Regression of Fibrosis and Ventricular Dysfunction.
To study the reversibility of the fibrotic process, mice underwent 7 and 28 days of I/R followed by a 30- and 60-day recovery period. Collagen area showed a significant decrease after 30 days of recovery (Fig. 3A) and further regression until 60 days, when collagen area was comparable to that in sham animals. The fractional shortening (data not shown) and the AW thickening demonstrated significant improvement after 60 days of recovery (Fig. 3B).
Figure 3.

Regression of fibrosis and ventricular dysfunction after discontinuation of repetitive I/R protocol. (A) Fibrosis regression in the left ventricular AW after discontinuation of the 7- and 28-day I/R protocol followed by 30- and 60-day recovery period. (B) AW thickening during the regression process. *, P < 0.05 vs. respective I/R groups.
Influence of Brief Repetitive I/R on Chemokines, Cytokines, and Angiogenic Factors.
The histological findings suggested that repetitive I/R induces an inflammatory response leading to macrophage infiltration and subsequent fibrotic interstitial remodeling. Accordingly, we examined myocardial mRNA expression of molecular signals associated with the inflammatory response. The C-C chemokine MCP-1 was strikingly induced and peaked at 3 days of I/R, which then diminished to the sham level after 14 days (Fig. 4). A modest increase of the mRNA expression of the C-C chemokines MIP-1β and MIP-1α was also noted (data not shown). Tumor necrosis factor α, IL-1β, and transforming growth factor β1 were not significantly induced above levels in sham hearts (data not shown). Angiogenic and fibrogenic factors, such as fibroblast growth factor 1 and 2, vascular endothelial growth factor, and flt-1 showed no difference between sham and I/R hearts (data not shown). However, the matricellular protein osteopontin, an index of active remodeling, was significantly up-regulated in ischemic hearts (data not shown).
Figure 4.

mRNA expression of chemokines in C57/BL6 mice undergoing repetitive I/R. (Upper) MCP-1 is highly induced after 3 days of I/R and normalizes after 14 days when compared with shams. (Lower) A representative transcript band with a corresponding housekeeping gene band. *, P < 0.05 vs. respective shams.
Influence of Overexpressed EC-SOD on Ischemic Myocardium.
We have demonstrated that chemokine induction after a single brief ischemic insult in the heart depends on free-radical generation (9). To investigate the role of reactive oxygen intermediates in this model, we used mice overexpressing the EC-SOD. Of total 127 EC-SOD-overexpressing mice, 118 underwent the protocol; two died and four had myocardial infarction and were therefore discarded. The histological evaluation after 5–7 days of I/R revealed that, in contrast to WT mice, EC-SOD-overexpressing mice exhibited only scattered macrophage infiltration in the ischemic area. Myofibroblast accumulation and tenascin deposition were also significantly reduced. The planimetric collagen evaluation revealed after 7 days of I/R much less fibrosis in EC-SOD than in C57/BL6 mice (Fig. 5A). At the same time, the fractional shortening in EC-SOD mice showed normal function, whereas the WT C57/BL6 had significant dysfunction (Fig. 5B). Simultaneously, the AW thickening in EC-SOD did not decrease, compared with regional dysfunction in WT C57/BL6 mice (Fig. 5C). In addition, the chemokine response was less pronounced and had a shorter duration in EC-SOD mice compared with their WT littermates. After 3 days of I/R, MCP-1 (Fig. 6) and MIP-1β mRNA synthesis was significantly lower in EC-SOD mice compared with WT C57/BL6. At day 7 chemokine mRNA synthesis in the ischemic EC-SOD was not different from the respective EC-SOD shams.
Figure 5.

Fibrosis and ventricular function in EC-SOD-overexpressing mice. (A) Collagen area of C57/BL6 mice compared with the EC-SOD mice revealed significantly less fibrosis in EC-SOD hearts after the repetitive brief I/R protocol. (B) WT mice exhibit decreased fractional shortening after 7 days of I/R, suggesting global dysfunction, whereas EC-SOD-overexpressing mice show normal function. (C) In addition, WT mice show diminished AW thickening, suggesting regional dysfunction, in contrast to EC-SOD mice, which demonstrate normal regional function. *, P < 0.05 in C57/BL6 vs. EC-SOD I/R groups.
Figure 6.

Comparison of chemokine induction in C57/BL6 and EC-SOD mice. (Upper) MCP-1 evaluation shows no difference between C57/BL6 and EC-SOD shams. The I/R protocol induced significantly less MCP-1 in EC-SOD than in C57/BL6 mice. MCP-1 expression in EC-SOD mice after 7 days of I/R is not significantly different from the corresponding shams. (Lower) A representative transcript band with corresponding housekeeping gene band. *, P < 0.05 in C57/BL6 vs. EC-SOD I/R groups.
Discussion
The hallmark of human hibernating myocardium is regional ventricular dysfunction, which is reversible after revascularization (7, 15). Numerous studies describe the structural alterations associated with myocardial hibernation in human myocardial biopsies from patients undergoing coronary bypass surgery (16, 17). However, understanding of the pathogenetic mechanisms responsible for myocardial hibernation has been hampered by the difficulty in developing reproducible animal models simulating viable dysfunctional myocardium. We describe a murine model, where repetitive brief I/R, induces fibrosis and regional ventricular dysfunction in the absence of myocardial infarction. The cardiac fibrosis and ventricular dysfunction are reversible after discontinuation of the ischemic insults. Our model provides direct evidence that an inflammatory response mediated by extracellular reactive oxygen generated from repetitive I/R may play a critical role in the pathogenesis of fibrotic remodeling and ventricular dysfunction in ischemic cardiomyopathy.
We have demonstrated that a single brief (15 min) I/R induces reactive oxygen-driven chemokine synthesis in the murine (9) and canine heart (10). However, in both animal models a single brief ischemic insult is not sufficient to induce leukocyte infiltration and structural changes in the myocardium. Repetitive brief murine I/R induced an inflammatory response in the ischemic myocardium, causing chemokine induction, macrophage infiltration, myofibroblast transformation, and progressive fibrotic changes. Expression of the matricellular proteins osteopontin and tenascin indicated an active interstitial remodeling process. After 14 days of repetitive I/R chemokine synthesis and macrophage infiltration subside, however, interstitial fibrosis persists and depends on continued I/R. Discontinuation of the I/R protocol leads to regression of the fibrotic process. Interstitial fibrotic remodeling of the murine heart is associated with regional systolic dysfunction in the ischemic territory, which is also reversible upon discontinuation of the I/R protocol. Thus, repetitive myocardial I/R in the mouse induced an inflammatory response, leading to a reversible cardiomyopathy associated with extensive interstitial fibrosis.
Our previous work suggested that chemokine induction after a single brief ischemic episode depends on oxidative stress. We hypothesized that free-radical generation may have a critical role in the pathogenesis of the cardiomyopathy associated with repetitive brief ischemia, by inducing chemokine synthesis in the myocardium. We used EC-SOD-overexpressing mice to examine the effects of attenuating oxidative stress on the cardiomyopathic process associated with repetitive myocardial I/R. The EC-SOD is overexpressed in all organs and its extracellular activity in the heart is ≈2.5-fold higher than in WT littermates (11). EC-SOD mice exhibited a significantly decreased chemokine response, diminished macrophage infiltration and myofibroblast accumulation, and much less extensive fibrotic remodeling than control mice. EC-SOD overexpression protected mice from developing systolic dysfunction after repetitive myocardial I/R, suggesting a crucial role for free-radical generation in the pathogenesis of the cardiomyopathy. In addition to their role in inducing chemokine expression, reactive oxygen intermediates may directly regulate interstitial remodeling and fibrosis through matrix metalloproteinase activation (18) and fibroblast stimulation (19).
The pathologic features observed in our murine model of ischemic cardiomyopathy resemble the structural alterations noted in human hibernating myocardium (3, 4, 20). Recent experiments using human myocardial biopsies suggested that reversible ischemic dysfunction is associated with an active inflammatory process (6), leading to interstitial remodeling (5). Our murine model demonstrates the importance of brief sublethal ischemic insults in the pathogenesis of ischemic cardiomyopathy and sheds light on the mechanisms responsible for cardiac fibrosis and dysfunction. Ischemia-induced oxidative stress has a crucial role in the development of the inflammatory response, fibrosis, and dysfunction. Interventions aiming at neutralizing this reactive oxygen-initiated process may be effective in reversing cardiac fibrosis and dysfunction.
Acknowledgments
We thank Thuy Pham, Alida Evans, Stephanie Butcher, and Kathryn Masterman for expert technical assistance and Conception Mata and Sharon Malinowski for editorial assistance with the manuscript. This work was supported by National Heart, Lung, and Blood Institute Grant HL-42550, the Medallion Foundation, the Methodist Hospital Foundation, and a grant-in-aid from the American Heart Association Texas affiliate (to N.G.F.). O.D. and P.K. are supported by the Deutsche Forschungsgemeinschaft (Grants DE 801/1-1 and KN 521/1-2).
Abbreviations
- I/R
ischemia and reperfusion
- MCP
monocyte chemoattractant protein
- EC-SOD
extracellular superoxide dismutase
- AW
anterior wall
- MIP
macrophage inflammatory protein
References
- 1.Rahimtoola S H. Circulation. 1985;72:V123–V135. [PubMed] [Google Scholar]
- 2.Kloner R A, Bolli R, Marban E, Reinlib L, Braunwald E. Circulation. 1998;97:1848–1867. doi: 10.1161/01.cir.97.18.1848. [DOI] [PubMed] [Google Scholar]
- 3.Vanoverschelde J L, Wijns W, Borgers M, Heyndrickx G, Depre C, Flameng W, Melin J A. Circulation. 1997;95:1961–1971. doi: 10.1161/01.cir.95.7.1961. [DOI] [PubMed] [Google Scholar]
- 4.Elsasser A, Schlepper M, Klovekorn W P, Cai W J, Zimmermann R, Muller K D, Strasser R, Kostin S, Gagel C, Munkel B, et al. Circulation. 1997;96:2920–2931. doi: 10.1161/01.cir.96.9.2920. [DOI] [PubMed] [Google Scholar]
- 5.Frangogiannis N G, Shimoni S, Chang S M, Ren G, Dewald O, Gersch C, Shan K, Aggeli C, Reardon M, Letsou G V, et al. J Am Coll Cardiol. 2002;39:1468–1474. doi: 10.1016/s0735-1097(02)01792-8. [DOI] [PubMed] [Google Scholar]
- 6.Frangogiannis N G, Shimoni S, Chang S M, Ren G, Shan K, Aggeli C, Reardon M J, Letsou G V, Espada R, Ramchandani M, et al. Am J Pathol. 2002;160:1425–1433. doi: 10.1016/S0002-9440(10)62568-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heusch G. Physiol Rev. 1998;78:1055–1085. doi: 10.1152/physrev.1998.78.4.1055. [DOI] [PubMed] [Google Scholar]
- 8.Nossuli T O, Lakshminarayanan V, Baumgarten G, Taffet G E, Ballantyne C M, Michael L H, Entman M L. Am J Physiol. 2000;278:H1049–H1055. doi: 10.1152/ajpheart.2000.278.4.H1049. [DOI] [PubMed] [Google Scholar]
- 9.Nossuli T O, Frangogiannis N G, Knuefermann P, Lakshminarayanan V, Dewald O, Evans A J, Peschon J, Mann D L, Michael L H, Entman M L. Am J Physiol. 2001;281:H2549–H2558. doi: 10.1152/ajpheart.2001.281.6.H2549. [DOI] [PubMed] [Google Scholar]
- 10.Lakshminarayanan V, Lewallen M, Frangogiannis N G, Evans A J, Wedin K E, Michael L H, Entman M L. Am J Pathol. 2001;159:1301–1311. doi: 10.1016/S0002-9440(10)62517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oury T D, Ho Y S, Piantadosi C A, Crapo J D. Proc Natl Acad Sci USA. 1992;89:9715–9719. doi: 10.1073/pnas.89.20.9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frangogiannis N G, Perrard J L, Mendoza L H, Burns A R, Lindsey M L, Ballantyne C M, Michael L H, Smith C W, Entman M L. Circulation. 1998;98:687–698. doi: 10.1161/01.cir.98.7.687. [DOI] [PubMed] [Google Scholar]
- 13.Hirsch S, Austyn J M, Gordon S. J Exp Med. 1981;154:713–725. doi: 10.1084/jem.154.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chromczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 15.Wijns W, Vatner S F, Camici P G. N Engl J Med. 1998;339:173–181. doi: 10.1056/NEJM199807163390307. [DOI] [PubMed] [Google Scholar]
- 16.Nagueh S F, Mikati I, Weilbaecher D, Reardon M J, Al-Zaghrini G J, Cacela D, He Z X, Letsou G, Noon G, Howell J F, et al. Circulation. 1999;100:490–496. doi: 10.1161/01.cir.100.5.490. [DOI] [PubMed] [Google Scholar]
- 17.Elsasser A, Schlepper M, Zimmermann R, Muller K D, Strasser R, Klovekorn W P, Schaper J. Mol Cell Biochem. 1998;186:147–158. [PubMed] [Google Scholar]
- 18.Rajagopalan S, Meng X P, Ramasamy S, Harrison D G, Galis Z S. J Clin Invest. 1996;98:2572–2579. doi: 10.1172/JCI119076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siwik D A, Pagano P J, Colucci W S. Am J Physiol. 2001;280:C53–C60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 20.Ausma J, Cleutjens J, Thone F, Flameng W, Ramaekers F, Borgers M. Mol Cell Biochem. 1995;147:35–42. doi: 10.1007/BF00944781. [DOI] [PubMed] [Google Scholar]