

Abstract
Two in vitro transcripts, one corresponding to the 5′ and central domains (residues 1–920) of 16S rRNA and the other corresponding to its 3′ domain (residues 922–1542), assemble efficiently in trans with 30S ribosomal proteins to form a compact ribonucleoprotein particle that cosediments with natural 30S subunits. Isolated particles are similar in appearance to natural 30S subunits with electron microscopy and contain a full complement of the small subunit ribosomal proteins. The particles have a reduced ability to bind tRNA (attributable to the location of the discontinuity in a conserved region of the rRNA) near features that have been implicated in tRNA binding. Association of these two halves of 16S rRNA in trans must be stabilized by either previously unidentified RNA–RNA contacts or interactions mediated by ribosomal proteins because there are no known direct interactions between them. The trans construct was used to probe the three-dimensional RNA neighborhood around position 922 of 16S rRNA by generating hydroxyl radicals from Fe(II) tethered to the 5′ end of the 3′ transcript. Hydroxyl radical-induced cuts in the 16S rRNA chain were localized by primer extension to nucleotides 923–929 and 1192–1198, providing evidence for the mutual proximity of the 920 and 1192 regions.
In the absence of a high-resolution crystal structure, our understanding of ribosome structure has relied on a variety of alternative approaches. One approach is based on interpretation of electron microscopic images (1–3) including the enhanced resolution of the more recent cryoelectron microscopy reconstruction studies (4, 5). A link between electron-microscopy morphology and molecular structure has been sought through model building (6–8) [based on the phylogenetically determined secondary structure of 16S rRNA (9) and the neutron-diffraction map for the positions of the ribosomal proteins (10)] using cross-linking and footprinting data (11–13) to constrain the folding of the rRNA. An important feature of the latter approach is that it has the potential to relate individual rRNA nucleotide positions to specific structural features of the ribosome. Although models for the 30S ribosomal subunit show a significant level of agreement, there are many discrepancies that can be ascribed to a lack of sufficient experimental constraints.
To this end, we have developed the use of site-directed hydroxyl radical probing of rRNA by using Fe(II) tethered to unique positions on individual ribosomal proteins (14, 15) via the reagent 1-(p-bromoacetamidobenzyl)-EDTA (BABE; ref. 15). Hydroxyl radicals attack the ribose moiety, resulting in cleavage of the RNA chain (17, 18). Because of the limited range of hydroxyl radicals, the positions of cleavage of the RNA provide information about the three-dimensional rRNA environment around the tethered Fe(II) probe. More recently, this approach has been extended to permit tethering to in vitro transcripts of tRNA and tRNA analogs via linkage to a 5′-phosphorothioate (19, 20).
In the experiments described here, this method has been adapted to allow probing from internal positions of the large rRNAs. First, we show that 30S ribosomal subunits can be reconstituted in vitro from two separate fragments of 16S rRNA prepared by in vitro transcription. These fragments comprise the 5′ and central domains (nucleotides 1–920) and the 3′ major and minor domains (nucleotides 922–1542), respectively. Particles were constructed previously from natural 16S rRNA containing a break in this region, generated by site-specific RNase H cleavage by Bogdanov and coworkers (21). Because there are no known direct interactions between these two halves of 16S rRNA, their association in trans must be stabilized either by previously unidentified RNA–RNA contacts or by interactions mediated by ribosomal proteins. These results support the idea that the structure of the 30S subunit is built from independently assembling domains. We used this 30S construct to probe the RNA environment around position 922 in the reconstituted 30S subunits by using Fe(II) tethered to the 5′ end of the 3′ fragment of 16S rRNA. The results of this study place two well-characterized functional sites, centered around positions 926 and 1192, respectively, in proximity to each other.
MATERIALS AND METHODS
Plasmid Construction.
A plasmid (pUC5′/C) encoding the 5′ and central domains of 16S rRNA (5′-central) was generated by using PCR cloning (22) from plasmid pBS16S.RS (23) with the following oligonucleotide primers: primer I (sequence for the T7 promoter in bold), 5′-GCTCTCTAGATAATACGACTCACTATAGGGAAATTGAAGAG-3′ and primer II, 5′-GATGGCATGCGATTCATTTGAGTTTTAACCTTGCGG-3′. The amplified DNA was digested with XbaI and SphI and cloned into pUC18 digested with the same enzymes to generate plasmid pUC5′/C. A plasmid (pUC3′D) encoding the 3′ domain of 16S rRNA (3′ domain) was generated similar manner by using another set of oligonucleotide primers, primer III (T7 promoter in bold), 5′-GCTCTCTAGATAATACGACTCACTATAGACGGGGGCCCGC-3′ and primer IV, 5′-GATCGCATGCCCTAAGGAGGTGATCCAACCG-3′. The amplified DNA was digested with the same enzymes mentioned above and cloned into pUC18 to generate plasmid pUC3′D. The resulting plasmids were checked by using dideoxy sequence analysis (24).
In Vitro Transcription.
Plasmids pUC5′/C and pUC3′D were linearized with TfiI and Bsu36I, respectively, and used for in vitro transcription (25, 26). The resulting transcripts, 5′-central and 3′-domain, respectively, were extracted at 4°C twice with phenol and twice with chloroform and recovered by ethanol precipitation. The RNA was further purified on a Pharmacia Sephadex G-50 column.
Reconstitution and Characterization of Ribonucleoprotein Particles.
In vitro reconstitution of 30S particles from natural or in vitro-transcribed 16S rRNA was performed as described (23, 27). Reconstitution of trans 30S (t30S) particles from an equimolar mixture of 5′-central and 3′-domain RNA (0.4 μM final concentration) was performed under the same conditions except that a MgCl2 concentration of 30 mM was required in the reconstitution buffer to obtain optimum yields. Reconstitution volumes varied between 0.25 and 1 ml. Resulting particles were purified by sucrose-gradient centrifugation and concentrated by using centrifugation through Microcon filters (Amicon) essentially as described (23). RNA was extracted with phenol and chloroform and analyzed by using 4% denaturing PAGE.
Subunits reconstituted from natural 16S rRNA or from 5′-central and 3′-domain RNA were assayed for tRNA-binding activity by using [32P]pCp-labeled tRNAPhe (28). Briefly, 2.5 pmol (0.25 μM final concentration) of 30S subunits, reconstituted 30S subunits, or t30S subunits were activated by incubation at 42°C for 30 min in binding buffer (80 mM potassium cacodylate, pH 7.5/30 mM MgCl2/100 mM NH4Cl) and then incubated at 37°C for 30 min. Next, trace amounts of [32P]pCp-labeled tRNAPhe (specific activity = 49,000 cpm/pmol) were diluted with 2.5 pmol of cold tRNAPhe (0.25 μM final concentration) and added to the ribosome mix, and the incubation was continued at 37°C for 30 min. The reaction mixtures were diluted in 500 μl of binding buffer and spotted on filter paper. The filters were washed with 3 ml of binding buffer, dried, and counted.
For protein analysis, sucrose gradient-purified t30S subunits or 30S subunits (75 μg) reconstituted from in vitro-transcribed 16S rRNA (23) were recovered by ethanol precipitation and resuspended in 10 μl of water. Proteins were extracted from these particles with acetic acid as described (29) and analyzed by two-dimensional gel electrophoresis (30).
Derivatization with Fe(II)–1-(p-bromoacetamidobenzyl)-EDTA (Fe(II)–BABE).
For 5′-derivatization with BABE, a 5′-phosphorothioate was introduced at the 5′ terminus of 3′-domain RNA by in vitro transcription in the presence of a 5-fold molar excess of 5′-guanosine-α-phosphorothioate (GMPS) over each dNTP, as described (19).
BABE modification of 5′-GMPS-3′-domain RNA was performed essentially as described (18). Briefly, 240 μg of 5′-GMPS-3′-domain RNA (11.8 μM final concentration) was incubated with preloaded Fe(II)–BABE (3 mM final concentration) in potassium phosphate buffer (pH 8.5; 40 mM final concentration) for 60 min at 37°C. The reaction was stopped by extracting twice with phenol to remove excess unreacted Fe–BABE followed by chloroform extraction and ethanol precipitation to recover 5′-Fe(II)–BABE-3′-domain RNA. The RNA was used in in vitro reconstitution experiments as described above.
Hydroxyl Radical Probing of Fe(II)-Tethered 30S Particles.
Hydroxyl radical formation was initiated by addition of ascorbate (6.3 mM final concentration) and hydrogen peroxide (0.06% final concentration) to 0.3 μM isolated t30S particles in 80 mM potassium cacodylate (pH 7.2), 20 mM magnesium acetate, and 150 mM ammonium chloride (final reaction volume 100 μl) followed by incubation for 10 min at room temperature. In control experiments, the particles were treated identically except for the omission of ascorbate and hydrogen peroxide. Reactions were stopped by addition of 600 μl of cold 100% ethanol and 3 M sodium acetate (0.3 M final concentration) and quick-freezing in dry ice/ethanol bath. Extraction of rRNA and localization of cleavage sites by using primer extension was performed as described (31).
RESULTS
Reconstitution of 30S Subunits from Two Synthetic Fragments of 16S rRNA.
In vitro transcripts corresponding to the 5′ and central domains (nucleotides 1–920) and the 3′ major and minor domains (nucleotides 922–1,542) of 16S rRNA were reconstituted with total 30S ribosomal proteins, and the resulting particles were isolated by sucrose-gradient centrifugation (Fig. 1). RNA was extracted from each of the three peaks and analyzed on a 4% denaturing polyacrylamide gel (Fig. 2, and data not shown). The fastest sedimenting major peak (Fig. 1, peak 3) cosedimented with 32P-labeled native 30S subunits and contained equimolar amounts of both 5′-central and 3′-domain RNAs (Fig. 2). We refer to these particles as trans 30S (t30S) particles. Peak 2 (Fig. 1) also contained both transcripts (data not shown) and most likely corresponds to partially assembled or unfolded 30S-like particles. Peak 1 (Fig. 1)consisted of particles containing solely 3′-domain rRNA (data not shown) and are likely to correspond to the previously described head particles (23). The overall yield of reconstituted t30S subunits was about 20% of input rRNA.
Figure 1.

Sucrose-gradient sedimentation of in vitro-transcribed t30S subunits. Complexes were loaded on 10-ml 10–40% sucrose gradients in 20 mM Hepes (pH 7.5), 100 mM NH4Cl, 30 mM MgCl2, and 6 mM B-mercaptoethanol, centrifuged for 13 hr in a Beckman SW41 rotor at 35,000 rpm at 4° and scanned with an ISCO density gradient fractionator (Model 183). Complexes were monitored by A260 (■). A trace amount of [32P]pCp-labeled 30S subunits, used as marker, was monitored by Cerenkov counting (○).
Figure 2.
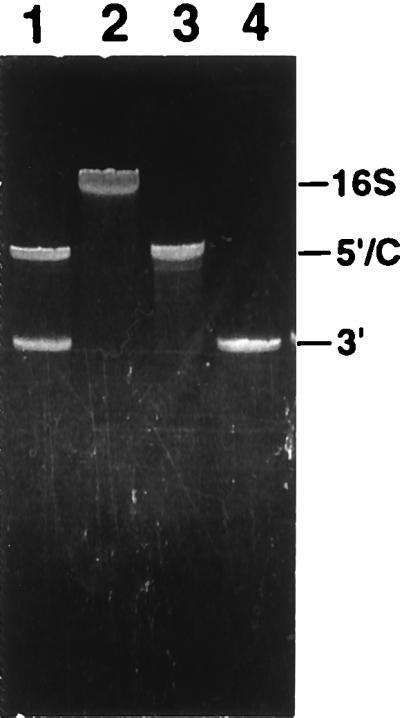
Analysis of rRNA extracted from sucrose gradient-purified t30S particles by using denaturing 4% polyacrylamide gel electrophoresis. Lane 1, t30S; lane 2, natural 30S; lane 3, 5′-central domain RNA; lane 4, 3′-domain RNA.
The protein composition of sucrose gradient-purified t30S subunits or subunits that were reconstituted from in vitro transcribed 16S rRNA was analyzed by two-dimensional gel electrophoresis (Fig. 3). A full complement of the small-subunit proteins is present in t30S particles (Fig. 3B) except for S1, which was absent in the total 30S ribosomal protein mixture used for reconstitution.
Figure 3.

Protein composition of gradient-purified 30S subunits reconstituted from in vitro-transcribed full-length 16S rRNA (A) or gradient-purified t30S particles analyzed by 2-dimensional gel electrophoresis (B) as in ref. 29.
Isolated t30S particles were visualized by electron microscopy using double-carbon film negative-stain preparations (32). t30S particles (Fig. 4B) appear similar in their gross morphology to natural 30S subunits (Fig. 4A). Distinctive features of natural 30S subunits (the platform, head, and body) are clearly evident. In addition, a smaller number of amorphous particles are observed.
Figure 4.

Electron microscopic analysis of natural 30S ribosomal subunits (A) and purified reconstituted t30S particles (B).
The ability of purified t30S subunits to bind tRNAPhe was assayed by using filter binding. The activity of these particles was severely reduced compared with that of 30S particles reconstituted in parallel experiments using natural 16S rRNA (Table 1). This reduction may be caused by the proximity of the internal break to parts of 16S rRNA that have been implicated in tRNA binding (33, 34) and/or to perturbation of higher-order structure near the break in the 16S rRNA backbone.
Table 1.
tRNA Binding Activity to 30S ribosomal subunits
| Particle | pmol of tRNA bound | Percent activity |
|---|---|---|
| Natural 30S | 0.8 | (100) |
| Reconstituted 30S | 0.21 | 26.7 |
| trans 30S | 0.03 | 3.8 |
Binding of 3′[32P] pCp-labeled tRNAPhe to 30S subunits in the presence of poly(U) mRNA as described in Materials and Methods. Background binding of tRNA (1,000 cpm) in the absence of ribosomes was substracted.
Site-Directed Probing of t30S Particles.
For site-directed hydroxyl radical probing, t30S particles were reconstituted by using a 3′ transcript derivatized at its 5′ end (position 922) with Fe(II)–BABE (15) via a 5′-phosphorothioate (19). Hydroxyl radical formation was initiated by addition of H2O2 and ascorbate, and the sites of cleavage of the 16S rRNA backbone were localized by primer extension using a set of DNA oligonucleotide primers (31). Two specific regions of cleavage were identified (Fig. 5). One region encompasses nucleotides 923–929 and is proximal to the site of attachment of the Fe(II) probe. The second set of cleavages includes positions 1192–1198 in the 3′-major domain. Both sets of cleavages depended on the presence of tethered Fe(II) and addition of ascorbate and H2O2 (Fig. 5, compare lanes 1 to 3 and 2 to 4). Some weak cleavage at positions 923–929 also was detected in the control samples in which no ascorbate and H2O2 were added (Fig. 5A, lane 2). A possible explanation for these cleavages is the presence of molecular oxygen and reducing agents in the reconstitution and primer-extension buffers, which could favor generation of hydroxyl radicals leading to self-cleavage of the derivatized RNA.
Figure 5.

Localization of positions of cleavage of 16S rRNA in t30S particles by hydroxyl radicals generated from Fe(II) tethered to position 922. The 3′-domain transcripts have either GMP (lanes 1 and 3) or BABE–GMPS (lanes 2 and 4) at their 5′ termini. Lanes 1 and 2 are control lanes, in which ascorbate and H2O2 were omitted from the probing reaction. Locations of the cleavage positions are indicated by vertical bars. A and G are sequencing lanes.
DISCUSSION
Small ribosomal subunits previously have been reconstituted from fragmented natural 16S rRNA generated by oligonucleotide-directed RNase H cleavage (21). One of the several cleavages that were tested was directed toward the 912–926 region of 16S rRNA—near the position of the interruption in the RNA chain described here. The 16S rRNA fragmented at this particular site was able to assemble efficiently with ribosomal proteins into a 30S subunit that retained the ability to associate with 50S subunits and translate poly(U) and natural mRNA, albeit with reduced efficiency. In contrast to the present study, the two pieces of rRNA that resulted from cleavage at position 920 were not separated before reconstitution but were copurified, possibly preserving intermolecular interactions.
In this paper we show that it is possible to reconstitute a 30S subunit-like particle in trans from two independently transcribed fragments of 16S rRNA. The reconstituted trans subunits cosediment with natural 30S subunits, have a similar ribosomal protein composition to their natural counterparts, and are similar in appearance to natural 30S subunits via electron microscopy. Their ability to bind tRNAPhe is severely reduced compared with subunits reconstituted from natural 16S rRNA, possibly because of the break in the rRNA chain close to features critical for tRNA binding. The 10- to 12-fold higher activity of the particles obtained by Bogdanov and coworkers (21) from fragmented natural 16S rRNA could be the result of possible preservation of intermolecular interactions in their protocol or to the generally higher activity observed for particles reconstituted from natural 16S rRNA (35).
Association of the 5′-central and 3′-domain rRNAs in trans must be stabilized by previously unidentified RNA–RNA contacts (21) or by interactions mediated by ribosomal proteins, because there are no known direct secondary-structure interactions between them. We have previously demonstrated that the isolated 3′ domain of 16S rRNA is capable of assembling into a compact, globular ribonucleoprotein particle that closely resembles the head of the 30S ribosomal subunit (23). In addition to containing a full complement of the small-subunit proteins that are known to bind to the 3′-domain of 16S rRNA, these particles also contained significant amounts of S4, a protein not known to interact with the 3′-domain. This finding hints at the possibility that the two fragments of 16S rRNA (or the independently assembled domains) in the t30S particles may be held together by protein–RNA or protein–protein interactions. Furthermore, studies with scanning transmission electron microscopy (36) have shown that in vitro assembly of 30S subunits proceeds through the formation of three independent domains that later converge and presumably interact via protein–protein contacts to form the final 30S subunit. Such a domain organization could facilitate the folding of a large ribonucleoprotein particle such as the 30S subunit.
We exploited the discontinuity at position 922 in the 16S rRNA chain for site-specific probing of the RNA neighborhood surrounding this position in the t30S particles. This was achieved by covalently tethering an Fe(II)–EDTA probe to the 5′ end of the 3′-domain transcript via a 5′-phosphorothioate at G922. Hydroxyl radicals generated from the Fe(II) probe at this position cleaved the 16S rRNA backbone in the 923–929 and 1192–1198 regions (Figs. 5 and 6). These findings place these two regions, which are distant from each other in the secondary structure of 16S rRNA, in proximity to each other in the 30S subunit. On the basis of the observed cleavage intensities and previous distance-calibration experiments (20), we estimate that positions 922 and 1196 lie within ≈30 ±10 Å of each other. This is consistent with previous results in which hydroxyl radicals generated from Fe(II) tethered to position 21 of ribosomal protein S5 resulted in cleavage of the 16S rRNA backbone at residues 921–925 and 1191–1197 (15). Both regions have been implicated in ribosomal function. Peptidyl-tRNA protects G926 from kethoxal attack (33), and the antibiotic streptomycin protects bases around position 915 (37). Mutations at residue C1192 confer resistance to the antibiotic spectinomycin (38), and spectinomycin protects N7 of G1,064, the Watson–Crick partner of C1192, from modification by dimethyl sulfate (37). Thus, the G1064–C1192 base pair is strongly implicated in spectinomycin binding and is likely part of the antibiotic binding site. Spectinomycin is believed to inhibit protein synthesis by blocking the EF-G-dependent translocation of tRNA (39). Our results raise the possibility that the effects of spectinomycin may in some way involve interactions between the 926 and 1192 regions of 16S rRNA. Binding of the drug, for example, could interfere with the relative movement of these two regions of 16S rRNA.
Figure 6.

Hydroxyl radical cleavages obtained from Fe(II) tethered to position 922 are summarized on a secondary-structure representation of 16S rRNA; the size of the circles represents the relative intensity of the cleavages. The location of the tethered Fe(II) is indicated.
By using this same approach to tether Fe(II) to other positions of the 16S rRNA chain, additional distance constraints can be obtained for use in constraining the folding of the RNA in structural models of the 30S subunit (40). Recently, a similar strategy has been used to incorporate a cross-linking reagent at internal breaks in 16S (41) and 23S rRNA (42) to obtain distance constraints for modeling the rRNAs. Finally, the system described here provides a possible approach to the identification of the molecular interactions involved in domain–domain contacts. For example, by omitting single proteins from the reconstitution of the trans particles, it should be possible to identify those proteins that are required to establish interdomain interactions.
Acknowledgments
We thank J. Moran, D. P. Greiner, and C. F. Meares for providing BABE and helping with synthesis. This investigation was supported in part by a California Division American Cancer Society Fellowship no. 1-38-97B to S.J.; by Grant GM-17129 from the National Institutes of Health to H.F.N.; and by a grant to the Center for Molecular Biology of RNA from the Lucille P. Markey Charitable Trust. This work was submitted in partial fulfillment of the requirements for the Ph.D. degree of R.R.S. (43).
ABBREVIATIONS
- TP30
total 30S ribosomal proteins
- BABE
1-(p-bromoacetamidobenzyl)-EDTA
- t30S
trans 30S
References
- 1.Lake J A. In: Ribosomes: Structure, Function and Genetics. Chambliss G, Craven G R, Davies J, Davis K, Kahan L, Nomura M, editors. Baltimore: University Park Press; 1980. pp. 207–236. [Google Scholar]
- 2.Stöffler G, Bald R, Kastner B, Lührman R, Stöffler-Melicke M. In: Ribosomes: Structure, Function and Genetics. Chambliss G, Craven G R, Davies J, Davis K, Kahan L, Nomura M, editors. Baltimore: University Park Press; 1980. pp. 171–206. [Google Scholar]
- 3.Boublik M, Mandiyan V, Tumminia S. In: The Ribosome: Structure, Function and Evolution. Hill W E, Moore P B, Dahlberg A, Schlessinger D, Garrett R A, Warner J A, editors. Washington, DC: Am. Soc. Microbiol.; 1990. pp. 114–122. [Google Scholar]
- 4.Stark H, Orlova E V, Rinke-Appel J, Jünke N, Mueller F, Rodnina M, Wintermeyer W, Brimacombe R, van Heel M. Cell. 1997;88:19–28. doi: 10.1016/s0092-8674(00)81854-1. [DOI] [PubMed] [Google Scholar]
- 5.Malhotra A, Penczek P, Agrawal R K, Gabashvili I S, Grassucci R A, Jünemann R, Burkhardt N, Nierhaus K H, Frank J. J Mol Biol. 1998;280:103–116. doi: 10.1006/jmbi.1998.1859. [DOI] [PubMed] [Google Scholar]
- 6.Mueller F, Brimacombe R. J Mol Biol. 1997;271:524–544. doi: 10.1006/jmbi.1997.1210. [DOI] [PubMed] [Google Scholar]
- 7.Stern S, Weiser B, Noller H F. J Mol Biol. 1988;204:447–481. doi: 10.1016/0022-2836(88)90588-8. [DOI] [PubMed] [Google Scholar]
- 8.Malhotra A, Harvey S C. J Mol Biol. 1994;240:308–340. doi: 10.1006/jmbi.1994.1448. [DOI] [PubMed] [Google Scholar]
- 9.Noller H F, Woese C R. Science. 1981;212:403–411. doi: 10.1126/science.6163215. [DOI] [PubMed] [Google Scholar]
- 10.Capel M S, Engelman D M, Freeborn B R, Kjeldgaard M, Langer J A, Ramakrishnan V, Schindler D P, Schneider D K, Schoenborn B P, Sillers I Y, et al. Science. 1987;238:1403–1406. doi: 10.1126/science.3317832. [DOI] [PubMed] [Google Scholar]
- 11.Stern S, Powers T, Changchien L-M, Noller H F. Science. 1989;244:783–790. doi: 10.1126/science.2658053. [DOI] [PubMed] [Google Scholar]
- 12.Powers T, Noller H F. RNA. 1995;1:194–209. [PMC free article] [PubMed] [Google Scholar]
- 13.Atmadja J, Brimacombe R. Nucleic Acids Res. 1985;13:6919–6936. doi: 10.1093/nar/13.19.6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heilek G M, Marusak R, Meares C F, Noller H F. Proc Natl Acad Sci USA. 1995;92:1113–1116. doi: 10.1073/pnas.92.4.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heilek G M, Noller H F. Science. 1996;272:1659–1662. doi: 10.1126/science.272.5268.1659. [DOI] [PubMed] [Google Scholar]
- 16.Rana T M, Meares C F. J Am Chem Soc. 1990;112:2458–2459. [Google Scholar]
- 17.Hertzberg R P, Dervan P B. J Am Chem Soc. 1982;104:313–314. [Google Scholar]
- 18.Tullius T D, Dombroski B A. Science. 1985;230:679–681. doi: 10.1126/science.2996145. [DOI] [PubMed] [Google Scholar]
- 19.Joseph S, Noller H F. EMBO J. 1996;15:910–916. [PMC free article] [PubMed] [Google Scholar]
- 20.Joseph S, Weiser B, Noller H F. Science. 1997;278:1093–1098. doi: 10.1126/science.278.5340.1093. [DOI] [PubMed] [Google Scholar]
- 21.Afonina E, Chichkova N, Bogdanova S, Bogdanov A. Biochimie. 1991;73:777–787. doi: 10.1016/0300-9084(91)90057-8. [DOI] [PubMed] [Google Scholar]
- 22.Saiki R K, Scharf S, Faloona F, Mullis K B, Horn G T, Erlich H A, Arnheim N. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 23.Samaha R R, O’Brien B, O’Brien T W, Noller H F. Proc Natl Acad Sci USA. 1994;91:7884–7888. doi: 10.1073/pnas.91.17.7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanger F, Nicklen S, Coulson A R. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Milligan J F, Uhlenbeck O C. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 26.Gurevich V T, Pokrovskaya I D, Obukhova T A, Zozulya S A. Anal Biochem. 1991;195:207–213. doi: 10.1016/0003-2697(91)90318-n. [DOI] [PubMed] [Google Scholar]
- 27.Cunningham P R, Nurse K, Ofengand J. FASEB J. 1993;7:177–180. doi: 10.1096/fasebj.7.1.7916699. [DOI] [PubMed] [Google Scholar]
- 28.England T E, Bruce A G, Uhlenbeck O C. Methods Enzymol. 1980;65:65–74. doi: 10.1016/s0076-6879(80)65011-3. [DOI] [PubMed] [Google Scholar]
- 29.Siegmann M, Thomas G. Methods Enzymol. 1987;146:362–369. doi: 10.1016/s0076-6879(87)46037-0. [DOI] [PubMed] [Google Scholar]
- 30.Geyl D, Böck A, Isono K. Mol Gen Genet. 1981;181:309–312. doi: 10.1007/BF00425603. [DOI] [PubMed] [Google Scholar]
- 31.Stern S, Moazed D, Noller H F. Methods Enzymol. 1988;164:481–489. doi: 10.1016/s0076-6879(88)64064-x. [DOI] [PubMed] [Google Scholar]
- 32.Lake J A, Pendergast M, Kahan L, Nomura M. Proc Natl Acad Sci USA. 1974;71:4688–4692. doi: 10.1073/pnas.71.12.4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moazed D, Noller H F. Cell. 1989;57:585–597. doi: 10.1016/0092-8674(89)90128-1. [DOI] [PubMed] [Google Scholar]
- 34.Von Ahsen U, Noller H F. Science. 1995;267:234–237. doi: 10.1126/science.7528943. [DOI] [PubMed] [Google Scholar]
- 35.Krzyzosiack W, Denman R, Nurse K, Boublik M, Gehrke C W, Agris P F, Ofengand J. Biochemistry. 1987;26:2353–2364. doi: 10.1021/bi00382a042. [DOI] [PubMed] [Google Scholar]
- 36.Mandiyan V, Tumminia S J, Wall J S, Hainfield J F, Boublik M. Proc Natl Acad Sci USA. 1991;88:8174–8178. doi: 10.1073/pnas.88.18.8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moazed D, Noller H F. Nature (London) 1989;342:142–148. doi: 10.1038/342142a0. [DOI] [PubMed] [Google Scholar]
- 38.Sigmund C D, Ettayebi M, Morgan E A. Nucleic Acids Res. 1984;12:4653–4663. doi: 10.1093/nar/12.11.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bilgin N, Richter A A, Ehrenberg M, Dahlberg A E, Kurland C G. EMBO J. 1990;9:735–739. doi: 10.1002/j.1460-2075.1990.tb08167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Newcomb, L. & Noller, H. F. (1998) Biochemistry, in press.
- 41.Baranov P V, Dokudovskaya S S, Oretskaya T S, Dontsova O A, Bogdanov A A, Brimacombe R. Nucleic Acids Res. 1997;25:2266–2273. doi: 10.1093/nar/25.12.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baranov P V, Gurvich O A, Bogdanov A A, Brimacombe R, Dontsova O A. RNA. 1998;4:658–668. doi: 10.1017/s1355838298980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samaha R R. Ph.D. thesis. Santa Cruz, CA: Univ. of California; 1995. [Google Scholar]