

Abstract
Objective
To determine if NF-κB regulates intestinal epithelial cell migration and if it has a role during bile salt-induced migration.
Summary Background Data
Mucosal restitution is an important repair modality in the gastrointestinal tract. The authors have shown that taurodeoxycholate (TDCA) increases intestinal epithelial cell migration. NF-κB regulates activation of a number of genes involved in inflammatory responses.
Methods
Studies were conducted in IEC-6 cells. IκB protein expression was determined by Western blot analysis. Sequence-specific NF-κB binding activity was measured by EMSA shift assays and nuclear localization by immunohistochemistry. Cell migration was examined by using an in vitro model that mimics the early cell division-independent stages of epithelial restitution.
Results
The process of cell migration over the wounded area was associated with a significant increase in NF-κB binding activity in IEC-6 cells. Immunohistochemistry revealed translocation of NF-κB into the nucleus. Western blot analysis showed that injury decreased IκB protein expression. Inhibition of the binding activity by treatment with a specific NF-κB inhibitor, MG-132, inhibited cell migration during restitution. Further, exposure to TDCA at the physiologic concentration that induces intestinal epithelial cell migration increased NF-κB binding activity, induced NF-κB translocation into the nucleus, and decreased IκB protein expression. MG-132 also inhibits bile salt-induced cell migration.
Conclusions
NF-κB regulates intestinal epithelial cell migration. Bile salts at physiologic concentrations increase cell migration by activation of NF-κB. These data show that bile salts may have a role in the maintenance of intestinal mucosal integrity.
Small intestinal digestive and secretory functions require an intact mucosa. The epithelium additionally serves as a barrier to a broad spectrum of noxious substances within the intestinal lumen. Injury to the mucosa is common and may be minor and superficial, or deep and extensive. Superficial erosions may be caused by luminal contents or medications; more extensive damage may result from ischemia, inflammatory conditions, or infections. Restitution is the process by which normal adjacent intestinal epithelial cells migrate over a denuded area to reseal and repair the epithelium. Migration of the cells occurs as a sheet to reform cell contacts and re-establish barrier function. Intestinal epithelial restitution occurs in a matter of hours following injury and does not require cell proliferation. 1–3 The stimuli and mediators of migration have been the subject of considerable investigation in recent years. Substances that have been identified as factors involved in this process include polyamines, growth factors, bile salts, and matrix components.
NF-κB is a ubiquitous transcription factor that regulates activation of a number of genes involved in proinflammatory responses, differentiation, and growth. 4–6 NF-κB is found in the cytoplasm bound to the endogenous inhibitors, known as IκBs. Activation of NF-κB occurs when IκB is phosphorylated, resulting in IκB degradation and cytosolic release of NF-κB. NF-κB then translocates into the nucleus and induces transcription of specific genes. 7–11 The NF-κB signaling cascade has been shown to be activated in the intestinal epithelium by tumor necrosis factor-alpha (TNF-α), bacterial lipopolysaccharides, interleukin-1 (IL-1), bacteria, and other agents. Once activated, the NF-κB signaling pathway transcriptionally regulates many cellular genes implicated in early immune, acute phase, and inflammatory responses, including TNF-α, IL-2, IL-6, IL-8, IL-12, inducible nitric oxide synthase, cyclooxygenase-2, intercellular adhesion molecule-1, and others. 6 Thus, the NF-κB signaling pathway is highly relevant to intestinal injury and repair.
Our previous studies have shown that the bile salt taurodeoxycholate (TDCA) increases intestinal epithelial migration during restitution. 12 Bile salts are normally found within the gastrointestinal succus entericus, where their primary function is to aid in the digestion of lipids and lipid-soluble vitamins. 13 In the lumen, most bile salts are present in micellar form, but some exist as free bile salts. Recent studies have shown that bile salts have various biologic effects independent of their role in digestion. The bile salt deoxycholic acid has been shown to induce apoptosis in a colon cancer cell line;14 taurodeoxycholic acid, TDCA, increases esophageal mucosal growth in a rabbit explant model. 15 Bile salts may also modulate gene expression; deoxycholic acid has been shown to modulate p53 gene expression in colonic adenoma cell lines. 16 These findings suggest that potent cellular effects of bile salts are operative at the gene expression and growth regulatory level.
Given our recent observations that TDCA augments intestinal epithelial restitution, we hypothesized that NF-κB could be a mediator in the process. The present studies asked if the NF-κB signaling pathway affects intestinal epithelial restitution and if the effect of TDCA on epithelial migration is regulated by NF-κB activation. First, we wanted to determine if injury to the epithelium activated the NF-κB signaling pathway. Second, we examined if inhibition of this pathway resulted in an inhibition of cell migration after injury. Third, we wished to elucidate if the effect of the bile salt TDCA on intestinal epithelial migration was regulated by the NF-κB signaling pathway.
METHODS
Materials
Disposable culture ware was purchased from Corning Glass Works (Corning, NY). Tissue culture media and dialyzed fetal bovine serum (FBS) were from GIBCO (Grand Island, NY). Biochemicals were purchased from Sigma (St. Louis, MO). The double-stranded oligonucleotides used in electromobility shift assay (EMSA) and antibodies against NF-κB were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). [τ-32P]ATP (3,000 Ci/mmol) was purchased from Amersham (Arlington Heights, IL).
Cell Culture and Experimental Protocol
The IEC-6 cell line was purchased from ATCC at passage 13. The cell line was derived from normal rat intestine and was developed and characterized by Quaroni et al. 17 IEC-6 cells originated from intestinal crypt cells as judged by morphologic and immunologic criteria. They are nontumorigenic and retain the undifferentiated character of epithelial stem cells.
Stock cells were maintained in T-150 flasks in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% heat-inactivated FBS with 1% antibiotic. Flasks were incubated at 37°C in a humidified atmosphere of 95% air/5% CO2. Stock cells were subcultured once a week at 1:2; medium was changed three times weekly. The cells were restarted from original frozen stock. Passages 16 to 19 were used in the experiments. There were no significant changes of biologic function and characterizations from passages 15 to 20.
Monolayer Wounding and Measurement of Migration
The protocol for the epithelial wounding model has been described previously. 18 Briefly, IEC-6 cells were plated at 6.25 × 104 cells/cm2 in DMEM plus 5% FBS on 60-mm plates thinly coated with Matrigel and grown until confluent. The epithelium was scraped using a sterile 6-mm-wide blade to create a smooth denuded wound, and cell migration was allowed to occur over the denuded area for 6 hours. Cell migration was measured by counting the cells in the denuded area in a randomized, blinded fashion. Results are reported as the number of cells per 1 mm of scratch.
Effect of TDCA on NF-κB Expression and Activation
IEC-6 cells were plated at 6.25 × 104 cells/mm2 in DMEM plus 5% FBS on 60-mm Matrigel-coated plates and grown until confluent. The epithelium was scraped using a sterile 6-mm-wide blade to create the wounded edge. Culture medium was changed to either control medium or medium containing 0.05 mmol/L TDCA. Cells were harvested 6 hours later for Western analysis and nuclear protein electrophoretic gel shift analysis.
Effect of TDCA and MG-132 on Intestinal Cell Migration
IEC-6 cells were grown as above. Following the standard wounding protocol, culture medium was changed immediately after wounding to either control medium or medium containing MG-132 at a concentration ranging from 1 to 10 μmol/L in the presence and absence of varying concentrations of TDCA. Cell migration was measured in a standard fashion by counting the cells in a denuded area in a randomized, blinded manner.
Western Blotting Analysis
Ten micrograms of cytoplasmic protein extracts was dissolved in SDS sample buffer, boiled for 5 minutes, and then subjected to electrophoresis on acrylamide gels according to Laemmli. 19 After SDS-PAGE, the gels were transferred to nitrocellulose membranes for 1 hour at 4°C. The blots were blocked with 5% nonfat dry milk in phosphate-buffered saline/0.1% Tween 20 (PBS-T) overnight at 4°C. Immunologic evaluation was performed for 1 hour in PBS-T containing 0.2 μg/mL affinity-purified polyclonal antibodies against NF-κB p65 subunit or IκBα. The blots were washed with PBS-T and incubated for 1 hour with goat antirabbit IgG antibody conjugated to peroxidase at a dilution of 1:3,000 in PBS-T. After extensive washing in PBS-T, the blots were developed for 30 to 60 seconds with enhanced chemiluminescence reagents.
Preparation of Nuclear Protein and Electrophoretic Shift Assays
Nuclear proteins were prepared by the procedure described previously, 20 and the protein contents in nuclear preparations were determined by the method described by Bradford. 21 Using the same cell culture methods, cells were harvested at 2 and 8 hours after wounding for nuclear protein extractions. The double-stranded oligonucleotides used in these experiments included 5′-AGTTGA GGGGACTTTCCC AGGC-3′, which contains a consensus NF-κB binding site that is underscored. These oligonucleotides were radioactively end-labeled with [τ-32P]ATP and T4 polynucleotide kinase. For EMSA, 0.035 pmol 32P-labeled oligonucleotides (∼30,000 cpm) and 10 μg nuclear protein were incubated in a total volume of 25 μL in the presence of 10 mmol/L Tris·HCl (pH 7.5), 50 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L dithiothreitol, 5% glycerol, and 1 μg poly(dI-dC). The binding reactions were allowed to proceed at room temperature for 20 minutes. Thereafter, 2 μL bromphenol blue (0.1% in water) was added, and protein–DNA complexes were resolved by electrophoresis on nondenaturing 5% polyacrylamide gels and visualized by autoradiography. The specificity of binding interactions was assessed by competition with an excess of unlabeled double-stranded oligonucleotide of identical sequence.
Gel supershift assays were accomplished by adding 1 μg (in 1 μL) p65 supershift antibody to the reaction mixture and incubating for an additional 30 minutes at room temperature.
Immunohistochemical Staining
Immunohistochemical staining for NF-κB staining was performed in IEC-6 cells by the indirect immunoperoxidase method as described previously. 22 The cells were incubated with rabbit polyclonal antibody against the p65 subunit of NF-κB at a dilution of 1:100 in PBS containing 1% FBS for 1 hour at room temperature, and then 1 hour of incubation with biotinylated goat antirabbit IgG at a dilution of 1:500. Nonspecific slides were incubated without antibody against NF-κB. The bound antibody was visualized with avidin–biotin complexes. The slides were counterstained with hematoxylin and mounted and viewed with a microscope.
Statistics
Values are given as means ± SE from six dishes. Autoradiographed results were repeated. The significance of the difference between means was determined by ANOVA. The level of significance was determined using Duncan’s multiple range test. 23
RESULTS
Effect of TDCA and Wounding on NF-κB Protein Expression and NF-κB Cellular Distribution
We have previously observed that bile salts enhance cell migration in our injury model. We hypothesized that NF-κB could be a mediator of this bile salt-stimulated effect. Figure 1 shows that both wounding the intestinal epithelial monolayer and the presence of a physiologic concentration of TDCA (0.05 mmol/L) increased NF-κB proteins levels. Further, the presence of TDCA after wounding the intestinal epithelial monolayer synergistically stimulated NF-κB protein expression versus TDCA or wounding alone. The increase in protein levels was noted 6 hours after injury or exposure to TDCA.
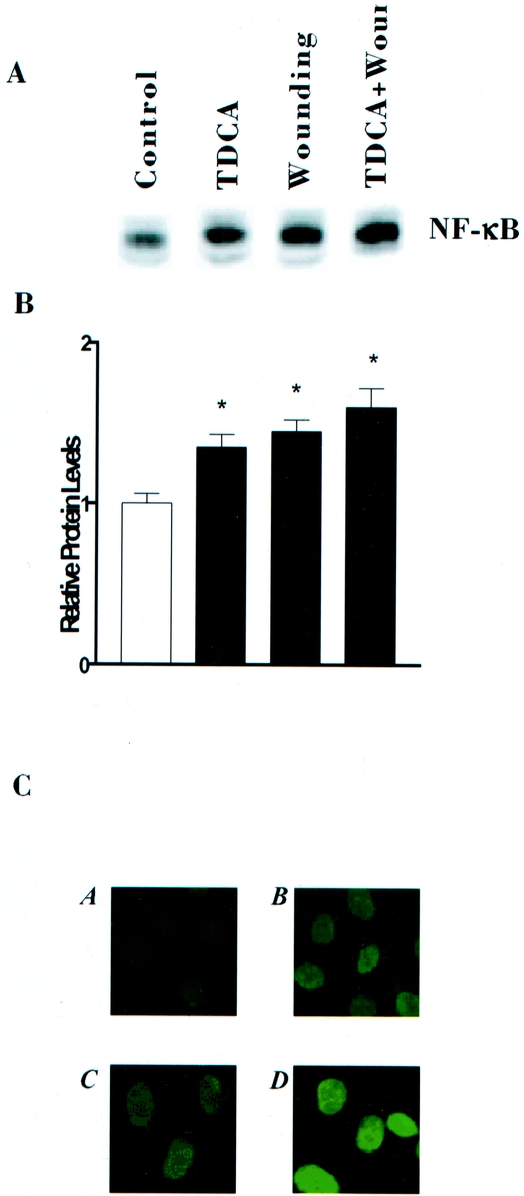
Figure 1. Levels and cellular distribution of NF-κB protein in control cells and cells treated with 0.05 mmol/L TDCA and wounding. (A) Representative autoradiograms from cells exposed to TDCA and wounding for 6 hours. Protein (10 υg) was applied to each lane and subjected to electrophoresis and Western immunoblotting. NF-κB protein was identified by probing nitrocellulose with specific antibody described in text and is indicated by NF-κB. (B) Quantitative analysis of Western blots by densitometry for cells described in (A). Relative levels of NF-κB were corrected for loading as measured by densitometry of β-actin. Blots were repeated in triplicate. P < .05 versus controls. (C) Cellular distribution of NF-κB 6 hours after treatment. Bound antibody (antip65 subunit) was visualized with a, control; b, wounding; c, 0.05 mmol/L TDCA: d, wounding and 0.05 mmol/L TDCA.
NF-κB activation was noted 6 hours after wounding or the addition of TDCA to media by nuclear localization of NF-κB protein by immunohistochemistry. Immunohistochemical staining showed that nuclear localization for the p65 subunit of NF-κB was synergistically increased in the presence of TDCA and wounding, relative to TDCA or wounding alone. These data show that both wounding and TDCA increase NF-κB protein expression and nuclear translocation in intestinal epithelial cells. TDCA functions synergistically with injury to increase NF-κB protein expression and nuclear translocation.
Changes in NF-κB Sequence-Specific Binding Activity in Wounded and Unwounded Cells
We studied the effect of intestinal injury on NF-κB activation by measuring sequence-specific NF-κB binding activity by EMSA. Cells harvested at 2 and 8 hours after wounding showed that the process of cell migration over the wounded area was associated with a significant increase in NF-κB binding activity in IEC-6 cells. Increased NF-κB binding occurred at 2 hours and continued to 8 hours after injury (Fig. 2A). These data are concordant with our immunohistochemical findings showing nuclear localization of the p65 subunit of NF-κB.
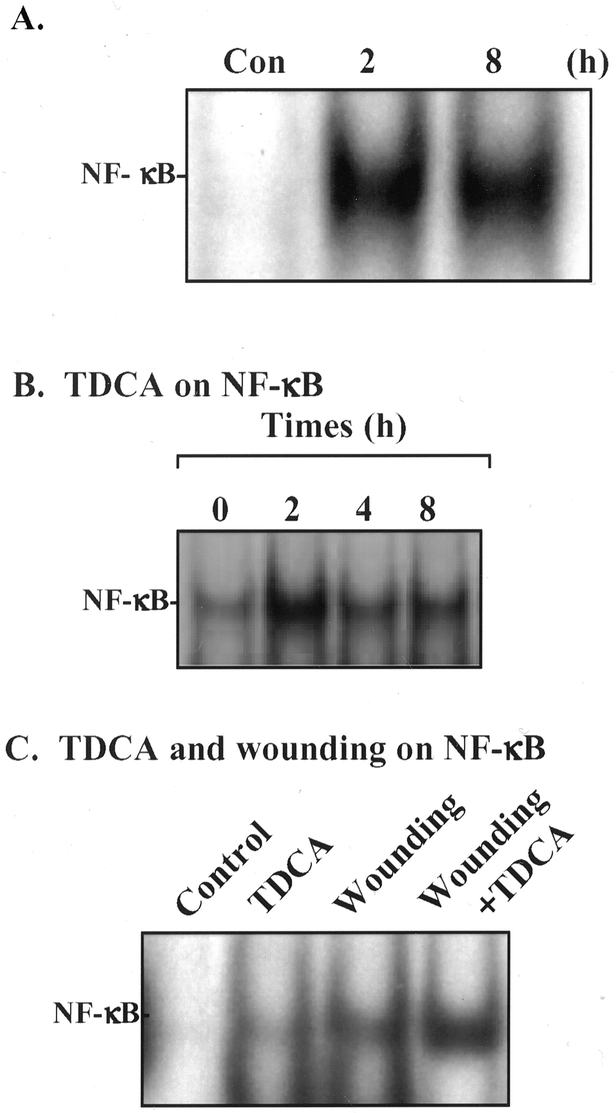
Figure 2. Changes in NF-κB sequence-specific binding activity in wounded IEC-6 cells in the presence and absence of bile salts. (A) Representative autoradiograms from control cells and cells 2 and 8 hours after wounding as described above. Nuclear extracts were prepared from cells after wounding. Electrophoretic mobility shift assay (EMSA) was performed using 10 υg nuclear protein and 0.035 pmol 32P-end-labeled oligonucleotides containing a single NF-κB. Positions of the specifically bound DNA-protein binding complex are indicated. (B) Effect of the addition of 0.05 mmol/L TDCA to the medium of IEC-6 cell on sequence-specific NF-κB binding activity measured by EMSA. (C) Effect of 0.05 mmol/L TDCA and wounding on sequence-specific NF-κB binding activity measured by EMSA 6 hours after the addition of TDCA and wounding of the IEC-6 monolayer.
We then characterized the effect of bile salt alone and in conjunction with wounding on NF-κB nuclear binding. Increased NF-κB activation occurred within 2 hours of exposure of the IEC-6 cells to the TDCA (see Fig. 2B) and persisted for 8 hours. Next we determined the effect of TDCA and wounding together on NF-κB activation. Figure 2C shows that there was a synergistic effect on NF-κB activation when wounding occurred in the presence of TDCA. Thus, we concluded that TDCA increases intestinal epithelial migration after injury by inducing NF-κB activation.
To confirm that the measured NF-κB binding activity was sequence-specific, competitive inhibition experiments were performed. As shown in Figure 3, NF-κB binding activity in wounded cells and cells exposed to TDCA was dose-dependently inhibited when graded concentrations of the unlabeled NF-κB oligonucleotide were added to the binding reaction mixture. We also examined the effect of the labeled oligonucleotide containing a mutated NF-κB binding site on NF-κB binding activity and found that the NF-κB-labeled, mutated oligonucleotide did not bind to the wounded IEC-6 cells or the IEC-6 cells exposed to TDCA. Further, the supershift of NF-κB using specific antibody against the p65 subunit confirmed NF-κB binding specificity.
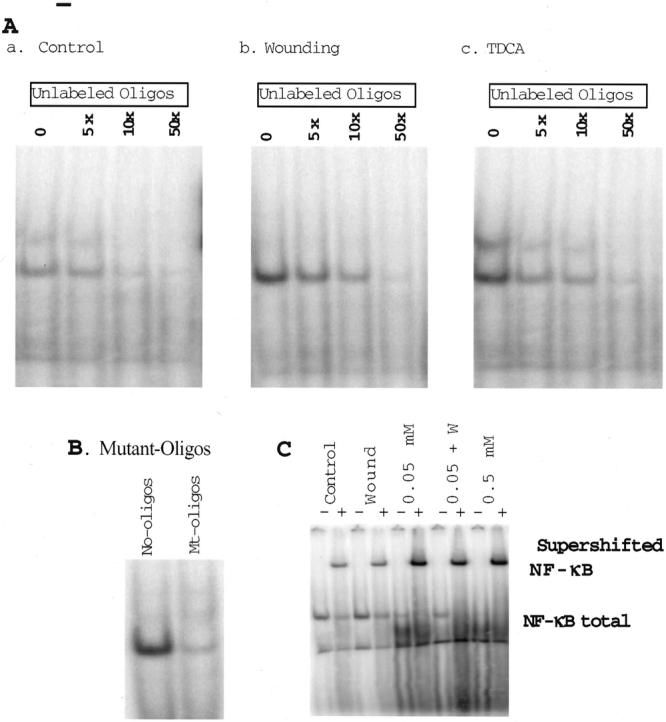
Figure 3. Changes in sequence-specific binding activity in IEC-6 cells exposed to unlabeled and mutant oligonucleotides containing a single NF-κB binding site and supershift of NF-κB using antibody to the p65 subunit of the NF-κB heterodimer. (A) Effects of unlabeled NF-κB oligonucleotide added to the binding reaction mixture as a cold competitor on NF-κB binding activity. a, Representative autoradiograms from control cells. b, Representative autoradiograms from cells 6 hours after wounding. c, Representative autoradiograms from cells treated with 0.05 mmol/L TDCA for 6 hours. (B) Sequence-specific binding of labeled normal versus labeled mutated oligonucleotides to the NF-κB binding site. (C) NF-κB supershift using the p65 antibody to control and wounded IEC-6 cells 6 hours after wounding. Supershift against unwounded IEC-6 cells (control), 6 hours after wounding (wound), 6 hours after the addition of 0.05 and 0.5 mmol/L TDCA (0.05 and 0.5, respectively), and 6 hours after the addition of 0.05 mmol/L TDCA and wounding (0.05 +W) with (+) and without (-) p65 supershift antibody.
Effect of Wounding and Bile Salt on IκB Expression
To determine how injury and the presence of bile salt induce activation of NF-κB, we studied the effect of wounding the intestinal epithelium in the presence and absence of bile salt on IκB protein expression. IκB protein expression was determined 6 hours after wounding with or without the addition of bile salt. Figure 4 shows that wounding the intestinal epithelial monolayer resulted in a decrease in IκB protein expression, and the physiologic concentrations of TDCA that increase intestinal cell migration also result in a decrease in IκB protein expression. Further, the presence of TDCA after wounding the intestinal epithelial monolayer synergistically inhibited IκB protein expression versus TDCA or wounding alone.
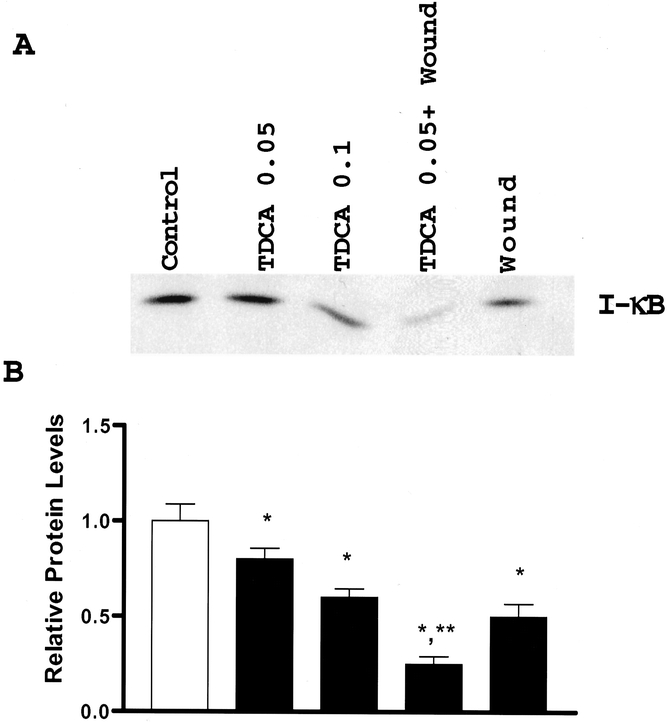
Figure 4. Levels of IκB protein in control cells and cells treated with 0.05 and 0.1 mmol/L TDCA and wounding. (A) Representative autoradiograms from cells exposed to TDCA and wounding for 6 hours. Protein (10 υg) was applied to each lane and subjected to electrophoresis and Western immunoblotting. IκB protein was identified by probing nitrocellulose with specific antibody to IκBα described in text and is indicated by IκB. (B) Quantitative analysis of Western blots by densitometry for cells described in (A). Relative levels of IκB were corrected for loading as measured by densitometry of β-actin. Blots were repeated in triplicate. *P < .05 versus controls, **P < .05 versus wounding alone or TDCA alone.
Effect of NF-κB Inhibition on IEC-6 Cell Migration
MG-132 is a specific proteosome inhibitor that has been used extensively to inhibit the NF-κB signaling pathway. 24–27 NF-κB is activated after degradation of phosphorylated cytosolic IκB by the proteosome complex. MG-132 inhibits the proteosome complex from degrading IκB after phosphorylation, preventing release of NF-κB from IκB, which in turn inhibits translocation of NF-κB into the nucleus. Thus, NF-κB cannot induce transcription of its target genes. To confirm the role of NF-κB activation in the process of IEC-6 cell migration after injury in the presence and absence of bile salts, this potent and specific inhibitor of NF-κB, MG-132, was used. Addition of MG-132 to the medium 1 hour before wounding inhibited NF-κB activation in a dose-dependent manner (Fig. 5A). Furthermore, histologic evaluation revealed there was no toxic effect to the IEC-6 cells in the presence of 1 and 10 μmol/L MG-132. Significantly, MG-132 at both 1 and 10 μmol/L also significantly inhibited IEC-6 cell migration in a dose-dependent manner (see Fig. 5B). These findings suggest that the NF-κB signaling pathway plays an important role in the regulation of cell migration after injury.
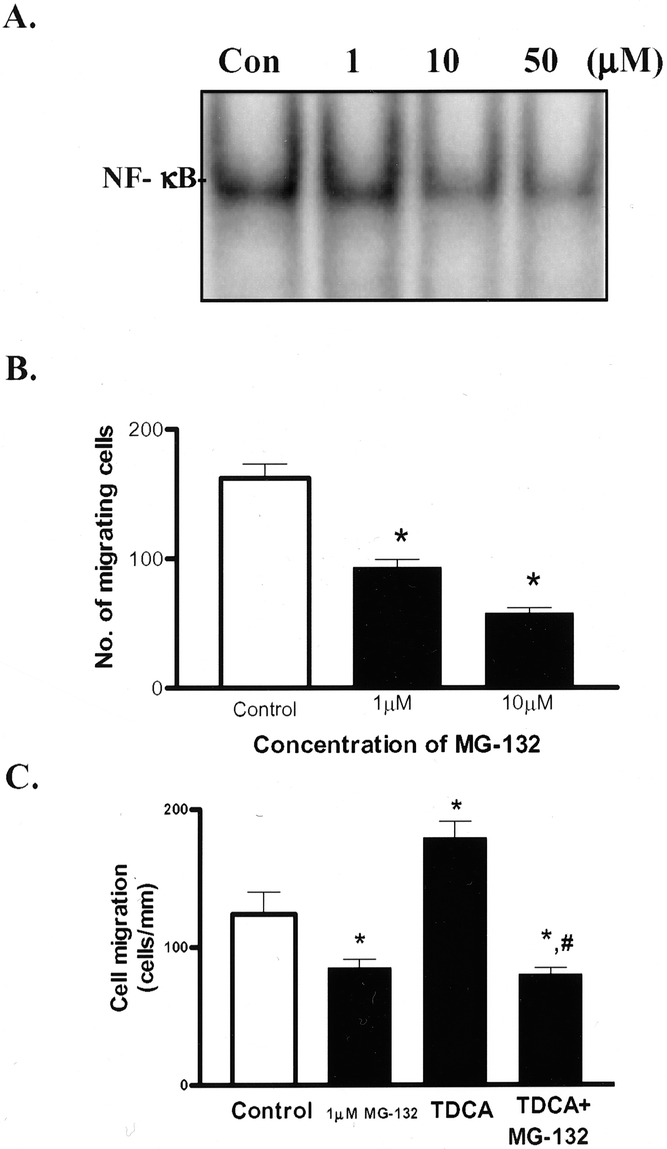
Figure 5. Effect of NF-κB inhibition on IEC-6 cell migration. (A) Effect of the proteosome inhibitor MG-132 added to the medium 1 hour before wounding on sequence-specific NF-κB binding activity using EMSA in IEC-6 cells 4 hours after wounding. (B) Effect of the proteosome inhibitor MG-132 on IEC-6 cell migration 6 hours after wounding. MG-132 was added 1 hour before wounding. Cell migration was assayed 6 hours after removal of part of cell layer. Values are mean ± SE from 6 dishes. *P < .05 compared with control group. (C) Effect of 1 υM MG-132 on IEC-6 cell migration 6 hours after wounding in the presence of 0.05 mmol/L TDCA. MG-132 was added 1 hour before wounding. Cell migration was assayed 6 hours after removal of part of cell layer. Values are mean ± SE from 6 dishes. *P < .05 compared with control group; #P < .05 compared with TDCA alone.
Last, we evaluated the effect of NF-κB blockade on bile salt-stimulated migration. MG-132 was added to the medium 1 hour before wounding IEC-6 cells in the presence of 0.05 mmol/L TDCA. Figure 5C shows that TDCA increased cell migration versus control and that this effect was inhibited in the presence of MG-132. These findings show that NF-κB activation is required for intestinal epithelial cell migration after injury and physiologic concentrations of the bile salt TDCA.
DISCUSSION
NF-κB is a ubiquitous transcription factor found in the intestinal epithelium that is activated by a variety of agents, including phorbol esters IL-1, TNF-α, lipopolysaccharide, double-stranded RNA, cAMP, bacteria, and viral transactivators. 6 After activation NF-κB is translocated into the nucleus and regulates transcription of cellular genes implicated in early inflammatory responses. In the present study we found that NF-κB is activated after intestinal epithelial injury, and inhibition of NF-κB activation by the proteosome inhibitor MG-132 significantly decreases cell migration after injury. Thus, NF-κB regulates intestinal restitution after injury.
While the primary role of bile salts is to facilitate lipid absorption, diverse cellular effects have also been reported. Bile salts are physiologically active components within the gastrointestinal lumen. Reflux of bile salts into the esophagus causes mucosal injury and is implicated in the development of reflux esophagitis. 28 Conversely, bile salts at low concentrations stimulate proliferation of the colonic epithelium and are involved in the regulation of colonic mucosal growth. 14,29–32 Previously, we showed that the bile salt TDCA at physiologic concentrations stimulates intestinal epithelial migration after injury. 12 In this study we have shown that TDCA activates NF-κB during this stimulation, as evidenced by a reduction of cytoplasmic IκB, translocation of NF-κB into the nucleus, and DNA binding. Furthermore, inhibition of NF-κB activation significantly inhibits intestinal cell migration in the presence of TDCA. Therefore, NF-κB regulates intestinal epithelial cell migration following injury and cell migration induced by bile salts.
Our data cannot address the question of signal transduction by which either TDCA or wounding initiates NF-κB activation. Hsu et al. 33,34 have characterized TNF-α-induced NF-κB activation in enterocytes. They identified scaffolding and adapter proteins that transmit extracellular signals inside the cells. TNF-α stimulates the TNF receptor-1 (TNFR1) to trimerize and recruit the TNF receptor-associated factor and the receptor interacting protein to the cytoplasmic portion of the TNFR1. The signal is transmitted to the NF-κB-inducing kinase, which activates the IKK, resulting in degradation of the NF-κB inhibitor IκB and allowing NF-κB activation. Similarly, IL-1β activates NF-κB through a signaling cascade that requires the participation of IL-1 receptor accessory protein, MyD88, and IL-1 receptor-associated kinase to associate with and activate TRAF-6, where the signal is transmitted to NIK, which activates the IKK complex. 35–38
Our studies have shown that both wounding the intestinal epithelium and bile salt at physiologic concentrations activate NF-κB and stimulate cellular migration. These data suggest a physiologic role for luminal bile salts in small intestinal mucosal repair in vivo, although our observations to date are limited to this in vitro model. Free bile salts, including the conjugated bile salts TDCA and chenodeoxycholic acid, as well as unconjugated bile salts such as deoxycholic acid are found in the lumen of the gastrointestinal tract throughout the small bowel. The precise concentrations of these compounds at the luminal surface are highly variable and difficult to quantitate, but they can range from near zero to 2 to 3 mmol/L. 13
Activation of NF-κB does not uniformly stimulate intestinal epithelial restitution. TNF-α has been shown to increase NF-κB activation in intestinal epithelial cells, 4,5 but TNF-α does not enhance intestinal epithelial migration after injury. 39 IL-1β, which also activates NF-κB, does increase intestinal epithelial cell migration after injury. 39 Clearly, NF-κB is involved in the regulation of intestinal epithelial cell migration, but activation of NF-κB alone does not augment intestinal migration after injury.
TGF-β is one growth factor that has been identified as necessary for intestinal cell migration. 40–45 Inhibition of TGF-β function with anti-TGF-β antibody inhibits intestinal cell migration after injury. We have shown previously that the bile salt TDCA at physiologic concentrations augments intestinal epithelial cell migration by increasing TGF-β expression, 12 an effect that also is inhibited by anti-TGF-β antibody. In this study, TDCA at the same concentration was noted to activate NF-κB, demonstrating that NF-κB is a signaling pathway for bile salt-stimulated epithelial migration. Further studies will define if the NF-κB signaling we have observed leads to TGF-β gene transcription in our model.
In summary, wounding the intestinal epithelium increases NF-κB activation, which stimulates intestinal epithelial cell migration after injury. Similarly, the bile salt TDCA augments intestinal epithelial cell migration at physiologic concentrations by activating NF-κB. Furthermore, the effect of bile salt on NF-κB expression, translocation into the nucleus, and DNA-specific binding acts synergistically with the effect of direct injury of the intestinal epithelium to activate NF-κB. Thus, bile salt has a beneficial effect during intestinal epithelial restitution at least partially through the activation of NF-κB. Further characterization of the role of bile salts and NF-κB during intestinal epithelial injury and repair may lead to novel strategies to enhance mucosal repair.
Footnotes
Correspondence: Eric D. Strauch, MD, Department of Surgery, University of Maryland School of Medicine, 22 South Greene Street, Baltimore, MD 21201.
E-mail: estrauch@umaryland.edu
Accepted for publication August 22, 2002.
References
- 1.Feil W, Wenzl E, Vattay P, et al. Repair of rabbit duodenal mucosa after acid injury in vivo and in vitro. Gastroenterology. 1987; 92: 1973–1986. [DOI] [PubMed] [Google Scholar]
- 2.Rutten MJ, Ito S. Morphology and electrophysiology of guinea pig gastric mucosal repair in vitro. Am J Physiol. 1983; 244: G171–G182. [DOI] [PubMed] [Google Scholar]
- 3.Silen W, Ito S. Mechanism for rapid-epithelialization of the gastric mucosal surface. Annu Rev Physiol. 1985; 47: 217–229. [DOI] [PubMed] [Google Scholar]
- 4.Baeuerle PA, Henkle P. Function and activation of NF-κB in the immune system. Annu Rev Immunol. 1994; 12: 141–179. [DOI] [PubMed] [Google Scholar]
- 5.Barnes PJ, Karin M. Nuclear factor-κB, a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997; 336: 1066–1071. [DOI] [PubMed] [Google Scholar]
- 6.Jobin C, Sartor RB. The IκB/NF-κB system: a key determinant of mucosal inflammation and protection. Cell Physiol. 2000; 278: 451–462. [DOI] [PubMed] [Google Scholar]
- 7.Brown KS, Gerstberger L, Carlson G, et al. Control of IκB∝ proteolysis by site-specific, signal-induced phosphorylation. Science. 1995; 267: 1485–1488. [DOI] [PubMed] [Google Scholar]
- 8.Beg AA, Finco TS, Nantermet PV, et al. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IκB∝: a mechanism for NF-κB activation. Mol Cell Biol. 1993; 13: 3301–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee FS, Hagler J, Chen ZJ, et al. Activation of the IκB∝ kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997; 88: 213–222. [DOI] [PubMed] [Google Scholar]
- 10.Mercurio FH, Zhu BW, Murray A, et al. IKK-1 and IKK-2: cytokine-activated IκB kinases essential for NF-κB activation. Science. 1997; 278: 860–866. [DOI] [PubMed] [Google Scholar]
- 11.Thompson JE, Phillips RJ, Erdjument-Bromage H, et al. IκBβ regulates the persistent response in a biphasic activation of NF-κB. Cell. 1995; 80: 573–582. [DOI] [PubMed] [Google Scholar]
- 12.Strauch ED, Wang JY, Bass BL. Bile salt stimulates intestinal epithelial migration through TGF-β after wounding. J Surg Res. 2001; 97: 49–53. [DOI] [PubMed] [Google Scholar]
- 13.Hoffman AF. Intestinal absorption of bile acids and biliary constituents. Physiol GI Tract. 1994; 2: 1845–1865. [Google Scholar]
- 14.Martinez JD, Stratagoules ED, LaRue JM, et al. Different bile acids exhibit distinct biological effects: the tumor promoter deoxycholic acid induces apoptosis and the chemopreventive agent ursodeoxycholic acid inhibits cell proliferation. Nutr Cancer. 1998; 31: 111–118. [DOI] [PubMed] [Google Scholar]
- 15.D’Addio V, Sayles JM, Alvarez C, et al. Bile: A trophic factor for rabbit esophageal epithelium in vitro. Surg Forum. 1996; 647: 125–127. [Google Scholar]
- 16.Palmer DG, Paraskeva C, Williams AC. Modulation of p53 expression in cultured colonic adenoma cell lines by the naturally occurring lumenal factors butyrate and deoxycholate. Int J Cancer. 1997; 73: 702–706. [DOI] [PubMed] [Google Scholar]
- 17.Quaroni AJ, Wands RL, Trelstad KJ, et al. Epithelial cell cultures from rat small intestine. J Cell Biol. 1979; 80: 248–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCormack SA, Wang JY, Johnson LR. Polyamine deficiency causes reorganization of F-actin and tropomyosin in IEC-6 cells. Am J Physiol. 1994; 267: C715–C722. [DOI] [PubMed] [Google Scholar]
- 19.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970; 227: 680–685. [DOI] [PubMed] [Google Scholar]
- 20.Ye ZS, Samuels HH. Cell- and sequence-specific binding of nuclear protein to 5′-flanking DNA of the rat growth hormone gene. J Biol Chem. 1987; 262: 6313–6317. [PubMed] [Google Scholar]
- 21.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72: 248–254. [DOI] [PubMed] [Google Scholar]
- 22.Li LJ, Li JN, Rao M, et al. Inhibition of polyamine synthesis induces p53 gene expression but not apoptosis. Am J Physiol. 1999; 276: C946–954. [DOI] [PubMed] [Google Scholar]
- 23.Harter JL. Critical values for Duncan’s new multiple range test. Biometrics. 1960; 16: 671–685. [Google Scholar]
- 24.Hilt W, Wolf DH. Proteosomes: destruction as a programme. Trends Biochem Sci. 1996; 21: 96–102. [PubMed] [Google Scholar]
- 25.Li L, Rao JN, Bass BL, Wang J-Y. NF-κB activation and susceptibility to apoptosis after polyamine depletion in intestinal epithelial cells. Am J Physiol. 2001; 280: G992–G1004. [DOI] [PubMed] [Google Scholar]
- 26.Fiedler MA, Wernke-Dollries K, Stark JM. Inhibition of TNF-α induced NF-κB activation and IL-8 release in A549 cells with the proteosome inhibitor MG-132. Am J Respir Cell Mol Biol. 1998; 19: 259–268. [DOI] [PubMed] [Google Scholar]
- 27.Zernecke A, Weber KSC, Weber C. Combined modulation of the mesangial machinery for monocyte recruitment by inhibition of NF-κB. Am J Physiol. 2001; 281: C1881–1888. [DOI] [PubMed] [Google Scholar]
- 28.Vaezi MF, Singh S, Richter JE. Role of acid and duodenogastric reflux in esophageal mucosal injury: a review of animal and human studies. Gastroenterology. 1995; 108: 1897–1907. [DOI] [PubMed] [Google Scholar]
- 29.Huang XP, Fan XT, Desjeux JF, et al. Bile acids, non-phorbol-ester-type tumor promoters, stimulate the phosphorylation of protein kinase C substrates in human platelets and colon cell line Ht29. Int J Cancer. 1992; 52: 444–450. [DOI] [PubMed] [Google Scholar]
- 30.Fitzer CJ, O’Brian CA, Guillem JG, et al. The regulation of protein kinase C by chenodeoxycholate, deoxycholate and several structurally related bile acids. Carcinogenesis. 1987; 8: 217–220. [DOI] [PubMed] [Google Scholar]
- 31.Craven PA, Pfanstiel J, DeRubertis FR. Role of activation of protein kinase C in the stimulation of colonic epithelial proliferation and reactive oxygen formation by bile acids. J Clin Inves. 1987; 79: 532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pongracz J, Clark P, Neoptolemos JP, et al. Expression of protein kinase C isoenzymes in colorectal cancer tissue and their differential activation by different bile acids. Int J Cancer. 1995; 61: 35–39. [DOI] [PubMed] [Google Scholar]
- 33.Hsu H, Huang J, Shu HB, et al. TNF-Dependent recruitment of the protein kinase RIP to the TNF receptor 1 signaling complex. Immunity. 1996; 4: 387–396. [DOI] [PubMed] [Google Scholar]
- 34.Hsu H, Shu HB, Pan MG, et al. TRADD-TRAF-2 and TRAD-FADD interactions define two distinct TNF receptor signal transduction pathways. Cell. 1996; 84: 299–308. [DOI] [PubMed] [Google Scholar]
- 35.Burns K, Martinon F, Esslinger C, et al. MYD88, and adaptor protein involved in interleukin-1 signaling. J Biol Chem. 1998; 273: 12203–12209. [DOI] [PubMed] [Google Scholar]
- 36.Cao Z, Henzel WJ, Gao X. IRAK: a kinase associated with the interleukin-1 receptor. Science. 1996; 271: 1128–1131. [DOI] [PubMed] [Google Scholar]
- 37.Cao Z, Xiong J, Takeuchi M, et al. TRAF6 is a signal transductor for interleukin-1. Nature. 383:443–446. [DOI] [PubMed]
- 38.Wesche H, Henzel WJ, Shillinglaw W, et al. MYD88: an adaptor that recruits IRAK to the IL-1 receptor complex. Immunity. 1997; 7: 837–847. [DOI] [PubMed] [Google Scholar]
- 39.Axel UD, Podolsky DK. Cytokine modulation of intestinal epithelial cell restitution: central role of transforming growth factor β. Gastroenterology. 1993; 105: 1323–1332. [DOI] [PubMed] [Google Scholar]
- 40.Armendariz-Borunda J, Katai H, Jones CM, et al. Transforming growth factor-β gene is transiently enhanced at a critical stage during liver regeneration after CCl4 treatment. Lab Invest. 1993; 69: 283–294. [PubMed] [Google Scholar]
- 41.Ignotz RA, Massague J. Transforming growth factor β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986; 261: 4337–4345. [PubMed] [Google Scholar]
- 42.Peitenpol JA, Holt JT, Stein RW, et al. Transforming growth factor β1 suppression of c-myc gene transcription: role in inhibition of keratinocyte proliferation. Proc Natl Acad Sci USA. 1990; 87: 3758–3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Basson JA, Modlin IM, Flynn SD. Independent modulation of migration and proliferation by growth factors, matrix proteins and pharmacological agents in an in vitro model of mucosal healing. Surgery. 1992; 112: 299–308. [PubMed] [Google Scholar]
- 44.Barnard JA, Lyons RM, Moses HL. The cell biology of transforming growth factor β. Biochim Biophys Acta. 1990; 1032: 79–87. [DOI] [PubMed] [Google Scholar]
- 45.Ciacci C, Lind SE, Podolsky DK. Transforming growth factor β regulation of migration in wounded rat intestinal epithelial monolayers. Gastroenterology. 1993; 105: 93–101. [DOI] [PubMed] [Google Scholar]