

Abstract
Objective
To study the effects of peritoneal resuscitation from hemorrhagic shock.
Summary Background Data
Methods for conventional resuscitation (CR) from hemorrhagic shock (HS) often fail to restore adequate intestinal blood flow, and intestinal ischemia has been implicated in the activation of the inflammatory response. There is clinical evidence that intestinal hypoperfusion is a major factor in progressive organ failure following HS. This study presents a novel technique of peritoneal resuscitation (PR) that improves visceral perfusion.
Methods
Male Sprague-Dawley rats were bled to 50% of baseline mean arterial pressure (MAP) and resuscitated with shed blood plus 2 equal volumes of saline (CR). Groups were 1) sham, 2) HS + CR, and 3) HS + CR + PR with a hyperosmolar dextrose-based solution (Delflex 2.5%). Groups 1 and 2 had normal saline PR. In vivo videomicroscopy and Doppler velocimetry were used to assess terminal ileal microvascular blood flow. Endothelial cell function was assessed by the endothelium-dependent vasodilator acetylcholine.
Results
Despite restored heart rate and MAP to baseline values, CR animals developed a progressive intestinal vasoconstriction and tissue hypoperfusion compared to baseline flow. PR induced an immediate and sustained vasodilation compared to baseline and a marked increase in average intestinal blood flow during the entire 2-hour post-resuscitation period. Endothelial-dependent dilator function was preserved with PR.
Conclusions
Despite the restoration of MAP with blood and saline infusions, progressive vasoconstriction and compromised intestinal blood flow occurs following HS/CR. Hyperosmolar PR during CR maintains intestinal blood flow and endothelial function. This is thought to be a direct effect of hyperosmolar solutions on the visceral microvessels. The addition of PR to a CR protocol prevents the splanchnic ischemia that initiates systemic inflammation.
Shock as a result of blood loss results in inadequate organ perfusion and tissue oxygenation, with subsequent tissue acidosis and lactic acidemia. Central hemodynamic endpoints for resuscitation from compensated hemorrhagic shock, when taken alone, fail to measure adequate organ perfusion and oxygen delivery. Two separate clinical studies by Scalea et al. 1 and Abou-Khalil et al. 2 document that despite normalization of blood pressure, heart rate, and urine output, tissue hypoperfusion persists in 80% to 85% of patients, as evidenced by lactic acidemia and decreased mixed venous oxygen saturation. Other clinical studies have shown that the level and the rate of normalization of serum lactate (indirect measures of tissue oxygen utilization) correlated with mortality both in degree of elevation and in the time-dependent rate of normalization. 3,4 Systemic base deficit (an indicator of tissue perfusion) also shows a similar predictive pattern of mortality. 5 However, interventions that focus on correction of this oxygen debt by driving oxygen transport variables, such as cardiac index or oxygen delivery index following resuscitation, to supernormal levels fail to reduce mortality in severely injured patients. 6,7
Taken as a whole, those studies indicate that systemic markers of tissue perfusion fail to account for the distribution of blood flow to different tissue beds. Certain tissues appear to benefit from a redistribution of flow (e.g., heart, brain, and striated muscle), while the splanchnic vascular bed shows a persistent hypoperfusion following conventional intravenous resuscitation (CR) from shock. Intestinal ischemia after hemorrhagic shock (HS) has been documented in both experimental 8,9 and clinical 10 studies. It appears that this splanchnic hypoperfusion results from local microvascular endothelial cell dysfunction, 11,12 and the resultant intestinal injury leads the gut-associated lymphatic tissue (GALT) to become a cytokine-generating organ. 13 Recent experimental data suggest that those proinflammatory cytokines cause remote organ injury through lymphatic drainage from the gut. 14
Standard therapies for hemorrhagic shock focus on intravascular volume replacement and preservation of cell membrane function. Additional systemic therapies that are designed to preserve endothelial cell function and splanchnic perfusion appear to have other adverse consequences. 15–19 Despite a better understanding of the physiologic events of fluid resuscitation regimens 20–22 that emphasize rapid hemodynamic stabilization, reversal of the pathophysiologic course in the splanchnic circulation after CR continues to constitute a major but elusive treatment goal. Investigators have adopted several approaches to alleviate the hemorrhage-induced host-stress response and gut hypoperfusion. Blockage of proinflammatory mediators, integrins, and other cell adhesion molecules or addition of radical scavengers to CR regimens gives marginal alterations in the inflammatory response and microvascular injury. 23,24 Pentoxifylline-supplemented resuscitation 8,17 or pretreatment with selective inhibitors of the complement system 9 or with heparin 16 can prevent the microvascular impairment to restore tissue perfusion after CR from HS.
Commercial peritoneal dialysis solutions possess a vasodilation effect on visceral and parietal arterioles. 25 Our laboratory has recently demonstrated a generalized and sustained dilation effect of these solutions on the intestinal microcirculation. 26 Thus, we hypothesized that initiation of peritoneal resuscitation (PR) with commercially available hyperosmolar dextrose-based solution (Delflex 2.5%) during CR from HS prevents the progressive intestinal vasoconstriction and hypoperfusion and the sequelae of impaired microvascular dilation.
METHODS
Animals and Surgery
Male Sprague-Dawley rats (205 ± 5 g) were used in all experiments, which conformed to National Research Council guidelines and were approved by the Institutional Animal Care and Use Committee. Anesthesia was induced with intraperitoneal pentobarbital (60 mg/kg) and supplemented with subcutaneous injections as needed. Body temperature was maintained at 37 ± 0.5°C. Tracheostomy was performed and the animal was allowed to breathe room air. The carotid artery was cannulated with a PE-50 catheter to provide online recording of arterial blood pressure and heart rate. The right femoral artery and vein were cannulated with PE-50 to allow for blood withdrawal and resuscitation.
Solution A was a modified non-vasoactive Krebs’ solution that contained 6.92 g/L sodium chloride, 0.44 g/L potassium chloride, 0.37 g/L calcium chloride, and 2.1 g/L sodium bicarbonate at a pH of 7.4 (285–300 mOsm/L). Solution B was a 2.5% dextrose-based commercial peritoneal dialysis solution (Delflex, Fresenius USA, Inc., Ogden, UT) that contained 0.567 g/L sodium chloride, 0.392 g/L sodium lactate, 0.0257 g/L calcium chloride, and 0.0152 g/L magnesium chloride at a pH of 5.5 (398 mOsm/L).
The technique of intestinal video microscopy has been previously described. 17 Briefly, the distal ileum with an intact neurovascular supply was exposed through a midline abdominal incision. A 3-cm segment was opened along the antimesenteric border and suspended, serosal side up, over a viewing port in a 60-mL bath. The bathing solution (solution A) was maintained at 37°C and bubbled with nitrogen and carbon dioxide to maintain the pH at 7.4. The animal board and tissue bath were positioned on a trinocular microscope for direct in vivo intravital microscopy. Microvascular images were transmitted through the microscope and optical Doppler velocimeter (Microcirculation Research Institute, Texas A&M University, College Station, TX) to measure center-line red blood cell velocity, which allowed the calculation of blood flow. The image was then transmitted to a digital camera (Hitachi Denshi, Models [K-P D51/D50]), which provided 30 images per second of streamline video to a computer, where the digitalized microvascular images were stored for later measurement of microvascular diameters.
Criteria for an acceptable data collection during intravital microscopy included a baseline mean arterial pressure (MAP) > 90 mmHg, a center-line red blood cell velocity in a first-order arteriole > 20 mm/s, and active vasomotion in the intestinal A3 arterioles. Nomenclature for intestinal microvessels, described by Bohlen and Gore, 27 was used. First-order arterioles (A1) arise from a mesenteric arcade artery and penetrate the muscle layers to the submucosal layer, where second-order arterioles (A2) arise to run along the longitudinal axis of the bowel. First- and second-order venules parallel the first- and second-order arterioles. A2 arterioles give rise to branching second-order arcade vessels and smaller third-order arterioles (A3). The A3 vessels branch at right angles from A2 arterioles and become distal A3 (dA3) vessels, which terminate in the mucosa as a central villus arteriole. Along their course, A3 arterioles also give rise to smaller proximal A3 (pA3) arterioles that supply the seromuscular layers of the bowel wall.
Each animal and tissue bath was allowed to equilibrate for 40 minutes with the exteriorized ileum continuously suffused with solution A. Blood pressure, heart rate, rectal and bath temperatures, and bath pH were continuously monitored (Digi-Med Signal Analyzers, Micro-Med, Louisville, KY) and recorded every 5 minutes. Microvascular data consisted of A1, A2, pA3, and dA3 arteriolar diameters and centerline red cell velocity in the inflow A1 arteriole and the outflow V1 venule. At each “data time point,” multiple measurements were made on each vessel type, and 1-minute averages were used as the value for each vessel at that data time point. At the conclusion of each experiment, one dose of acetylcholine (ACh, 10−5 mol/L) was topically administered in the tissue bath and microvascular data were taken at 1-minute intervals over 10 minutes to assess endothelial-dependent vasodilation. Endothelial-independent relaxation to the nitric oxide (NO) donor sodium nitroprusside (SNP, 10−4 mol/L) was also assessed at the end of each animal protocol to determine the maximal dilation capacity for each microvessel.
The dilation that occurred as a result of Ach or Delflex in the tissue bath was calculated as a percentage of the NO-dependent dilation capacity (NDC) by:
% of NDC = [(x – BL)/(NDC – BL)] × 100
where x = measured vessel diameter during Delflex dwell or for the given ACh dose; NDC = the maximum recorded diameter with sodium nitroprusside; and BL = initial baseline value during solution A dwell. Percent of NO dilation capacity was used for this analysis to account for variations in dilator capacity and baseline among different vessel types.
The measured A1 red blood cell velocity was converted to blood flow (Jv) by the formula: Jv = (V/1.6) (πr2) (0.001), where V = red blood cell velocity (mm/s), r = vessel radius (μm), 1.6 is a factor to convert centerline velocity to average velocity across the vessel cross-sectional area, and 0.001 is the conversion factor to give flow in nL/s.
Hemorrhage was done to 50% of baseline MAP by withdrawing blood from the femoral artery at a maximum rate of 1 mL/min into a syringe prerinsed with 0.02 mL heparin (1,000 units/mL). The target blood pressure was maintained for 60 minutes by withdraw or reinfusion of blood as needed. Intravascular resuscitation was then performed with the return of shed blood and two volumes of normal saline over 30 minutes. Hemodynamic and microvascular data were then collected for 120 minutes.
Experimental Protocols
After the stabilization period, animals were placed in three experimental groups. Group 1 consisted of sham controls that had no HS/RES with solution A in the tissue bath. Group 2 underwent HS/CR with solution A in the tissue bath. Group 3 had HS/CR, but solution A was replaced with solution B at the time that CR was initiated (PR group). Each group consisted of 10 animals.
Statistics
All data are presented as mean ± SEM unless stated otherwise. Percentage change from baseline for each group, and differences among groups for response to ACh and SNP were compared by two-way ANOVA and Tukey, Dunnett, or Bonferroni multiple-comparison tests. A result was considered to be significant if the probability of a type-one error was P < .05.
RESULTS
There were no significant differences among measured baseline values between the sham and experimental groups. As expected, HS significantly reduced MAP (Fig. 1) and increased heart rate (data not shown) in groups 2 and 3 compared to their consecutive baseline values and to group 1. Resuscitation restored hemodynamics to baseline values and maintained them during the entire 2-hour post-resuscitation observation period without any further administration of fluids. As anticipated, the pH of the bathing solution in the PR group was significantly lower than the baseline value, and it remained lower during the 2-hour post-resuscitation period (Fig. 2) because commercial peritoneal dialysis solutions typically have a pH between 5 and 6.

Figure 1. Mean arterial blood pressure (MAP) data. BL, baseline; H, hemorrhagic shock; R, resuscitation. CR-treated, conventional resuscitation; PR-treated, peritoneal resuscitation. Hemorrhage showed a decrease in MAP. §P < .001 vs. sham by two-way ANOVA and Tukey posttest. *P < .001 vs. baseline by repeated measures ANOVA and Dunnett posttest.

Figure 2. Bathing solution pH data. BL, baseline; H, hemorrhagic shock; R, resuscitation. CR-treated, conventional resuscitation; PR-treated, peritoneal resuscitation. pH was lower during the resuscitation and post-resuscitation periods in the PR-treated group. *P < .001 vs. sham or CR-treated groups by two-way ANOVA and Tukey posttest. §P < .001 vs. baseline and repeated measures ANOVA and Dunnett posttest.
Microvascular Data
The responses of the intestinal arterioles to the events of HS and resuscitation are shown in Figure 3. Vessel diameters of intestinal arterioles were highly stable over the 6 hours of the experiment in the sham animals. There was a significant vasoconstriction of the A1 arterioles during HS in groups 2 and 3. This vasoconstriction was not seen in A3 vessels. Initially, resuscitation restored A1 diameters to baseline values in both hemorrhage groups. However, progressive vasoconstriction developed in all vessels in group 2 animals during the post-resuscitation period. In contrast, vessel diameters in group 3 PR animals demonstrated a significant and sustained vasodilation during the post-resuscitation period. Because different-level arterioles might possess differences in vascular tone depending on arteriolar size, and because these arterioles respond much differently to HS, post-resuscitation-mediated vascular diameter changes were expressed as a percentage of the NO maximal dilation capacity (Fig. 4). Arteriolar size had no significant effect on the percentage amount of post-resuscitation-induced changes in microvessel diameter. However, PR treatment in group 3 not only reversed the progressive vasoconstriction that occurred during the 2-hour post-resuscitation period in the CR group 2 but also caused an immediate and sustained generalized vasodilation during the entire 2-hour post-resuscitation period in all vessels.
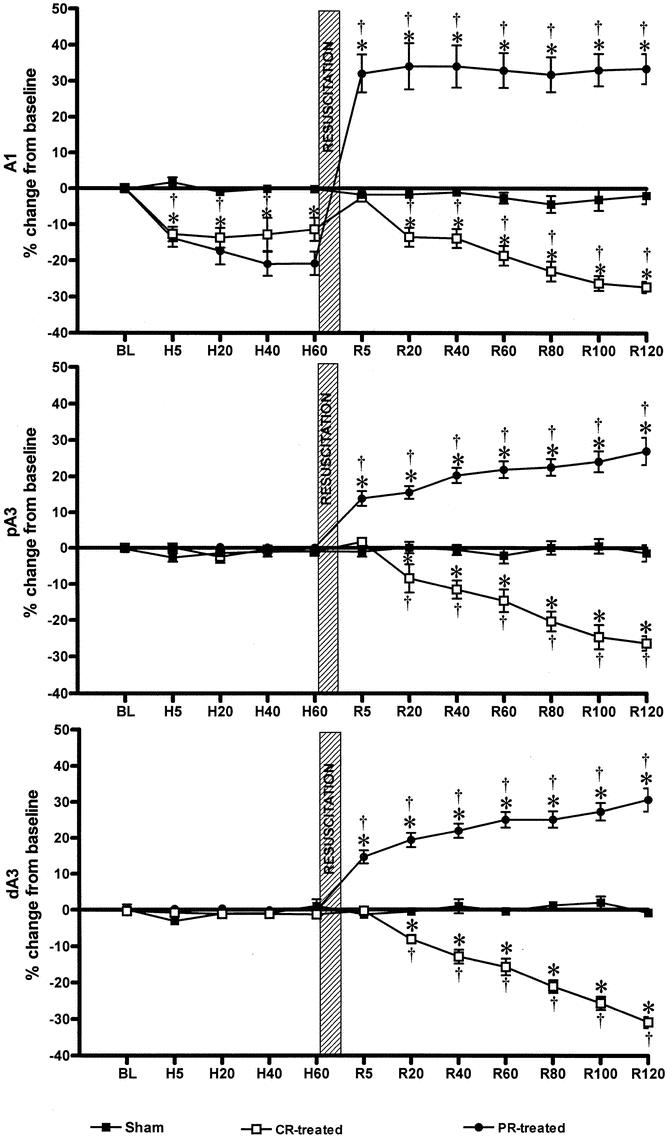
Figure 3. Microvascular diameter data. BL, baseline; H, hemorrhagic shock; R, resuscitation. CR-treated, conventional resuscitation; PR-treated, peritoneal resuscitation. *P < .001 vs. sham by repeated measures two-way ANOVA and Bonferroni posttest. †P < .001 vs. baseline by two-way ANOVA and Dunnett test.
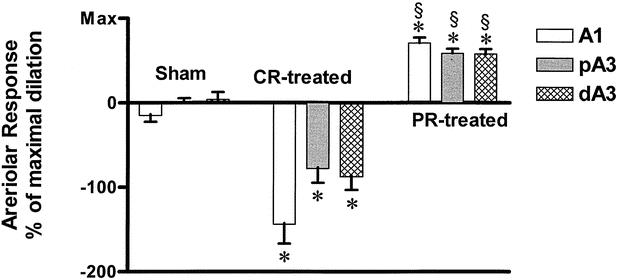
Figure 4. Post-resuscitation arteriolar diameter. A1, inflow first-order arteriole; pA3, proximal third-order arteriole; dA3, distal third-order arteriole; CR-treated, conventional resuscitation; PR-treated, peritoneal resuscitation; Max, maximum dilation response to SNP. *P < .001 vs. sham by two-way ANOVA and Tukey posttest. §P < .0001 vs. CR-treated group by two-way ANOVA and Bonferroni posttests.
HS caused a significant decrease in baseline diameters in V1 (−9.9 ± 2.1%), V2 (−13.97 ± 2.7%), and V3 (−15.85 ± 4.55%) venules. Unlike the arteriolar response to hemorrhage, the venular response was generalized and significantly greater (P < .05) in the smaller venules. Resuscitation initially restored this decrease in venular diameter to slightly above baseline values, followed by a progressive decrease in all venule sizes during the post-resuscitation period in group 2. However, in the PR group 3, the increase from baseline in venular diameter was greater (P < .01) in smaller venules: V1 (+11.49 ± 2.7%), V2 (+15.79 ± 3.36%), and V3 (+39.09 ± 7.08) at 120 minutes after resuscitation.
Blood Flow
Blood flow was stable throughout the observation period in group 1 (sham) animals (Fig. 5). Baseline A1 blood flow in the CR group 2 was 564 ± 20 nL/s, decreased to 138 ± 23 nL/s (P < .0001) during HS, and returned to baseline levels (507 ± 33 nL/s) after CR but then progressively decreased to less than 200 nL/s (P < .0001) within 40 minutes of CR. In contrast, A1 baseline flow was 568 ± 20 nL/s, decreased to 204 ± 23 nL/s during HS, and increased above the baseline to 754 ± 50 nL/s (P < .01) during the entire 2 hours in PR group 3 animals. A similar pattern was observed in venules, where V1 baseline outflow decreased from 392 ± 41 to 147 ± 53 nL/s (P < .01). Resuscitation restored V1 to the baseline value initially, but flow decreased during the post-resuscitation period in the CR group 2. In the PR group 3, V1 outflow was maintained above baseline at 463 ± 168 nL/s during the 2-hour post-resuscitation period.

Figure 5. A1 arteriolar blood flow data. BL, baseline; H, hemorrhagic shock; R, resuscitation. CR-treated, conventional resuscitation; PR-treated, peritoneal resuscitation. Hemorrhage significantly reduced A1 blood flow compared to sham. *P < .001 vs/ baseline by two-way ANOVA and Bonferroni posttest. †P < .01 vs. baseline by repeated measures ANOVA and Dunnett posttest. §P < .001 between groups by two-way ANOVA and Bonferroni posttest.
Endothelial Cell Function
At 2 hours after resuscitation, the arteriolar response to the endothelial-dependent agonist (ACh, 10−5 mol/L) showed a significant endothelial cell dysfunction in the CR group 2 compared to group 1 (sham). Direct peritoneal resuscitation in the PR group 3 reversed this endothelial cell dysfunction (P < .0001) (Fig. 6). There was no significant difference in arteriolar response to ACh between the group 1 and group 3 animals. No difference was observed in endothelial-independent dilation that was elicited by the addition of sodium nitroprusside among any of the animal groups. This indicates that the vascular dilator machinery remained intact in all three regardless of the experimental group. The vasodilation in the PR group 3 was similar in all levels of arterioles (Fig. 7). However, this generalized arteriolar dilation was less than the maximal dilation that was obtained with the endothelial-independent dilator sodium nitroprusside (SNP, 10−4 mol/L) or the endothelial-dependent dilator ACh. This suggests that the site of action of direct PR is more likely the vascular endothelial cell, not the vascular smooth muscle cell.
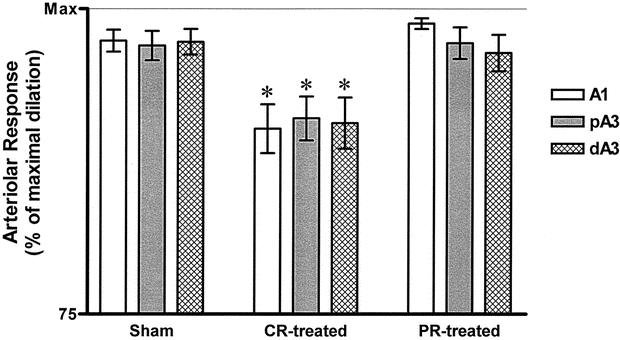
Figure 6. Endothelial-dependent relaxation to acetylcholine (Ach, 10−5 mol/L). A1, inflow first-order arteriole; pA3, proximal third-order arteriole; dA3, distal third-order arteriole; CR-treated, conventional resuscitation; PR-treated, peritoneal resuscitation; Max, maximum dilation response to SNP. Data presented as percentage of Max. CR from hemorrhagic shock caused impaired endothelial-dependent vasodilation. *P < .01 by two-way ANOVA followed by Tukey posttest.
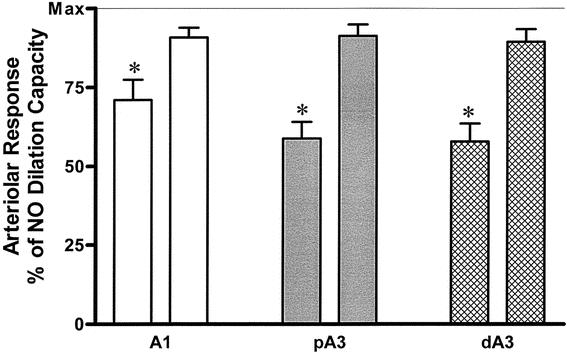
Figure 7. Magnitude of intestinal arteriolar response. A1, inflow first-order arteriole; pA3, proximal third-order arteriole; dA3, distal third-order arteriole; Max, maximum dilation response to SNP. At 2 hours after resuscitation, PR (first bar of each pair) produced a significant vasodilation at all arterioles examined. The magnitude of this dilation was significantly less than the ACh-mediated dilation (second bar of each pair). *P < .001 by two-way ANOVA and Bonferroni posttest.
DISCUSSION
Traditional methods of resuscitation from HS often fail to restore adequate intestinal blood flow in both the experimental 8,17 and clinical settings. 28 Persistent intestinal ischemia has been implicated as a cause for activation of the inflammatory response following CR from HS. It appears that the systemic inflammatory response is launched through lymphatic drainage of the intestinal tract 14 and that intestinal hypoperfusion is a major factor in the progressive organ failure that occurs following injury and HS. Our study presents a novel technique of direct intestinal resuscitation that uses a commercially available dialysis solution to improve visceral perfusion.
Our study confirms previous observations that in intestine 11,16,18 and other splanchnic organs, 29 CR from HS results in progressive intestinal microvascular vasoconstriction and hypoperfusion despite adequate restoration of baseline central hemodynamics. This microvascular vasoconstriction is associated with endothelial cell dysfunction, as reflected by impairment of the endothelial-dependent dilation response. The mechanism responsible for the post-resuscitation microvascular impairment is unclear. Some investigators have suggested that endothelial cell dysfunction accounts for the progressive post-resuscitation vasoconstriction. 15,16 Based on in vitro studies, 12 a reduced release of NO occurs very early after trauma hemorrhage and persists after CR; and this was interpreted to indicate endothelial cell dysfunction. Our previous in vivo studies have documented a reduction in NO release in the small intestine vasculature after HS/CR, and this appeared to be the result of endothelial cell dysfunction. 30,31
Among the various resuscitation regimens used to treat HS, few prevent post-resuscitation vasoconstriction. Pretreatment with heparin before HS/RES can preserve endothelial cell function. 16 This was attributed to the inhibitory effect of heparin on the complement system. 18 Subsequently, we have shown that specific inhibition of complement activation in vivo before initiation of CR preserves endothelial cell function and restores baseline diameter and blood flow in the intestinal vasculature. 9 Our present study demonstrates that initiation of PR simultaneously with intravenous CR not only preserves endothelial cell function and restores baseline diameters and blood flow, but also produces a sustained and generalized dilation throughout the intestinal vasculature to increase both arterial inflow and venous outflow. These effects indicate that the deleterious pathophysiologic events of HS/CR that produce tissue damage and release of proinflammatory mediators can be moderated by PR. Therefore, PR appears to be a simple treatment that prevents the progression of intestinal ischemia after HS/CR, a progression that otherwise leads to systemic inflammation and subsequent organ failure. Furthermore, PR is also a technique that could be adapted as a diagnostic tool to detect internal bleeding or as a therapeutic irrigation.
The sustained post-resuscitation vasodilation in our PR-treated group (group 3) is an effect of the dilation properties of commercial peritoneal dialysis solutions. These properties are attributed to hyperosmolality and the lactate buffer anion of the solution, 25 but could also be an effect of the glucose content. 32 Dilation is not restricted to the intestinal microcirculation, but also involves other parietal and visceral tissues (unpublished data). It is conceivable that PR treatment of hemorrhagic shock might halt ongoing ischemia and reverse the progress of systemic inflammation that originates from GALT tissue. 13 Other potential therapeutic effects of PR could include correction of acidosis and reduction of edema formation. Peritoneal dialysis has been shown to correct the low-flow state associated with HS, attenuates leukocyte adhesion, 32 and improves blood rheology in the microcirculation and therefore might halt or prevent the course of these white cell-initiated cascades. As noted before, we think that dialysis solutions preserve endothelial function primarily through an osmotic effect at the endothelial cell membrane.
In a recent study, 26 we demonstrated that ex vivo exposure of intestinal arterioles to conventional peritoneal dialysis solutions produces a sustained and generalized vasodilation, which is independent of arteriolar level and solution pH. Our present ex vivo model uses a very small segment of the terminal ileum (∼1 cm2), which is positioned in a relatively large tissue bath (60 mL). This means that the sustained vasodilation observed during the 2-hour post-resuscitation period in the ex vivo model might not apply to the in vivo clinical peritoneal dialysis situation, where there is a continuous tissue equilibration to dissipate the dilation components in the peritoneal solution. It is generally acknowledged that the vasoactive components of the commercial peritoneal dialysis solutions are hyperosmolality, lactate buffer anion, and low pH. Of these three components, hyperosmolality and lactate appear to be more potent vasoactive substances than pH. 26 However, despite the daily clinical use of commercial peritoneal dialysis solutions, the mechanisms of their medicated dilation are not fully understood. At this time we propose the possibility that extrusion of cellular water by osmosis through the “water only” transendothelial aquaphorin-1 pathways (aquaporin-1) leads to cell shrinkage (deformation) and an associated relative increase in intracellular ionic contents, especially Ca2+ and K+. These changes in intracellular ions could be the signal that initiates an endothelial-mediated vascular relaxation, largely mediated via an endothelial-derived hyperpolarizing factor. Alternatively, in the absorbing intestine, glucose mediates NO release via adenosine A2b receptors to cause significant relaxation of premucosal arterioles. 33 However, hyperosmolality-mediated dilation is not specific for glucose. 34 The NO pathway might not contribute to hyperosmolar-mediated dilation, 34 or depending on the rate and timing of NO production, NO might act as a direct signaling messenger for cytoprotection or as an indirect cytotoxic molecule. 35
The vascular endothelium plays an active role in physiologic processes such as hemostasis, regulation of vessel vascular tone, and vascular permeability. Events that disrupt endothelial cell wall integrity, and thus endothelial permeability, result in alteration of the normal physiologic function of these cells. In HS, neurohumoral reflexes promote vasoconstriction in certain vascular beds to ensure that an adequate fraction of the cardiac output supplies oxygen and nutrients to vital organs. 36 Another adaptive response to HS is the compulsory translocation of body water, electrolytes, and proteins to restore an effective blood volume and suppress baroreceptor activity. This results in cell swelling, which is associated with an increase in cellular H2O, Na+, K+, Ca++, and ATP, 37 together with accumulation of K+ in the interstitium. 38 Although these pathophysiologic events and the host-stress response are incited during shock and tissue injury, their sequence of occurrence and the consequences of these changes are still to be determined.
Recently, there has been an increasing interest in small-volume RES with intravenous administration of hypertonic saline. This solution was reported to preserve immunologic function compared to traditional lactated Ringer’s. 19 Hypertonic saline has the added advantage of lower infusion volumes to restore cardiac output and blood pressure, 39 but these beneficial effects are transient. 40 Furthermore, clinical trials with hypertonic intravenous saline resuscitation have shown a higher incidence of renal failure, impaired oxygenation, and prolonged number of days on a ventilator, without improved survival. 41 Other potential limitations for use of intravenous hypertonic saline resuscitation are sodium overload and increases in plasma osmolality. These problems are not encountered when dextrose-based peritoneal dialysis solutions are introduced in the peritoneal cavity, and thus these problems are not anticipated when used for direct peritoneal resuscitation.
In conclusion, we have shown that resuscitation from HS with conventional intravenous solutions resulted in progressive intestinal microvascular vasoconstriction and hypoperfusion despite restoration and maintenance of systemic hemodynamics. This vasoconstriction is associated with endothelial cell dysfunction manifested by impaired endothelial-dependent dilation in the intestinal microcirculation. Exposure of the intestine to a commercial dialysis solution concomitantly with intravenous resuscitation preserved endothelial cell function (PR group) and changed the progressive post-resuscitation vasoconstriction to a generalized and sustained vasodilation. This vasodilation is associated with a marked increase in intestinal arterial blood flow and venous outflow without adverse effects on systemic hemodynamic parameters. Thus, the addition of PR to a conventional resuscitation protocol prevents the progressive splanchnic ischemia that is thought to initiate systemic inflammation after injury and shock.
Discussion
Dr. Timothy C. Fabian (Memphis, TN): This is really a nice experiment: it is pretty straightforward and it is even easy for me to understand. It seems like an ideal area for translational research. Most of my comments revolve around that.
This was an ex vivo experiment. It would be very interesting to do in vivo experiments, and I suspect you may have already begun. If you have not, I am going to start. The first group of experiments could address functional and architectural visceral changes from the present experiments—we see flow changes and nice changes in diameter of the blood vessels, but, of course, for a difference to be a difference, it has to make a difference. Have you thought about looking at the histopathology of the intestinal mucosa, and especially the villous architecture? One would assume that if it makes a difference that in fact you would get alterations in the villous architecture.
What about other experiments looking at long-term survival with secondary bacterial challenges looking at organ failure and the like, and perhaps increasing the severity of shock. You mentioned that this is a 100% survival experiment. What about increasing the degree of shock?
The second group of questions is: What about using other hypertonic solutions? In the manuscript you suggested that this was in fact affected by the hypertonicity of the peritoneal dialysate. Have you thought about looking at urea, hypotonic saline, or other solutions?
Then finally, relating back again to translational research and clinical trials, previously your laboratory has done some really pretty work with complement blockade as well as pentoxifylline usage in a similar model. Those drugs of course aren’t used clinically, so I can see why we have not progressed with clinical work. But with the novel concepts presented today, it really would be pretty easy to translate this into the clinical environment. Millions of people receive peritoneal dialysis. We know it is completely safe. In this area, would it be effective? I really applaud the idea of doing it, and would suggest that you begin looking at a clinical trial to see if this would in fact work. Of course, some of the previously mentioned in vivo laboratory experiments need first to be done.
I would like to thank the Association for the privilege of discussing this interesting paper.
Dr. Lewis M. Flint, Jr. (Tampa, FL): Of the many attractive features of the Southern Surgical Association is not only a willingness but an encouragement of presenters and discussers to reflect on how we got to where we are today. I think it is worth noting that 20 years ago I was the first beneficiary of the relationship between the Department of Surgery at the University of Louisville and the Microcirculation Research Group at that university, a relationship that grew out of a conversation between Dr. Hiram Polk and Dr. Patrick Harris. Now there are several generations of faculty and residents who owe a debt of gratitude to Dr. Polk for having that conversation and maintaining that relationship, which has been remarkably productive over a number of years.
I enjoyed your paper, Dr. Garrison. I have two methodologic questions and one question that will ask you to speculate. The methodologic questions are these.
The intraperitoneal resuscitation solution was used as an adjunct to conventional resuscitation. I know that Dr. Harris, being the stickler for detail that he is, has probably demanded that you use the intraperitoneal solution alone and look at the results in the animal that both is continuing to be in hypotensive shock and after recovery. If you have that data, would you share it with us?
The other methodologic question that I would ask you about is this. There is a relapse of the microcirculation to a vasoconstrictor environment following conventional resuscitation for hemorrhagic shock. What that means and what one should do about it is a question that has not been answered by any research. David Hoyt has recently observed that patients and animals that revert into the hypotensive shock state after conventional resuscitation can be further resuscitated by additional volume therapy. I wonder, could you have obtained this same result by just giving additional volume?
Finally, since we have not been able to establish definitely a link between intestinal blood flow and intestinal oxygenation and subsequent multiple systems organ failure, where do you think we ought to go from here to look to establish that link?
Dr. Loring W. Rue, III (Birmingham, AL): I think the most appealing aspect of the concept presented here today is that this resuscitation potentially prevents the initiation of the adverse inflammatory responses after injury. Because of its complexity, developing a “magic bullet” to block various elements of inflammatory cascade seems unlikely. Hence, prevention, like much of what we do in trauma, seems like a very logical solution.
I do suggest that there is probably more work to be done in this field. The authors have convinced me of the vasodilatory effects of the resuscitation regimen; they haven’t demonstrated that this results result in a diminution in proinflammatory mediator generation. Additionally, I think we could get some more information in terms of delivery and consumption data to help enhance our appreciation of its effect.
As candidly admitted in the manuscript, the ex vivo model may not necessarily reflect the in vivo clinical scenario. Will the absorptive capacity of the parietal peritoneum influence the response to peritoneal resuscitation or just magnify it to the volume necessary to achieve the desired therapeutic endpoint?
Since the authors provide peritoneal resuscitation simultaneous with conventional resuscitation, is there any data available in the setting of conventional resuscitation preceding peritoneal resuscitation, which would seem to be a more likely clinical scenario?
I am concerned about one aspect of the manuscript in that the authors speculate that this approach may provide diagnostic value to detect intraperitoneal bleeding. In years past DPL perhaps forced the hand of many surgeons to operate on patients with injuries that we now know can be safely managed nonoperatively. Do you think we might risk a pendulum swing in the opposite direction?
I really enjoyed the presentation, and it really whets the appetite for future studies which will come from the surgeons at the University of Louisville which will further enhance our understanding of the injury biology.
Dr. Basil A. Pruitt, Jr. (San Antonio, TX): I wish you would speculate a little more on the mechanism involved. If the hypertonic ion is absorbed, one might expect that circulating blood volume would increase. Do you have blood volume measurements that could explain this? If the ion is not absorbed, then one would expect that the intraperitoneal volume would increase.
Dr. John A. Mannick (Boston, MA): Dr. Pruitt has asked a question that was on my mind; namely, isn’t there a possibility that you will be sucking fluid out of the circulatory system with this technique in vivo and therefore making the intravascular circulating volume lower, which I can’t imagine would really be good for the intestine? And I wonder what the original rationale for this was. I am absolutely thrilled with the results reported, but I must confess I don’t understand why this should work. I assume Dr. Garrison can enlighten us.
Dr. Gregory B. Bulkley (Baltimore, MD): I am fascinated with their approach to this. We know that the mesenteric vasoconstrictor response to shock is a physiologically advantageous response of the animal to redirect blood flow to, quote, more vital organs. This is Walter B. Cannon’s “fight or flight” response. What we assume is, is that because of modern medicine we are resuscitating and making animals and patients survive that wouldn’t have otherwise survived and then these animals or patients go on to live, and these problems that have been speculated on about cytokines and so forth then become a problem as a result of this. The point of all this is that it seems like it is important to ask the question of the effect of this on the systemic circulation, not just in the ways that Dr. Pruitt and Dr. Mannick spoke about, but if you are balancing mesenteric blood flow against systemic blood flow, wouldn’t it be nice to measure the two together to find out if you are robbing from Peter to pay Paul, and how exactly that is going to benefit you?
If I were going to ask that question, I am not sure I would use a microvasculature approach. I might instead measure the total flow and the A-VO2 difference, which would tell you oxygen consumption, and then you could measure with the gut in the animal the total systemic hemodynamic effect as well as the other.
I am just wondering if the authors would consider complementing these really excellent studies that are looking at a small area of the intestine with something that would compare those results in a microcosm, if you will, with what is happening in the whole animal.
The second quick question is, we have known from a lot of work from many laboratories that the pharmacologic mechanism to this is the hypersensitivity of the mesenteric vasculature resistant to angiotensin, and you can easily prevent this by just blocking angiotensin in a whole host of ways. Even just taking out the kidneys will do it. So I wonder if they have ever compared this with a pharmacologic approach, which seems a lot simpler.
Dr. H. Biemann Othersen, Jr. (Charleston, SC): That was an elegant study, beautifully presented. In 1949 Dr. Melvin Knisely came from Chicago to South Carolina, bringing with him his microvascular studies on sludging of blood and direct observation of blood vessels. My question is, you were looking at the diameter of the blood vessels and the flow through them. While looking through the microscope, was there any evidence of intravascular problems, such as sludging of blood, or change in the rheology of the blood flow?
Dr. F. Charles Brunicardi (Houston, TX): I would like to congratulate Drs. Garrison and Richardson for a superb physiologic study. The NIH is encouraging medical scientists to develop unifying hypotheses on how organs function. With our experience, surgeons have a golden opportunity to develop such hypotheses. There are different levels of regulation of any organ, such as hormonal, nutrient, neural, nitric oxide, the microcirculation, and most recently molecular mechanisms. I have two questions about your model.
I assume that you regulated the systemic blood pressure and temperature; please comment on those two important factors. Second, I would like you to speculate on the level of regulation of the effect you are seeing.
Dr. R. Neal Garrison (Louisville, KY): This experiment was in a nonlethal model. Although we have not done it, I would suspect that the histopathology would be normal in these animals.
We wanted to prove the effect of the treatment so that mortality or animal death was not a factor. With the most severe model, there has been shown in conventional resuscitation that there are white cells sticking, as well as other things. As Dr. Deitch pointed out 4 years ago, there are changes in the lymph flow and in the pattern of the cytokine release, and there are subsequent problems in the lung. A more severe model, which measures outcome a little bit later than 120 minutes after resuscitation, would show changes in histopathology. I don’t have any data, but I do not suspect that lung function in this model would actually change. Therefore, we would need to do this experiment in a more severe model, as you suggested.
There are certainly other hypertonic solutions that have been placed intravascularly into the bloodstream. But there are problems with those solutions, as you know, of hypertonicity and other systemic problems which we do not see compared to peritoneal dialysis patients. Sodium shifts and all of the things that happen when you use intravascular hypertonic solutions are not seen in dialysis patients. There is some absorption of the glucose. And in this model the glucose goes up to about 120 mg/dL at 1 hour into it and then returns to normal at 2 hours after the resuscitation.
There are certainly other systemic drugs—pentoxifylline, complement inhibition, magnesium chloride—a whole host of drugs that have been utilized, which do show that there is preservation of microvascular flow. However, none of these drugs are used clinically or for human experimentation. We simply wanted to use a solution that will vasodilate and is readily available clinically, to show the effect. The other drugs certainly would have to have FDA approval to be used, before they could be used in resuscitations from shock in a clinical trial.
We are hoping to do some beginning clinical trials. We want to show the effect in a small group of patients well studied and well controlled. In clinical patients, the problem is how to measure blood flow to the intestine. It is very hard to do. We can measure the gastric pH, but that is a relatively indirect method of measuring intestinal blood flow.
Dr. Flint, I appreciate your comments about Dr. Polk and Dr. Harris. Dr. Harris has certainly been my scientific mentor and colleague for a number of years, and he had significant input into the data analysis of this manuscript.
If you add additional intravascular volume to the animal, you can maintain the hemodynamics in a more severe model. In this model, the animal survives. If you push the tolerance of the animal past shock to almost unsustainable shock, you can return heart rate and blood pressure to normal, but it will deteriorate. You can maintain it by increasing infusions of saline solution. In this model, you don’t have to.
We have resuscitated animals with up to five volumes of saline, and the microcirculation deteriorates regardless of the amount of intravascular saline. We cannot resuscitate the microvascular bed with intravascular volume replacement alone.
We have not established a link to organ system failure. Those are future studies that we need to address.
Dr. Rue, I thank you for your comments.
We do not know what the effect of this solution is on the cytokines. We do have studies ongoing. We are trying to reduplicate Dr. Deitch’s experiment, measuring the cytokines in the lymph to see if they are improved, but we have no data to support that.
We do have some interesting data, which we haven’t completed, which show that the peritoneal resuscitation preserves the flow into other microcirculatory beds. It is very interesting and leads to the question of how it occurs. I am not really sure. I believe it is the link where you downregulate or decrease the intestinal inflammatory cytokines, which then ultimately have the effect on the parietal or on the extraparietal microcirculation. I do not know how or why that occurs.
Concerning the DPL infusion, we did not mean to imply that this would take us back to the old days of diagnostic peritoneal lavage and how it fits into the concept of nonoperative management of splenic and hepatic trauma. I am not sure where this is going to fit in clinically. That remains for clinicians to determine. We were simply trying to speculate that it possibly could be used as a peritoneal lavage solution or diagnostic solution.
Dr. Pruitt, I don’t know what the mechanism of it is. I believe it is maintaining the endothelial cell membrane function, and that it is simply a matter of maintaining the ability of the endothelial cell to include potassium and exclude sodiums. Therefore, you maintain the ability of that cell membrane to function. I say that because acetylcholine, when placed on the endothelial cell, maintains its ability to react. Outside that, I have no idea what its mechanism might be.
Concerning the issue of blood volume changes, I have no data as to blood volume measurements now or about the volume of peritoneal fluid that remains. Dr. Mannick asked similar questions about how it works, and I have no data to indicate a mechanism at present.
Dr. Bulkley, you asked about the effect on systemic resuscitation and A-VO2 differences and other mechanisms. Those are future studies. This is a small animal. It is hard to measure A-VO2 differences in a rat; we would have to go to a larger animal model. Those are speculative procedures that we think we could probably do if we had the money to do it. Dr. Brunicardi’s suggestion that we ask the NIH for funding would help us to do those experiments, but right now we are on a pretty thin budget.
Finally, I was asked the question about the sludging of the blood flow. In this model, you can see sludging of the red cells as they go by. You can actually watch white cells bounce along the microcirculation. Peritoneal resuscitation eliminates all of that white cell stickiness and the sludging that you see. You mostly see this on the venular side, because the arterial flow occurs rather promptly. But in the venules, you can see the white cells sticking. The peritoneal resuscitation prevents all of that white cell stickiness and all the rolling and transgression of the white cells through the venular walls.
References
- 1.Scalea TM, Maltz S, Yelon J, et al. Resuscitation of multiple trauma and head injury: role of crystalloid fluids and inotropes. Crit Care Med. 1994; 20: 1610–1615. [PubMed] [Google Scholar]
- 2.Abou-Khalil B, Scalea TM, Trooskin SZ, et al. Hemodynamic response to shock in young trauma patients: need for invasive monitoring. Crit Care Med. 1994; 22: 633–639. [DOI] [PubMed] [Google Scholar]
- 3.Broder G, Weil MH. Excess lactate: an index of reversibility of shock in human patients. Science. 1964; 143: 1457. [DOI] [PubMed] [Google Scholar]
- 4.Abramson D, Scalea TM, Hitchcock R, et al. Lactate clearance and survival following injury. J Trauma. 1993; 35: 584–589. [DOI] [PubMed] [Google Scholar]
- 5.Rutherford EJ, Morris JA, Reed GW, et al. Base deficit stratifies mortality and determines therapy. J Trauma. 1992; 33: 417–422. [DOI] [PubMed] [Google Scholar]
- 6.Heyland DK, Cook DJ, King D, et al. Maximizing oxygen delivery in critically ill patients: a methodologic appraisal of the evidence. Crit Care Med. 1996; 24: 517–524. [DOI] [PubMed] [Google Scholar]
- 7.Dunham CM, Siegel JH, Weireter L, et al. Oxygen debt and metabolic acidemia as quantitative predictors of mortality and the severity of the ischemia insult in hemorrhage shock. Crit Care Med. 1991; 19: 231–243. [DOI] [PubMed] [Google Scholar]
- 8.Flynn WJ, Cryer HG, Garrison RN. Pentoxifylline restores intestinal microvascular blood flow during resuscitated hemorrhagic shock. Surgery. 1991; 110: 350–356. [PubMed] [Google Scholar]
- 9.Fruchteman TM, Spain DA, Wilson MA, et al. Complement inhibition prevents gut ischemia and endothelial cell dysfunction after hemorrhage/resuscitation. Surgery. 1998; 124: 782–791. [DOI] [PubMed] [Google Scholar]
- 10.Ivatury RR, Simon RJ, Islam S, et al. A prospective randomized study of endpoints of resuscitation after major trauma: global oxygen transport indices versus organ-specific gastric mucosal pH. J Am Coll Surg. 1996; 183: 145–154. [PubMed] [Google Scholar]
- 11.Fruchterman TM, Spain DA, Wilson MA, et al. Selective microvascular endothelial cell dysfunction in the small intestine following resuscitated hemorrhagic shock. Shock. 1998; 10: 417–422. [DOI] [PubMed] [Google Scholar]
- 12.Wang P, Ba ZF, Chaudry IH. Endothelial cell dysfunction occurs very early following trauma-hemorrhage and persists despite fluid resuscitation. Am J Physiol. 1993; 265: 973–979. [DOI] [PubMed] [Google Scholar]
- 13.Mainous MR, Ertel W, Chaudry IH, et al. The gut: A cytokine generating organ in systemic inflammation. Shock. 1995; 4: 193–199. [PubMed] [Google Scholar]
- 14.Magnotti LJ, Upperman JS, Xu DZ, et al. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and promotes lung injury after hemorrhage shock. Ann Surg. 1998; 228: 517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flynn WJ, Pilati D, Hoover EL. Xanthine oxidase inhibition after resuscitated hemorrhagic shock restores mesenteric blood flow without vasodilation. Shock. 1997; 8: 300–304. [DOI] [PubMed] [Google Scholar]
- 16.Wang P, Ba ZF, Chaudry IH. Endothelial cell dysfunction occurs after hemorrhage in nonheparinized but not in preheparinized models. J Surg Res. 1993; 54: 499–506. [DOI] [PubMed] [Google Scholar]
- 17.Waxman K, Clark L, Soliman MH, et al. Pentoxifylline in resuscitation of experimental hemorrhagic shock. Crit Care Med. 1991; 19: 728–731. [DOI] [PubMed] [Google Scholar]
- 18.Watkins JM, Spain DA, Krystopik RJ, et al. Heparin preserves intestinal perfusion after hemorrhage and resuscitation. J Surg Res. 1996; 66: 154–158. [DOI] [PubMed] [Google Scholar]
- 19.Angle N, Hoyt DB, Coimbra R, et al. Hypertonic saline resuscitation diminishes lung injury by suppressing neutrophil activation after hemorrhagic shock. Shock. 1998; 9: 164–170. [DOI] [PubMed] [Google Scholar]
- 20.Velmahos GC, Demetriades D, Shoemaker WC, et al. Endpoints of resuscitation of critically injured patients: normal or supranormal? A prospective randomized trial. Ann Surg. 2000; 232: 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solomonov E, Hirsh M, Yahiya A, et al. The effect of vigorous fluid resuscitation in uncontrolled hemorrhagic shock after massive splenic injury. Crit Care Med. 2000; 28: 749–754. [DOI] [PubMed] [Google Scholar]
- 22.Jonas J, Heimann A, Strecker U, et al. Hypertonic/hyperoncotic resuscitation after intestinal superior mesenteric artery occlusion: early effects on circulation and intestinal reperfusion. Shock. 2000; 14: 24–29. [DOI] [PubMed] [Google Scholar]
- 23.Childs EW, Smalley DM, Moncure M, et al. Effect of LFA-1beta antibody on leukocyte adherence in response to hemorrhagic shock in rats. Shock. 2000; 14: 49–52. [DOI] [PubMed] [Google Scholar]
- 24.Liaudet L, Soriano FG, Szabo E, et al. Protection against hemorrhagic shock in mice genetically deficient in poly(ADP-ribose) polymerase. Proc Natl Acad Sci USA. 2000; 97: 10203–10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller FN, Nolph KD, Joshua IG, et al. Hyperosmolality, acetate, and lactate: dilatory factors during peritoneal dialysis. Kidney Int. 1981; 20: 397–402. [DOI] [PubMed] [Google Scholar]
- 26.Zakaria ER, Spain DA, Harris PD, et al. Generalized dilation of the visceral microvasculature by peritoneal dialysis solutions. Perit Dial Int. 2002; 22: 593–601. [PubMed] [Google Scholar]
- 27.Bohlen HG, Gore RW. Preparation of rat intestinal muscle and mucosa for quantitative microcirculatory studies. Microvas Res. 1976; 11: 103–110. [DOI] [PubMed] [Google Scholar]
- 28.Porter JM, Ivatury RR. In search of the optimal end points of resuscitation in trauma patients: A review. J Trauma. 1998; 44: 908–914. [DOI] [PubMed] [Google Scholar]
- 29.Bauer M, Marzi I, Ziegenfuss T, et al. Comparative effects of crystalloid and small volume hypertonic hyperoncotic fluid resuscitation on hepatic microcirculation after hemorrhagic shock. Circ Shock. 1993; 40: 187–193. [PubMed] [Google Scholar]
- 30.Fruchtermann TM, Spain DA, Matheson PJ, et al. Small intestinal production of nitric oxide is decreased following resuscitated hemorrhage. J Surg Res. 1998; 80: 102–109. [DOI] [PubMed] [Google Scholar]
- 31.White R, Ram S. Peritoneal dialysis solution attenuates microvascular leukocyte adhesion induced by nitric oxide synthesis inhibition. Adv Perit Dial. 1996; 12: 53–56. [PubMed] [Google Scholar]
- 32.Matheson PJ, Wilson MA, Spain DA, et al. Glucose-induced intestinal hyperemia is mediated by nitric oxide. J Surg Res. 1997; 82: 146–154. [DOI] [PubMed] [Google Scholar]
- 33.Matheson PJ, Spain DA, Harris PD, et al. Glucose and glutamine gavage increase portal vein nitric oxide metabolite levels via adenosine A2b activation. J Surg Res. 1999; 84: 57–63. [DOI] [PubMed] [Google Scholar]
- 34.Massett MP, Koller A, Kaley G. Hyperosmolality dilates rat skeletal muscle arterioles: role of endothelial K(ATP) channels and daily exercise. J Appl Physiol. 2000; 89: 2227–2234. [DOI] [PubMed] [Google Scholar]
- 35.Liaudet L, Soriano FG, Szabo C. Biology of nitric oxide signaling. Crit Care Med. 2000; 28: 37–52. [DOI] [PubMed] [Google Scholar]
- 36.Mackway-Jones K, Foex BA, Kirkman E, et al. Modification of the cardiovascular response to hemorrhage by somatic afferent nerve stimulation with special reference to gut and skeletal muscle blood flow. J Trauma. 1999; 47: 481–485. [DOI] [PubMed] [Google Scholar]
- 37.Illner H, Shires GT. The effect of hemorrhagic shock on potassium transport in skeletal muscle. Surg Gynecol Obstet. 1980; 150: 17–25. [PubMed] [Google Scholar]
- 38.McKinley BA, Houtchens BA, Janata J. Continuous monitoring of interstitial fluid potassium during hemorrhagic shock in dogs. Crit Care Med. 1981; 9: 845–851. [DOI] [PubMed] [Google Scholar]
- 39.Cone JB, Wallace BH, Caldwell FT Jr, et al. Beneficial effects of a hypertonic solution for resuscitation in the presence of acute hemorrhage. Am J Surg. 1987; 154: 585–588. [DOI] [PubMed] [Google Scholar]
- 40.Prough DS, Whitley JM, Taylor CL, et al. Small-volume resuscitation from hemorrhagic shock in dogs: effects on systemic hemodynamics and systemic blood flow. Crit Care Med. 1991; 19: 364–372. [DOI] [PubMed] [Google Scholar]
- 41.Lucas CE. The water of life. A century of confusion. Bull Am Coll Surg. 2001; 192: 86–93. [DOI] [PubMed] [Google Scholar]
Footnotes
Presented at the 114th Annual Session of the Southern Surgical Association, December 1–4, 2002, Palm Beach, Florida.
Supported by VA Merit Review grants of Dr. Garrison and Dr. Spain.
Correspondence: R. Neal Garrison, MD, Department of Surgery, University of Louisville, Louisville, KY 40292.
E-mail: rngarr01@gwise.louisville.edu
Accepted for publication December 2002.