

Abstract
Objective
To evaluate the efficacy and safety of an investigational fibrin sealant (FS) in a randomized prospective, partially blinded, controlled, multicenter trial.
Summary Background Data
Upper extremity vascular access surgery using polytetrafluorethylene (PTFE) graft placement for dialysis was chosen as a reproducible, clinically relevant model for evaluating the usefulness of FS. The FS consisted of pooled human fibrinogen (60 mg/mL) and thrombin (500 NIH U/mL). Time to hemostasis was measured, and adverse events were monitored.
Methods
Consenting adult patients (n = 48) undergoing placement of a standard PTFE graft were randomized in a 2:1:1 ratio to the treatment group using FS (ZLB Bioplasma AG, Bern, Switzerland), oxidized regenerated cellulose (Surgicel, Johnson & Johnson, New Brunswick, NJ), or pressure. Patients received heparin (3,000 IU IVP) before placement of vascular clamps. If the treatment was FS, clamps were left in place for 120 seconds after the application of study material to permit polymerization. If treatment was Surgicel, clamps were left in place until the agent had been applied according to manufacturer’s instructions. If the treatment was pressure, clamps were released as soon as the investigator was ready to apply compression. Immediately after release of the last clamp, the arterial and venous suture lines were evaluated for bleeding. The time to hemostasis at both the venous and arterial sites was recorded.
Results
Significant (P ≤ .005) reduction in time to hemostasis was achieved in the FS group. Thirteen (54.2%) patients randomized to FS experienced immediate hemostasis at both suture lines following clamp removal compared to no patients using Surgicel or pressure. Only one patient (7.1%) in the Surgicel group and no patients in the pressure group experienced hemostasis at 120 seconds from clamp removal, compared to 13 (54.2%) patients for FS. Adverse events were comparable in all groups. There were no seroconversions.
Conclusions
FS achieved more rapid hemostasis than traditional techniques in this peripheral vascular procedure. FS use appeared to be safe for this procedure.
Peripheral vascular operations are frequently performed with systemic heparinization that may contribute to intraoperative bleeding. In addition, polytetrafluorethylene (PTFE) grafts secured with polypropylene suture are often used in these procedures. This technique may be associated with suture line or anastomotic bleeding. 1 Because operating time is expensive (approximately $25/min), an effective, rapidly performing hemostatic agent could not only enhance patient care but also reduce costs. Fibrin sealant, a two-component surgical tissue adhesive and hemostatic agent derived from pooled human plasma, has been employed to reduce bleeding in a variety of surgical specialities 2–19 and could be useful in this setting.
Approximately 30,000 to 40,000 PTFE shunts for dialysis access are done annually for the more than 250,000 people requiring dialysis treatment for renal failure in the United States. This upper extremity vascular access shunt procedure is desirable and useful for a clinical hemostatic research trial. The operation itself is challenging but reproducible. Patients with end-stage renal disease have an additional obstacle to effective hemostasis, as there is an inherent coagulopathy from platelet dysfunction associated with uremia. A previous pilot study 19 supported the usefulness of the PTFE arteriovenous shunt clinical model. The study suggested the potential hemostatic benefits of fibrin sealant relative to frequently used active comparators, including oxidized regenerated cellulose and pressure. The present larger randomized, prospective, multicenter study was initiated to evaluate the efficacy and safety of a new investigational form of fibrin sealant containing no bovine products in this clinical setting. 20
METHODS
Protocol
This study was designed to evaluate an investigational form of fibrin sealant in a randomized, prospective, partially blinded, controlled, multicenter trial meeting FDA approval for phase II clinical trials. The trial was sponsored by ZLB Bioplasma AG (Bern, Switzerland). The study population consisted of patients requiring dialysis for end-stage renal failure who were to undergo placement of PTFE upper extremity arteriovenous shunts. Criteria for inclusion included adult patients who were:
Undergoing PTFE graft placement for dialysis access using a standard-wall graft, diameter 5.0 to 7.0 mm, or a flare or taper graft, diameter 4.0 to 7.0 mm
Undergoing new end-to-side shunts, or undergoing new end-to-side (at arterial site) and end-to-end (at venous site) shunts, or undergoing revision of an existing shunt by a jump shunt (end-to-end graft-to-graft anastomosis closer to the arterial side and new end-to-side or end-to-end vein-to-graft anastomosis) or with a standard-wall PTFE patch on the venous or arterial side only or on both sides, where at least in part the anastomosis line had to be between the graft and patch
Undergoing graft placement using monofilament nonabsorbable suture (polypropylene 6-0) and a BV-1 needle
Criteria for exclusion included history of hypersensitivity, allergy, or anaphylactic reaction to any plasma-derived product, including fibrin sealant; treatment with any investigational agent in past 30 days; undergoing a procedure involving graft material other than PTFE with monofilament nonabsorbable suture; clinical signs of acquired immune deficiency syndrome or current malignancy; pregnancy; condition that might, in the investigators’ judgment, increase risk to patient or reduce chance of obtaining meaningful data; previous enrollment in this study.
This study was conducted in accordance with FDA, Good Clinical Practices, the Declaration of Helsinki, and all other applicable ethical, legal, and regulatory requirements. Institutional review board approval was required at each study center before beginning study conduct.
Eligible patients who provided written informed consent underwent the clinical trial operative procedure. A PTFE graft was placed between artery and vein in the upper arm or forearm. Anastomoses were performed with 6-0 polypropylene sutures on BV-1 needles. All patients in the study received heparin (3,000 IU IVP) before placement of the vascular clamps required for performance of the anastomoses. The blinding for the surgeon was maintained until after completion of the anastomoses; then the surgeon was informed to which of the three study groups the patient had been assigned based on a 2:1:1 randomization scheme.
Group A patients received the investigational fibrin sealant (FS) consisting of fibrinogen (60 mg/mL), thrombin (500 NIH U/mL with 40 mmol/L calcium chloride) applied as a bead (approximately 2 mL in total per subject) to the arterial and venous anastomoses using a dual-lumen tip applicator system (Micromedics, Eagan, MI). A period of 120 seconds for polymerization of the FS was allowed to pass before removal of the vascular clamps, venous side first. For group B patients, one or two single layers of oxidized regenerated cellulose (Surgicel, Johnson & Johnson, New Brunswick, NJ) were applied to the anastomoses, and then the vascular clamps were removed. For group C patients, pressure was applied to the anastomoses with surgical gauze and the vascular clamps were removed. Time to hemostasis at each anastomosis was determined; this was facilitated by the removal of excess blood using the edge of a sterile gauze pad. Hemostasis was defined as the point at which the surgeon determined that no additional hemostasis measures were indicated and the wound could be closed. No additional hemostatic treatments at each suture line were permitted until 10 minutes after clamp release unless required by urgent medical necessity.
The three treatment groups were compared in an intent-to-treat analysis in terms of primary and secondary endpoints reflecting efficacy and safety. The primary endpoint was time to hemostasis at both the arterial and venous anastomosis sites. Secondary efficacy variables included occurrence of hemostasis at 120 seconds after clamp release, time to hemostasis separately at the arterial and venous anastomotic sites, time of application plus time to hemostasis, need for rescue treatment, failure to achieve hemostasis within 10 minutes of clamp release, and duration of graft implant procedure. Safety evaluation was performed based on adverse event evaluation and determination of seroconversions.
The statistical methods used were applied to the intent-to-treat population (n = 48). Continuous data were summarized by treatment group using descriptive statistics. Categorical data were summarized by treatment group using frequency tables. All statistical tests were two-sided using 0.025 as the significance level (i.e., a result was considered statistically significant at P < .025). There was no interim analysis.
For the primary efficacy endpoint and the secondary endpoints involving time to hemostasis, each control group was compared to the FS group by the Wilcoxon test, using the intent-to-treat population. The P value by the log-rank test was also determined. The median time to hemostasis and its 95% confidence interval for each treatment group were calculated. The success rates in achieving hemostasis were analyzed by the Fisher exact test, using the intent-to-treat population.
The incidence of adverse events was compared between treatment groups using logistic regression. Changes in laboratory parameters and other safety data were compared using the two-sample t test or Wilcoxon rank sum test for continuous data, logistic regression for binary data (e.g., present/absent), and the row effects model, a log-linear model, for original data (e.g., improved/unchanged/worsened).
Assignment
Randomization was done by study center using a permuted-block design. Patients were randomly assigned to the FS, Surgicel, or pressure groups in a 2:1:1 ratio within each block. When a patient was ready to receive randomized treatment, the study coordinator phoned a 24-hour randomization call center. Confirmation questions were asked and a randomization number and treatment group (A, B, or C) were provided. Randomization was then confirmed by fax.
Blinding (Partial) Procedures
While PTFE graft implantation was being performed, a member of the study center staff, working out of view of the investigator, contacted a central randomization service (Applied Clinical Concepts Inc., Durham, NC) to determine which treatment the patient was to receive. All patients had vials used and applicators prepared to simulate preparation of FS. However, the vials for groups B and C (controls) contained normal saline solution and not FS, while those for group A contained the FS. When the investigator was ready to apply the study treatment, the applicator was presented to the investigator and the investigator was told which treatment the patient was to receive.
FS and control saline solution were prepared and reconstituted out of view of the investigator. The applicator was identical for all patients and was presented to the investigator already loaded. Packets of Surgicel for patients in group B were kept out of view of the investigator until the time of unblinding. Thus, the investigator remained blinded to study treatment until it was time to apply the treatment. When the investigator had completed both the venous and the arterial anastomoses and was ready to apply the study treatment, he or she was told which treatment the patient was receiving. Unblinding (resulting in an only partially blinded trial) at this point was necessary because the application procedures were different for the three treatments.
Patients could be screened and enrolled in this trial up to 21 days before the operative procedure. Efficacy data were obtained in the operating room on the day of the procedure. Baseline laboratory tests, including serology for anti-HAV, anti-HCV, anti-HBc, HBsAg, anti-HIV1 and 2, and parvovirus B19 Ab IgG and IgM, biochemistry, coagulation, and hematology, were obtained before the operation. Follow-up examinations of the patients’ grafts, wounds, adverse events, biochemistry, hematology, and coagulation as well as parvovirus B19 studies were performed on day 14 (± 4) after the procedure. All other follow-up viral serology tests were performed 6 months (± 2 weeks) after the procedure.
This trial was performed at four centers (each randomizing 37, 6, 3, and 2 patients, respectively, in the intent-to-treat analysis) that were each allowed two practice FS patients before beginning the randomized trial.
RESULTS
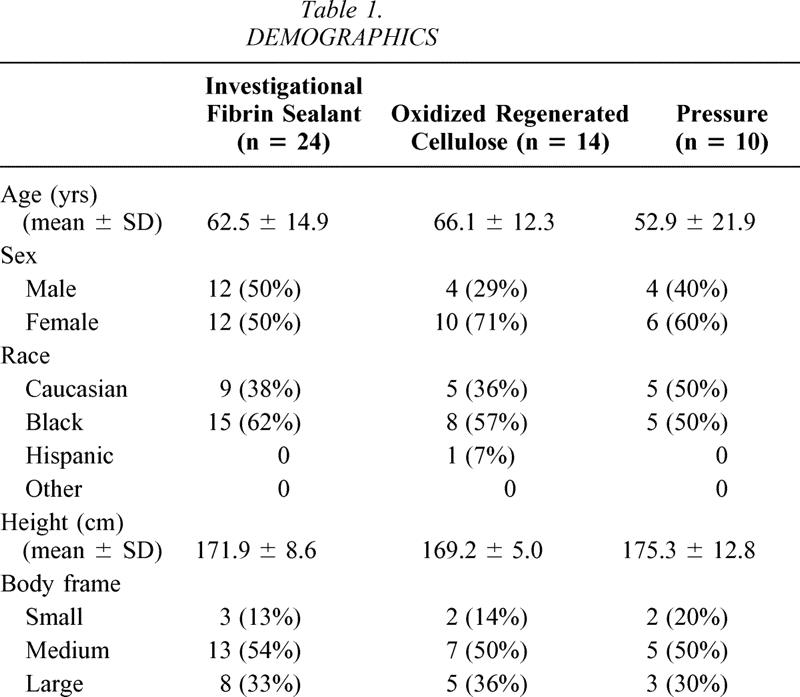
The demographics of the three treatment groups are provided in Table 1. Mean age of the population was 61.5 ± 16.2 years. Patients in the pressure group tended to be younger than in the other two groups, but this was not statistically different. Overall, the majority of patients (28 [58%]), were female and the majority were also black (28 [58%]). Distribution of ethnic groups, height, and weight were similar in all three groups. Forty-eight patients were available for the intent-to-treat analysis. There were 24, 14, and 10 patients, respectively, in groups A (FS), B (Surgicel), and C (pressure).
Table 1. DEMOGRAPHICS
With respect to the primary endpoint in the intent-to-treat population (Fig. 1, Table 2), time to hemostasis (mean ± SE) was significantly (P ≤ .0001) shorter for the FS group (56.3 ± 14.9 seconds) compared to the Surgicel group (772.9 ± 113.6 seconds) and the pressure group (1,269.6 ± 224.1 seconds). With respect to the secondary endpoint, complete hemostasis was significantly (P ≤ .005) higher in the FS group (54% [13 patients]) compared to either the Surgicel or pressure group (0% for both groups at clamp release and 7% [1 patient] and 0%, respectively, at 120 seconds after clamp release). Time of application plus time to hemostasis was significantly less (P ≤ .0001) in the FS group (216.1 ± 15.8 seconds) compared to the Surgicel group (786.4 ± 122.2 seconds) or the pressure group (1,269.6 ± 224.1 seconds). Time to hemostasis at both the arterial and venous sites was also significantly (P ≤ .0001) shorter in the FS group. Finally, the incidence of achieving hemostasis at 10 minutes was significantly (P ≤ .0005) better in the FS group (24/24 patients [100%]) than in either the Surgicel group (7/14 [50%]) or the pressure group (3/10 [30%]). With respect to the procedure duration, both overall duration of procedure (P ≤ .02) and duration of procedure from start of treatment application (P ≤ .01), respectively, were significantly shorter in the FS group (125.6 ± 24.0 minutes and 27.4 ± 7.2 minutes) compared to the pressure group (145.6 ± 19.7 minutes and 39.2 ± 12.4 minutes). The same duration of procedure measurements comparing the FS group (as above) and the Surgicel group (130 ± 22.3 minutes and 34.4 ± 14.3 minutes), while shorter, did not reach statistical significance.
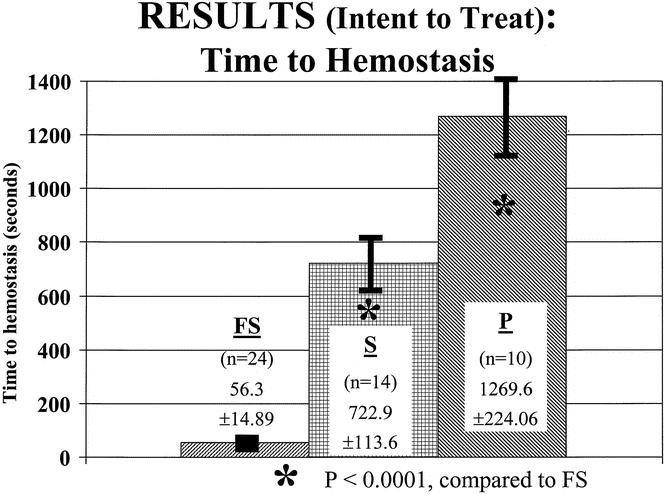
Figure 1. Primary endpoint, time to hemostasis, demonstrating significant (P < .001) reduction in patients treated with investigational fibrin sealant (FS) versus controls of Surgicel (S) and pressure (P).
Table 2. EFFICACY RESULTS IN INTENT-TO-TREAT POPULATION

With respect to safety data, there were no significant changes or differences between any group for biochemistry, coagulation, and hematology results. There were no seroconversions for anti-HAV, anti-HCV, anti-HBc, HBsAg, anti-HIV1, and anti-HIV2 at 6 months and no evidence of new parvovirus B19 infection in any group at 14 days.
There were no significant differences in the intent-to-treat population for adverse events or serious adverse events between the treatment groups. Occurrences were 19 (79.2%) and 3 (12.5%) for the FS group, 11 (78.6%) and 10 (14.3%) for the Surgicel group, and 10 (100%) and 2 (20%) for the pressure group. With respect to adverse events, 83% of the intent-to-treat population suffered one or more adverse events, with nonspecific pain, pain at the incision, and evidence of edema of the upper arm being most frequent. Other events occurring in two or more patients included erythema, nausea, limb pain, cellulitis, ecchymosis, skin irritation, subclavian steal, vomiting, and wound drainage. No events were categorized by the investigator as related to the FS.
With respect to serous adverse events, 15% of the intent-to-treat population suffered one or more serious adverse events, including most frequently vascular graft disorders and occlusion or hypotension. None was judged to be related to the FS. There were two deaths in the intent-to-treat population, one due to sepsis and one due to respiratory failure. One was in the FS group and the other in the pressure group. Graft occlusion occurred in three (12.5%) of the FS patients, one (7.1%) of the Surgicel patients, and two (20%) of the pressure patients. All grafts were patent at the day 14 evaluation. Two patients in the intent-to-treat population underwent early thrombectomy for graft thrombosis with no evidence of fibrosis secondary to the FS.
DISCUSSION
The efficacy and safety of the FS were supported by this vascular access shunt trial. The agent achieved hemostasis faster than two other commonly used means for bleeding control in vascular procedures using PTFE.
Significant benefit from this agent was demonstrated in terms of the primary endpoint, time to hemostasis. A mean improvement of more than 10 minutes was documented by using the FS. Secondary efficacy endpoints including incidence of complete hemostasis at clamp release, at 120 seconds, and at 10 minutes, as well as time of application plus time to hemostasis, and time to hemostasis at the arterial and venous anastomoses showed statistically significant benefit using the FS. Duration of procedure data showed statistically significant changes. There was a 20-minute reduction for overall length of operation and a 12-minute reduction for duration of agent application to end of operation for the FS group versus the control pressure group. Differences in these same parameters between the FS and Surgicel on the order of 4 and 7 minutes, respectively, were smaller and did not reach statistical significance.
There were no significant differences in the incidence of adverse events of any type between the groups. Serologic data also confirmed that there was no evidence of viral disease transmission.
These data support the usefulness of the FS to facilitate more rapid hemostasis in vascular operations involving the use of heparin, PTFE, and monofilament polypropylene suture. A previous animal study 21 using liquid FS and a small human pilot study 19 suggested a benefit to the hemostatic agent in a similar setting. A randomized human clinical trial 22 recently supported a hemostatic benefit to using collagen patches coated with FS to shorten time to hemostasis and reduce blood loss when using PTFE grafts for vascular reconstruction. Bleeding at vascular anastomotic sites between native tissue and PTFE can produce intraoperative delays as well as increased blood loss and other complications. The hemostatic advantages of FS versus the control comparators of Surgicel and pressure were strongly supported in this trial. The present study also suggests a significant duration of procedure benefit in favor of the FS compared to pressure alone. A 20-minute benefit, assuming an operative cost of $25/min, more than pays for the cost of 2 mL of currently available commercial fibrin sealants. The operative time reductions for the investigational FS versus Surgicel were not statistically significant. On the other hand, if mean time of application plus time to hemostasis data alone are considered with respect to the investigational FS, there were significant time savings of just under 6 minutes and over 17.5 minutes versus Surgicel and pressure, respectively. These latter data suggest a possible cost benefit to the investigational FS.
The new investigational FS used in this trial may have several advantages. It eliminates an allergic risk, as it contains no bovine aprotinin 20 and is thus composed entirely of human proteins. It uses solvent/detergent cleansing to reduce the risk of human viral disease transmission. Although this issue has not appeared to be a major clinical concern in the literature, it is reported that symptomatic parvovirus transmission can occur from using FS. 23 In this study parvovirus evaluation using IgM and IgG antibody detection was employed, and no evidence of disease transmission was detected.
The other significant safety concern in the literature is the risk of microvascular anastomotic thrombosis as a result of using FS. Reports in animals 24,25 suggest that concentrations of thrombin greater than 500 units in FS can result in an increase in microvascular venous anastomotic thrombosis. The agent used in this present study had a thrombin concentration equal to 500 units. An increased incidence of graft thrombosis in the FS group was not noted relative to the two active control groups.
This study achieved its goals even though it was terminated early by the sponsors. The study was stopped because of slower-than-desired enrollment and because the majority of patients (37) were enrolled at only one site, suggesting the potential for systematic errors entering the analysis. Therefore, the study was terminated based on its lower-than-anticipated enrollment and the fact that a majority of patients were randomized at one site. In addition, sites were only partially blinded, as the investigators were unblinded at the time of application of the three treatments to the already completed PTFE suture lines. Thus, the possibility for surgical investigator bias could not be eliminated from this trial.
In all other respects, the study model itself appeared to be advantageous in that a limited, reproducible, but challenging operation that could benefit from improved hemostasis was used. No unusual disadvantages to the model other than the difficulty of partial blinding were noted.
CONCLUSIONS
This randomized, prospective, partially blinded trial evaluating an investigational FS in PTFE vascular access procedures strongly supports the efficacy and safety of this material. The benefits documented in this trial may be useful in other vascular procedures involving the use of heparin and PTFE.
Acknowledgments
The authors thank Kristen Faircloth and Sylvie Martinez for their help in the preparation of this manuscript. Statistical analyses were provided by K.F. Yee, MSc, principal biostatistician at PAREXEL International.
Discussion
Dr. Ali F. Aburahma (Charleston, WV): I would like to congratulate Dr. Schenk and his group on a well-designed and nicely presented study. This randomized prospective multicenter trial is analyzing efficacy and safety of the new fibrin sealant to achieve hemostasis in patients who need PTFE dialysis graft, which is a rather common procedure in North America.
One unique thing about this fibrin sealant which makes it different than others is that it does not contain the bovine element which many others do contain. As indicated by Dr. Schenk, this fibrin sealant achieved a shorter hemostasis time than any other conventional method being tried in the past several years.
I have the following questions for Dr. Schenk. Do you see any long-term effect of this fibrin sealant in regard to initiating some late fibrosis which might make revising these grafts somewhat more difficult than usual? How many of the early graft failures can be attributed to the fibrin sealant, if it did? And did you observe any abnormalities when you explored these grafts when you noticed they were thrombosed earlier? Do you have the late outcome of these grafts? And do you anticipate any late effect on the graft patency in the long run? Finally, since overall saving time was less than 6 minutes, as indicated in the manuscript, when you compare fibrin sealant to the conventional Surgicel method, which is commonly done by many surgeons, do you see there was any cost benefit by cutting down only 6 minutes in this using the fibrin sealant?
Dr. Worthington G. Schenk, III (Charlottesville, VA): With regard to long-term results and whether this results in fibrosis that could interfere with either long-term patency or other resuscitative efforts, we do not yet have long-term comparative results to tell you about. The fibrin sealant material itself is in fact physically gone within a relatively short period of time, but that doesn’t mean it couldn’t incite an inflammatory response which could be deleterious. We simply don’t know that at this point.
I don’t know the data on all of the early graft failures. But for most of those, particularly the ones at my institution, I know that there was another obvious indication, etiology, for the early failure. For example, the graft which when we reopened is found to have an unexpected total occlusion of the outflow vein further downstream would be an obvious reason why it would have failed, and in fact those grafts tended to fail again within a relatively short period of time, and we felt that those were not related to the fibrin sealant product itself.
What about the cost-effectiveness and the time savings? The 2-mL package of the fibrin sealant costs about $100. In my institution, OR time, depending on how you want to amortize it, costs between $18 to $24 a minute, so that if you save 4 minutes of OR time you have exceeded the break-even point.
To make this truly practical, I think it is probably helpful to have enough experience with it to be able to predict ahead of time which cases are likely to benefit from it, so that you would be able to stratify this somewhat yourself by your clinical experience. Patients who have a great deal of atherosclerotic degeneration of the native vessel, patients who gets PTFE patches, for example, and patients who either have an underlying coagulopathy or whom you intend to anticoagulate would be good candidates to use this and enjoy the cost savings.
References
- 1.Baker JD. Bleeding through PTFE grafts. Eur J Vasc Surg. 1987; 1: 41–43. [DOI] [PubMed] [Google Scholar]
- 2.Bayfield MS, Spotnitz WD. Fibrin sealant in thoracic surgery. Pulmonary applications, including management of bronchopleural fistula. Chest Surg Clin North Am. 1996; 6: 576–583. [PubMed] [Google Scholar]
- 3.Dunn CJ, Goa KL. Fibrin sealant; a review of its use in surgery and endoscopy. Drugs. 1999; 58: 863–886. [DOI] [PubMed] [Google Scholar]
- 4.Detweiler MB, Detweiler JG, Fenton J. Sutureless and reduced suture anastomosis of hollow vessels with fibrin glue: a review. J Invest Surg. 1999; 12: 245–262. [DOI] [PubMed] [Google Scholar]
- 5.Crawford RW, Giangrande P, Murray D. Fibrin sealant reduces blood loss in total hip arthroplasty. Hip Intl. 1999; 9: 127–132. [Google Scholar]
- 6.Holcomb JB, Pusateri AE, Hess JR, et al. Implications of new dry fibrin sealant technology for trauma surgery. Surg Clin North Am. 1997; 77: 943–952. [DOI] [PubMed] [Google Scholar]
- 7.Jackson MR, Alving BM. Fibrin sealant in clinical and preclinical studies. Curr Opin Hematol. 1999; 6: 415–419. [DOI] [PubMed] [Google Scholar]
- 8.Levy OL, Martinowitz U, Oran A, et al. The use of fibrin tissue adhesive to reduce blood loss and the need for blood transfusion after total knee arthroplasty: a prospective, randomized, multicenter study. J Bone Joint Surg [Am]. 1999; 81: 1580–1586. [DOI] [PubMed] [Google Scholar]
- 9.Martinowitz U, Varon D, Heim M. The role of fibrin tissue adhesives in surgery of haemophilia patients. Haemophilia. 1998; 4: 443–448. [DOI] [PubMed] [Google Scholar]
- 10.McCarthy PM. Fibrin glue in cardiothoracic surgery. Transfus Med Rev. 1993; 7: 173–179. [DOI] [PubMed] [Google Scholar]
- 11.Selesnick S, al-Rawi M. Adhesives in otology and neurotology. Am J Otolaryngol. 1997; 18: 81–89. [DOI] [PubMed] [Google Scholar]
- 12.Spotnitz WD. Fibrin sealant in the United States: Clinical use at the University of Virginia. Thromb Haemost. 1995; 74: 482–485. [PubMed] [Google Scholar]
- 13.Spotnitz WD, Welker RL. Clinical used of fibrin sealant. In: Mintz PD, ed. Transfusion Therapy: Clinical Principles and Practice. Bethesda, MD: AABB Press, 1999: 199–220.
- 14.Tholpady SS, Schlosser JR, Spotnitz WD, et al. Repair of an osseous facial critical-sized defect using augmented fibrin sealant. Laryngoscope. 1999; 109: 1585–1588. [DOI] [PubMed] [Google Scholar]
- 15.Stoeckli SJ, Moe KS, Huber A, et al. A prospective randomized double-blind trial of fibrin glue for pain and bleeding after tonsillectomy. Laryngoscope. 1999; 109: 652–655. [DOI] [PubMed] [Google Scholar]
- 16.Greenhalgh DG, Gamelli RL, Lee M, et al. Multicenter trial to evaluate the safety and potential efficacy of pooled human fibrin sealant for the treatment of burn wounds. J Trauma Inj Infect Crit Care. 1999; 46: 433–440. [DOI] [PubMed] [Google Scholar]
- 17.Brennan M. Fibrin glue. Blood Rev. 1991; 5: 240–244. [DOI] [PubMed] [Google Scholar]
- 18.Clark RA. Fibrin sealant in wound repair: a systematic surgery of the literature. Exp Opin Invest Drugs. 2000; 9: 2371–2392. [DOI] [PubMed] [Google Scholar]
- 19.Schenk WG, Goldswaite Jr CA, Burks S, et al. Fibrin sealant facilitates hemostasis in arteriovenous polytetrafluorethylene grafts for renal analysis access. Am Surg. 2002;68:728–732. [PubMed]
- 20.Schoenecker J, Johnson R, Fields R, et al. Relative safety of hemostatic agents. J Am Coll Surg. 2002; 195: S22. [Google Scholar]
- 21.Dickneite G, Metzner H, Nicolay U. Prevention of suture hole bleeding using fibrin sealant: benefits of Factor XIII. J Surg Res. 2000; 93: 201–205. [DOI] [PubMed] [Google Scholar]
- 22.Czerny M, Verrel F, Weber H, et al. Collagen patch coated with fibrin glue components: treatment of suture hole bleedings in vascular reconstruction. J Cardiovasc Surg. 2000; 41: 553–557. [PubMed] [Google Scholar]
- 23.Hino M, Ishiko O, Honda KI, et al. Transmission of symptomatic parvovirus B19 infection by fibrin sealant used during surgery. Br J Haematol. 2000; 108: 194–185. [DOI] [PubMed] [Google Scholar]
- 24.Drake DB, Faulkner BC, Amiss Jr LR, et al. Thrombogenic effects of a non thrombin-based fibrin sealant compared with thrombin-based fibrin sealant in microvenous anastomoses in a rat model. Ann Plast Surg. 2000;45:520–524. [DOI] [PubMed]
- 25.Frost-Arner L, Spotnitz WD, Rodeheaver GT, et al. Comparison of the thrombogenicity of internationally available fibrin sealants in an established microsurgical model. J Plastic Reconstruct Surg. 2001; 108: 1655. [DOI] [PubMed] [Google Scholar]
Footnotes
Presented at the 114th Annual Session of the Southern Surgical Association, December 1–4, 2002, Palm Beach, Florida.
Clinical research trial funding provided by ZLB Bioplasma, AG Bern, Switzerland.
Correspondence: Dr. William D. Spotnitz, Director, Surgical Therapeutic Advancement Center, University of Virginia Health System, P.O. Box 801370, Charlottesville, VA 22908-1370.
E-mail: spotnitz@surgery.ufl.edu
Accepted for publication December 2002.