

Abstract
Lysyl-tRNA synthetases (LysRSs) are unique amongst the aminoacyl-tRNA synthetases in being composed of unrelated class I and class II enzymes. To allow direct comparison between the two types of LysRS, substrate recognition by class I LysRSs was examined. Genes encoding both an archaeal and a bacterial class I enzyme were able to rescue an Escherichia coli strain deficient in LysRS, indicating their ability to functionally substitute for a class II LysRS in vivo. In vitro characterization showed lysine activation and recognition to be tRNA-dependent, an attribute of several class I, but not class II, aminoacyl-tRNA synthetases. Examination of tRNA recognition showed that class I LysRSs recognize the same elements in tRNALys as their class II counterparts, namely the discriminator base (N73) and the anticodon. This sequence-specific recognition of the same nucleotides in tRNALys by the two unrelated types of enzyme suggests that tRNALys predates at least one of the LysRSs in the evolution of the translational apparatus. The only observed variation in recognition was that the G2⋅U71 wobble pair of spirochete tRNALys acts as antideterminant for class II LysRS but does not alter class I enzyme recognition. This difference in tRNA recognition strongly favors the use of a class I-type enzyme to aminoacylate particular tRNALys species and provides a molecular basis for the observed displacement of class II by class I LysRSs in certain bacteria.
The aminoacyl-tRNA synthetases have long been upheld as a paradigm of molecular evolution. This is because their products, aminoacyl-tRNAs, are essentially the same in all living organisms. The role of aminoacyl-tRNA synthetases is to interpret the genetic code in terms of amino acids, providing the essential link between RNA and protein without which translation would be impossible. This highly conserved function has been assumed to place constraints on the evolutionary variation of aminoacyl-tRNA synthetases beyond those enforced on most other protein families. A broader evolutionary interest in the aminoacyl-tRNA synthetases stems from their biological function being one of the core requirements for progression from the RNA world to the universal common ancestor in numerous schemes for the origin of life (reviewed in refs. 1 and 2). Thus, it could reasonably be assumed that in depth investigations of the aminoacyl-tRNA synthetases would provide insights into the origins of the genetic code and contemporary translation. Although studies of the aminoacyl-tRNA synthetases have provided a wealth of information in numerous areas, including specificity and catalysis in biological systems, protein-RNA molecular recognition, and diversification within protein families, they have not provided the expected bounty of evolutionary information such as has been garnered, for example, from ribosomal RNA. One of the main reasons underlying this has been the limited range of complete families of genes encoding aminoacyl-tRNA synthetase available, namely those from Escherichia coli, yeast, and human. However, the recent boom in whole genome sequencing has vastly expanded the number of known synthetase families. This has revealed an unexpected diversity among the aminoacyl-tRNA synthetases, which now clearly can be seen to have evolved independently of each other (see, for example, ref. 3). The starkest examples of this are the replacement of asparaginyl- and glutaminyl-tRNA synthetases by tRNA-dependent transamidation pathways (4, 5) and the existence of unrelated lysyl-tRNA synthetases (LysRSs) with identical activities (6).
The aminoacyl-tRNA synthetases can be divided into two classes (I and II) of 10 members each based on the presence of mutually exclusive amino acid sequence motifs (7–9). This division reflects structurally distinct topologies within the active site; class I aminoacyl-tRNA synthetases contain a Rossmann fold, and class II synthetases contain a unique anti-parallel β fold. In addition, it has been observed that class I enzymes bind the acceptor helix of tRNA on the minor groove side whereas class II enzymes bind the major groove side (10). It generally is assumed that an aminoacyl-tRNA synthetase of particular substrate specificity will always belong to the same class regardless of its biological origin, reflecting the ancient evolution of this enzyme family (11, 12). The only known exceptions to this rule are the LysRSs, which are composed of two unrelated families, namely class I enzymes in certain archaea and bacteria and class II enzymes in all other organisms (13, 14). The LysRS family now provides an unprecedented opportunity to study the determinants of specificity during the evolution of molecular recognition because both class I and class II LysRSs are able to use the same amino acid and highly similar tRNA substrates. In addition, the unusual phylogenetic distribution of the class I LysRS-encoding lysS genes may reveal insights into the molecular basis of horizontal gene transfer (15) and displacement.
MATERIALS AND METHODS
General.
In vitro transcription of tRNA genes and subsequent purification and refolding of transcripts were performed as detailed (16), and mature E. coli tRNALys was purchased from Sigma. Plasmid preparation for transcription of tRNA genes was performed by standard techniques using precipitation with polyethylene glycol (17), and the tRNA gene sequences were confirmed by sequencing before transcription. Template DNA for transcription by T7 RNA polymerase was digested with BstNI. Media for bacterial growth and molecular biology protocols were standard unless otherwise noted (17). Methanococcus maripaludis genomic DNA was kindly provided by W. H. Whitman (University of Georgia), and Borrelia burgdorferi genomic DNA was provided by D. R. Akins and J. D. Radolf (University of Texas).
Strains and Plasmids.
The strain PALΔSΔUTR/pMAK705lysU and the plasmid pKS-lysS have been described (18). The pBR322-derived vector pCBS1-BstrpS, which contains the Bacillus stearothermophilus trpS gene under the control of the E. coli trpS promoter, was kindly provided by M. Kitabatake (Yale University). To allow the constitutive expression of the B. burgdorferi and M. maripaludis lysS genes in E. coli, they were subcloned behind the trpS promoter domain in pCBS1 to give pCBS1-BBlysS and pCBS1-MmlysS, respectively. The genes encoding tRNALys from B. burgdorferi and M. maripaludis were amplified by PCR and were subcloned behind a synthetic T7 RNA polymerase promoter in the vector pUC119. These constructs then were transformed into the E. coli strain DH5α, and their sequences were confirmed. The gene encoding E. coli tRNALys G2⋅U71 was generated by the same procedure by using a mutant oligonucleotide.
Cloning of the Gene Encoding M. maripaludis tRNALys.
Methanococcus vannielli (19) and Methanococcus jannaschii (20) contain identically ordered gene clusters encoding several tRNAs (including tRNALys) and 5S ribosomal RNA. Based on the assumption that a similar cluster would exist in M. maripaludis, oligonucleotide primers complementary to conserved regions flanking the tRNALys gene were used to amplify the corresponding region from genomic DNA. The primers used were MMtRNAK1 (GCGGACTGTAGATCCGCATGTCGCTG) and MMtRNAK2 (GAACCCGAGTCAC[AG]GGAGTGACAGTC). The resulting fragment was subcloned into the pBluescript II KS (Stratagene)-derived T-vector (21) and was used to transform the E. coli strain DH5α. The cloned fragment was sequenced from two independent clones on both strands, all of which contained an identical sequence encoding tRNALys.
Sequencing of the Gene Encoding Aeropyrum pernix LysRS.
The complete genome of A. pernix (type strain: K1; JCM9820; ref. 22) was sequenced by using a modified whole genome shotgun strategy. Analysis of the nucleotide sequence showed the presence of an ORF encoding a class I- but not a class II-type LysRS. A detailed description of the sequencing protocol together with the complete analysis of the A. pernix genome can be found elsewhere (Y.K. and H.K., unpublished work).
Protein Purification.
B. burgdorferi LysRS was purified as described (13). M. maripaludis LysRS was purified as before (14) except that, in the final purification step, the sample was applied to a Mono-Q column, which then was developed with a 0–500 mM NaCl gradient in standard buffer. E. coli LysRS was purified partially from the E. coli strain BL21 (DE3). A cell-free extract was prepared as described (13) and was applied to a Q-Sepharose Fast Flow column, which was developed in a 0–500 mM NaCl gradient. The fractions containing LysRS activity were pooled, were precipitated with ammonium sulfate, were resuspended in 20 mM Hepes, (pH 7.2), 1 mM MgCl2, 10 mM NaCl, and 5 mM DTT, and finally were dialyzed against the same buffer containing 30% glycerol. The concentration of LysRS in this partially purified fraction then was determined by active site titration. T7 RNA polymerase was purified as described (16).
Kinetic Analyses.
The pyrophosphate exchange reaction was performed at 37°C as described (23) in the presence of 100 mM Hepes (pH 7.2), 25 mM KCl, 10 mM MgCl2, 4 mM DTT, 2 mM lysine, 100 nM LysRS, and 1.5 μM tRNALys. Aminaocylation assays were performed as described (13, 14) with the concentration of the substrate under investigation varied over the range 0.2–5 × KM.
Cyanogen Bromide Treatment of tRNA.
tRNA (2,000 pmols) was resuspended in 90 μl STE (100 mM NaCl/10 mM Tris⋅Cl, pH 8.0/1 mM EDTA, pH 8.0) with 100 mM NaHCO3 (pH 8.9), and the mixture was incubated with 225 μg CNBr for 10 minutes at room temperature with shaking. The pH was adjusted to 6.0 with HCl, and the reaction mixture was dialyzed against 0.5 mM EDTA (pH 6.0) (24). The KM for tRNALys was determined by varying the concentration of CNBr-treated tRNA from 0.3–6 × KM.
Synthesis of Diadenosine Tetraphosphate (Ap4A).
Samples were incubated in 100 mM Hepes (pH 7.2), 5 mM ATP, 10 mM MgCl2, 20 mM KCl, 100 μM Lysine, 4 mM DTT, and 50 nM LysRS. After incubation at 37°C, the reaction was quenched with 10% perchloric acid and was neutralized with KOH. After removing the precipitate by centrifugation, ATP was depleted with 50 units of alkaline phosphatase (Boehringer–Mannheim) for 30 minutes at 37°C. Alkaline phosphatase then was inactivated by freeze/thaw. Luciferase (Photinus pyralis) was added to a final concentration of 1.2 μg/ml, and luciferin was added at a final concentration of 120 μM. Ten minutes after the addition of luciferin/luciferase, allowing for consumption of any remaining ATP, snake venom phosphodiesterase I was added to a final concentration of 8 μg/ml, and the light produced was quantified in a liquid scintillation counter at room temperature (25, 26). E. coli S100 was prepared in the same manner as described above and then was treated with RNase at a final concentration of 20 μg/ml for 5 min on ice.
Complementation of an E. coli lysS lysU Double Mutant.
The E. coli strain PALΔSΔUTR/pMAK705lysU was transformed at 30°C with either pKS-lysS, pCBS1-EctrpS, pCBS1-BblysS, or pCBS1-MmlysS. The resulting transformants then were tested for growth on Luria–Bertani agar (100 μg/ml ampicillin) at 30°C, 40°C, and 42°C with and without the addition of l-lysine (5 mM).
RESULTS
Molecular Phylogeny of Class I LysRS and tRNALys.
The 12 known predicted amino acid sequences of class I LysRS proteins were aligned and used to construct a phylogenetic tree (Fig. 1). The class I LysRS is found in both Euryarchaeota (two species of Methanococcus, Pyrococcus horikoshii, Pyrococcus furiosus, Methanobacterium thermoautotrophicum, and Archaeoglobus fulgidus) and in Crenarchaeota (a species of Cenarchaeum, the ubiquitous marine archaeon, and A. pernix, an obligately aerobic hyperthermophile). The known bacterial class I lysS genes are found in two highly disparate taxa, the spirochetes [B. burgdoeferi and Treponema pallidum (13)] and the α-proteobacteria (Rickettsia prowazekii and Rhodobacter capsulatus). The euryarchaeal versions of the enzyme form a distinctly coherent phylogenetic grouping in agreement with the 16s rRNA phylogeny (27, 28), from which the crenarchaeal examples are quite distant. In contrast, the bacterial LysRS proteins do not form a single grouping. This suggests that the bacterial class I LysRS proteins may have arisen through two distinct gene transfer events from the Archaea to Bacteria: LysRS in α-proteobacteria arising from the crenarchaeal LysRS and that in the spirochetes arising from the pyrococcal LysRS.
Figure 1.

Unrooted phylogeny of class I LysRSs. Phylogenies were constructed by using the maximum likelihood method (1,000 puzzling steps) implemented in the program puzzle 4.0 (48) and protein parsimony methods (200 bootstrap replicates) implemented in the Phylip package version 3.5c (J. Felsenstein, University of Washington). Numbers represent the percentage occurrence of nodes from maximum likelihood and protein parsimony methods, respectively. Sequences were aligned by using the program clustal x (49).
To compare the tRNA substrates of class I and class II LysRS enzymes, the tRNALys gene of M. maripaludis was cloned and sequenced. Comparison with other tRNALys species (Fig. 2) indicates a high degree of conservation, particularly at nucleotides 1–3, 34–36, and 70–73, all of which are known to be important during interaction with LysRS (see below). In contrast to the LysRS encoding genes, there appear to be no obvious divisions at the primary sequence level between the lysine tRNAs. The only exception is the G2⋅U71 wobble pair, which is found exclusively in spirochete lysine tRNA species, which are substrates of class I LysRS enzymes.
Figure 2.

Predicted secondary structure of unmodified tRNALys from B. burgdorferi, E. coli, and M. maripaludis. The anticodon and discriminator base are circled; the G2⋅U71 wobble pair is boxed.
Biochemical Phylogeny of Class I LysRS.
Sequence analysis indicates that a subgroup of the known LysRS enzymes are class I aminoacyl-tRNA synthetases. Detailed comparison of these sequences with those of other class I aminoacyl-tRNA synthetases suggests that they are related, albeit distantly, to CysRS, ArgRS, GluRS, and GlnRS (M.I. and D.S., unpublished results; ref. 29). The last three enzymes are unique among the known aminoacyl-tRNA synthetases in that they require the presence of tRNA to perform the first step of the aminoacylation reaction, aminoacyl-adenylate synthesis (ref. 30 and Eq. 1).
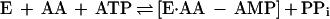 |
1 |
where AA equals amino acid and E equals aminoacyl-tRNA synthetase.
We therefore tested the ability of class I LysRS enzymes to catalyze aminoacyl-adenylate synthesis using the pyrophosphate exchange reaction (Fig. 3A). In the absence of tRNA, no significant turnover of 32P from PPi to ATP was observed whereas the addition of tRNA stimulated label exchange at a rate comparable to that observed for other aminoacyl-tRNA synthetases. These results provide experimental support for the classification of spirochete and archaeal LysRS proteins as class I aminoacyl-tRNA synthetases. To further investigate this classification, we tested the ability of the class I LysRS to support the synthesis of the alarmone Ap4A, a compound produced both in vivo and in vitro by class II LysRS enzymes through subversion of the aminoacyl-adenylate biosynthesis pathway (26). After RNase treatment to inhibit Lys-tRNALys biosynthesis, only an E. coli cell-free extract, but not B. burgdorferi or M. maripaludis LysRS, was able to synthesize Ap4A (Fig. 3B). These clear differences in the process of aminoacyl-adenylate synthesis between the class I and class II LysRS enzymes provide phenotypic support for their classification within different divisions of the aminoacyl-tRNA synthetases.
Figure 3.

(A) Pyrophosphate exchange by M. maripaludis LysRS. The reaction was incubated for 20 min at 37°C in the absence of various components as indicated. Values represent micromoles of ATP in a 20-μl aliquot. (B) In vitro Ap4A biosynthesis. Reactions were performed in the presence of E. coli cell-free extract (•), M. maripaludis LysRS (○), or B. burgdorferi LysRS (■). Values represent picomoles of Ap4A in a 50-μl aliquot.
Protein-tRNA Molecular Recognition by Class I LysRS.
To investigate the ability of class I LysRS enzymes to recognize a tRNALys, which is normally a substrate for a class II LysRS, we attempted to complement an E. coli strain in which the chromosomal copies of the lysS and lysU genes, which encode two isoforms of class II LysRS, have been disrupted [growth is maintained at permissive temperatures by a plasmid borne copy of lysU (ref. 18 and Table 1)]. Under normal growth conditions, only the B. burgdorferi lysS gene was able to rescue growth at a restrictive temperature, albeit less efficiently than E. coli lysS. Addition of exogenous lysine was necessary for phenotypic rescue by the M. maripaludis lysS gene, probably as a result of tRNA-dependent reduction in the lysine affinity of the enzyme (see below). These results indicate that in vivo class I LysRS enzymes can functionally substitute for their class II counterparts.
Table 1.
Complementation of E. coli lysS lysU double mutants by genes encoding class I LysRS proteins
| Plasmid | Growth conditions*
|
||||
|---|---|---|---|---|---|
| 30°C | 40°C | 40°C + Lys | 42°C | 42°C + Lys | |
| pCBS1-Bs trpS | ++ | − | − | − | − |
| pKS-Ec lysS | ++ | + | + | ± | ± |
| pCBS1-Mm lysS | ++ | − | ± | − | − |
| pCBS1-Bb lysS | ++ | + | + | − | − |
++, growth after 24 h; +, growth after 48 h; ±, growth after 72 h; −, no growth.
Growth was on Luria–Bertani agar (100 μg/ml ampicillin) with the addition of 5 mM l-lysine where indicated.
The molecular basis of tRNA recognition by class I LysRS was investigated by using both the archaeal enzyme from M. maripaludis and the bacterial enzyme from B. burgdorferi. It has been shown that M. vanielli tRNALys contains the modified nucleotide methyl-aminomethyl-2-selenouridine (mnm5Se2U) at position 34 (31). Because this organism is the closest known relative of M. maripaludis, with which it shares an identical gene sequence for tRNALys, we assumed that M. maripaludis tRNALys was modified similarly. In addition, it has been shown that E. coli tRNALys contains an analogous modification ofU34, methyl-aminomethyl-2-thiouridine (mnm5S2U). The reactivity of the thio- and selenomoities toward cyanogen bromide is well documented (Eq. 2; refs. 24 and 31) and was used as the basis for probing anticodon recognition by M. maripaludis LysRS (Table 2).
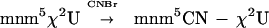 |
2 |
where X equals S or Se. Modification of both M. maripaludis and E. coli tRNA led to small reductions in the kcat for Lys-tRNALys synthesis whereas modification of the E. coli tRNA caused a significant fall in the KM for tRNA. This improvement in the apparent affinity for tRNALys on modification is in contrast to the loss of recognition by E. coli LysRS of the same substrate (data not shown), indicating that, although both the class I and class II LysRS enzymes recognize the anticodon, they do so differently. Changes also were observed in the KM for lysine, which depended strongly on both the source of the tRNA and modification of the anticodon. Anticodon recognition has been implicated in tRNA-dependent amino acid activation in E. coli GlnRS (32) and correlates with the requirement for tRNA during lysyl-adenylate synthesis reported above.
Table 2.
Recognition of tRNA by M. maripaludis LysRS
| tRNA | KM tRNA, μM | KM Lys, μM | kcat, S−1 |
|---|---|---|---|
| M. maripaludis* | 0.11 ± 0.03 | 2.2 ± 0.2 | 0.2 ± 0.05 |
| M. maripaludis-CNBr† | 0.08 ± 0.01 | 25.0 ± 8.0 | 0.05 ± 0.01 |
| E. coli‡ | 3 ± 0.8 | 350 ± 47 | 0.004 ± 0.0004 |
| E. coli-CNBr† | 0.25 ± 0.05 | 230 ± 48 | 0.002 ± 0.0001 |
M. maripaludis total tRNA (40 pmol tRNALys/A260).
tRNA was treated with CNBr as described.
E. coli tRNALys.
The sequence-specific recognition of tRNALys was investigated in vitro by using unmodified RNA transcripts of wild-type and mutant tRNA genes. These studies were confined to the B. burgdorferi LysRS because the transcript of M. maripaludis tRNALys could not be aminoacylated by its cognate synthetase under normal conditions (data not shown). To allow direct comparison of tRNA recognition by class I and class II LysRS enzymes, the tRNALys mutant set previously studied in the E. coli system was investigated (ref. 33; Table 3). The class I LysRS shows a nearly identical recognition pattern to its class II counterpart. The anticodon is recognized primarily at U35 and U36 and, to a slightly lesser extent, at U34. This correlates with the observation that B. burgdorferi contains two tRNALys isoacceptors with the anticodons UUU and CUU and thus would not be expected to specifically recognize U34. The identity of the discriminator base (N73) also was shown to be important during interaction with tRNALys, although mutation of this position was less detrimental to recognition by class I than by class II LysRS. Of interest, this increased ability to tolerate mutation of the discriminator base also has been described for the human class II-type LysRS (34). The ability of B. burgdorferi LysRS to aminoacylate a transcript of one of its cognate tRNA genes (tRNALys) also was investigated (Table 4). In contrast to the M. maripaludis and E. coli tRNALys transcripts, B. burgdorferi tRNALys was an excellent substrate for aminoacylation, indicating that modified nucleotides do not play a significant role in the recognition of this tRNA. Furthermore, the 160-fold difference in the catalytic efficiency (kcat/KM) of aminoacylation between the E. coli and B. burgdorferi tRNA transcripts indicates that nucleotides, in addition to the anticodon and discriminator base, play a role in recognition by the bacterial class I LysRS.
Table 3.
Recognition of tRNALys by B. burgdorferi LysRS
| tRNALys | B. burgdorferi LysRS Vmax/KM, relative* | E. coli LysRS Vmax/KM, relative† |
|---|---|---|
| Wild-type | 1 | 1 |
| Anticodon | ||
| UCU | <0.04 | <0.01 |
| UAC | <0.04 | <0.01 |
| UUA | <0.04 | <0.01 |
| UUG | 0.08 | 0.22 |
| GUU | 0.34 | 0.059 |
| Discriminator | ||
| G73 | 0.28 | 0.11 |
| C73 | 0.28 | 0.10 |
| U73 | 0.29 | 0.12 |
For the wild-type tRNALys, kcat = 0.22 ± 0.03 min−1 and KM = 1.5 ± 0.6 μM.
Values taken from ref. 33 (for wild-type, kcat = 3.32 min−1 and KM = 1.9 μM).
Table 4.
Recognition of in vitro transcribed tRNALys variants by B. burgdorferi and E. coli LysRS
| LysRS | tRNA | KM tRNA, μM | kcat, min−1 | kcat/KM, R* |
|---|---|---|---|---|
| B. burgdorferi | B. burgdorferi | 1.98 ± 0.32 | 47.4 ± 4.2 | 1 |
| E. coli wild type | 1.5 ± 0.6 | 0.22 ± 0.003 | 160 | |
| E. coli G2⋅U71 | 3.9 ± 0.6 | 0.21 ± 0.02 | 432 | |
| E. coli | B. burgdorferi | 4.3 ± 0.4 | 0.21 ± 0.01 | 490 |
| E. coli wild type† | 1.9 | 3.32 | 14 | |
| E. coli G2⋅U71 | 6.3 ± 0.8 | 0.15 ± 0.008 | 1022 |
Relative value expressed as fold decrease in kcat/KM compared to aminoacylation of B. burgdorferi tRNA1Lys transcript by B. burgdorferi LysRS.
From ref. 33.
Rejection of tRNALys by Class II LysRS.
All tRNALys isoacceptors encoded in the genomes of B. burgdorferi and T. pallidum contain a G2⋅U71 wobble pair, in contrast to all other known lysine tRNAs, which contain a canonical Watson–Crick pair at this position. To probe the function of this base pair, position 71 of E. coli tRNALys was mutated, resulting in a change from G2⋅C71 → G2⋅U71. The in vitro recognition of a transcript of this mutant gene then was investigated by using B. burgdorferi and E. coli LysRS (Table 4). Although the presence of the wobble pair has little effect on recognition by B. burgdorferi class I LysRS, it acts as a strong antideterminant for the E. coli class II LysRS. The G2⋅U71 variant shows a 73-fold reduction in the catalytic efficiency for aminoacylation compared with wild-type, which corresponds to a difference in Gibbs’ free energy for tRNA binding (ΔΔGtRNA) of 2.7 kcal⋅mol−1.
DISCUSSION
Molecular Phylogeny Shows an Archaeal Origin of Bacterial Class I LysRS.
The phylogenetic analysis of class I LysRS amino acid sequences shows a relationship between the archaeal examples, which reiterates phylogenies deduced from small subunit rRNA sequences. There is a clear separation of the crenarchaeal and euryarchaeal kingdoms, with the Pyrococci forming a coherent group within the latter (35). The bacterial class I LysRS proteins do not group together, instead grouping with the pyrococcal sequences in the case of the spirochetes and with Cenarchaeum symbiosum in the case of the α-proteobacteria. This separation of the bacterial sequences, which are not deeply rooted in the class I LysRS phylogeny, indicates that they may have arisen relatively recently after two separate horizontal gene transfer events from archaea. The absence of class I LysRS sequences from the genomic sequences of more deeply rooted bacteria such as Aquifex aeolicus (36) and Thermotoga maritima indicates that the class I LysRS was absent during the early evolution of bacteria, supporting an archaeal origin for the contemporary bacterial examples of this protein. Furthermore, preliminary investigation of additional partial genomic sequences indicates the presence in other bacteria (e.g., Streptomyces coelicolor) of class I LysRS proteins, suggesting that they may be distributed even more widely than originally anticipated (13).
Given that the molecular phylogeny shows an archaeal origin for bacterial class I LysRS proteins, this now raises the question of the origin of the enzyme itself. Two possibilities exist (13): either the class I and class II LysRS proteins coexisted in the universal ancestor (37) or the class I LysRS arose early in the archaeal lineage after duplication and diversification of another class I aminoacyl-tRNA synthetase. Comparison of LysRS with other class I aminoacyl-tRNA synthetases shows that these enzymes form a separate group that is not closely related to any other single group of synthetases (M.I. and D.S., unpublished results; ref. 29). If the class I LysRS had arisen in the archaeal lineage, then it would be expected to be related to an archaeal cluster of synthetases of particular amino acid specificity, in much the same way that all glutaminyl-tRNA synthetases are most closely related to eukaryotic, but not bacterial, glutamyl-tRNA synthetases (38). The lack of a close homology between LysRS and other class I aminoacyl-tRNA synthetases clearly indicates that the class I-type LysRS was present in the universal ancestor of extant life whereas phylogenetic data suggests that it was only retained during the evolution of the archaeal lineage.
Proteobacteria Can Accommodate a Class I LysRS.
The horizontal gene transfer scenario described above is strongly supported by the observation that class I LysRS encoding-genes can substitute functionally for their class II counterparts in E. coli. The archaeal gene from M. maripaludis was considerably less efficient at rescuing the growth phenotype of the E. coli mutant than the bacterial gene from B. burgdorferi. Because both the B. burgdorferi and M. maripaludis lysS genes can be expressed in E. coli to comparable levels (data not shown), this results from better recognition of E. coli tRNALys by the spirochete enzyme, as confirmed by in vitro analyses. This difference in recognition is accentuated by the tRNA-dependent recognition of lysine by class I LysRS, which renders the E. coli mutant transformed with the archaeal enzyme auxotrophic for lysine. Nevertheless, the observation that, under appropriate conditions in vivo, an archaeal class I-type LysRS can functionally replace a bacterial class II-type LysRS provides experimental support for the horizontal gene transfer hypothesis outlined above.
Molecular Basis for Gene Displacement in Bacteria.
The completion of a number of microbial genomic sequences recently has shown that horizontal gene transfer is widespread and thus may have contributed significantly to the evolution of many organisms (e.g., ref. 39). Horizontal gene transfer generally is regarded as an event that initially neither advantages nor handicaps the recipient cell but simply increases the size of the host’s gene pool. The presence of two LysRS proteins in the ancestor of spirochetes after horizontal gene transfer would mimic both the universal ancestor and contemporary bacteria such as E. coli, which contain two isoforms of the class II enzyme (40). Whether the maintenance of two LysRS enzymes offers a selective advantage is unclear but may relate to an improvement in cadaverine resistance under certain conditions (41). Examination of the molecular determinants of tRNALys recognition by class I and class II LysRS enzymes suggests that loss of the class II enzyme-encoding gene could have resulted from a point mutation in the recipient’s tRNALys gene. If this mutation gave rise to a noncanonical G2⋅U71 wobble pair in the acceptor helix of the folded tRNA (which to date has been found in lysine tRNAs exclusively from spirochetes), then recognition by the class II LysRS would be impaired severely. In contrast, such a mutation would have no significant effect on recognition by the class I LysRS enzyme, which then would be preferred by the cell to act as the provider of lysyl-tRNA, the substrate for translation of lysine codons. Under these conditions, the class II LysRS would be functionally displaced and there would be no selective advantage to retaining an accurate copy of the gene that encoded it, resulting in its degeneracy and eventual loss. Thus, differences in tRNA recognition between class I and class II LysRS enzymes provide a molecular basis for gene displacement. The recruitment of a heterologous aminoacyl-tRNA synthetase by a mutated tRNA is analogous to the “tRNA gene recruitment” hypothesis in which tRNAs with anticodon mutations are proposed to become substrates for homologous aminoacyl-tRNA synthetases of different amino acid specificity (42).
Substrate Conservation Leads to Functional Convergence of Divergent Enzymes.
In vitro analysis of molecular recognition clearly indicates that both class I and class II LysRS proteins recognize the discriminator base (N73) and anticodon of tRNALys. However, the marked differences in their abilities to recognize a tRNA with a chemically modified anticodon suggest that they bind tRNA differently. This was confirmed by the strikingly different effects on recognition of introducing a G2⋅U71 wobble pair into the acceptor helix of E. coli tRNALys. Although this mutation had no significant effect on aminoacylation by the class I enzyme, it led to a 73-fold drop in the efficiency (kcat/KM) of the class II LysRS. An explanation for these effects can be found by comparison to the alanine system. All known alanine tRNA species contain a G3⋅U70 wobble pair whose function is to distort the acceptor helix such that functional groups in the minor groove can be specifically recognized by alanyl-tRNA synthetase, a class II enzyme (43, 44). This correlates with observations from structural (8, 10) and biochemical (45) data that class I and class II synthetases approach the RNA acceptor helix from different sides. This is apparently also the case for the two types of LysRS, in which the ability of the class II LysRS to recognize tRNALys is impaired by the exposure of functional groups in the minor groove of the G2⋅U71 mutant. Because the class I LysRS approaches the opposite side of the acceptor stem, its aminoacylation efficiency is not impaired by this mutation. Thus, although the class I and class II LysRS enzymes recognize the same sites in their RNA substrates, they do so differently (similarly, they differ in their exact mechanisms of lysine activation). Only small changes are observed in lysine tRNA sequences from organisms in different kingdoms; thus, there are no obvious gross differences that distinguish the substrates of class I and class II LysRS enzymes. (M.I. and D.S., unpublished results; ref. 29).
The functional convergence of two unrelated LysRS enzymes strongly suggests that the evolution of specificity in the aminoacyl-tRNA synthetases was driven by conserved, preexisting substrates. The possibility that tRNALys existed before one class of LysRS is consistent with earlier suggestions that tRNAs are derived from more primitive structures not involved in translation (46) and that tRNAs in general predate aminoacyl-tRNA synthetases (47). In view of the experimental data presented here, it now seems reasonable to assume that transfer RNAs did indeed appear before at least some of the aminoacyl-tRNA synthetases during the evolution of the contemporary translational apparatus.
Acknowledgments
We thank Sylvain Blanquet, Pierre Plateau, and Koji Tamura for kindly providing strains and plasmids and Siv Andersson for access to the R. prowazekii lysS sequence before publication. We are greatly indebted to Alan Curnow, Alan Weiner, and Carl Woese for critical reading of the manuscript. Work in the author’s laboratory was supported by a grant from the National Institute of General and Medical Sciences (to D.S.).
ABBREVIATIONS
- LysRS
lysyl-tRNA synthetase
- Ap4A
diadenosine tetraphosphate
Footnotes
References
- 1.Gesteland R F, Atkins J F. The RNA World. Plainview, NY: Cold Spring Harbor Lab. Press; 1993. [Google Scholar]
- 2.Schimmel P, Ribas de Pouplana L. Cell. 1995;81:983–986. doi: 10.1016/s0092-8674(05)80002-9. [DOI] [PubMed] [Google Scholar]
- 3.Shiba K, Motegi H, Schimmel P. Trends Biochem Sci. 1997;22:453–457. doi: 10.1016/s0968-0004(97)01135-3. [DOI] [PubMed] [Google Scholar]
- 4.Ibba M, Curnow A W, Söll D. Trends Biochem Sci. 1997;22:39–42. doi: 10.1016/s0968-0004(96)20033-7. [DOI] [PubMed] [Google Scholar]
- 5.Curnow A W, Tumbula D, Pelaschier J, Min B, Söll D. Proc Natl Acad Sci USA. 1998;95:12838–12843. doi: 10.1073/pnas.95.22.12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doolittle R F. Nature (London) 1998;392:339–342. doi: 10.1038/32789. [DOI] [PubMed] [Google Scholar]
- 7.Eriani G, Delarue M, Poch O, Gangloff J, Moras D. Nature (London) 1990;347:203–206. doi: 10.1038/347203a0. [DOI] [PubMed] [Google Scholar]
- 8.Cusack S. Curr Opin Struct Biol. 1997;7:881–889. doi: 10.1016/s0959-440x(97)80161-3. [DOI] [PubMed] [Google Scholar]
- 9.Nagel G M, Doolittle R F. J Mol Evol. 1995;40:487–498. doi: 10.1007/BF00166617. [DOI] [PubMed] [Google Scholar]
- 10.Arnez J G, Moras D. Trends Biochem Sci. 1997;22:211–216. doi: 10.1016/s0968-0004(97)01052-9. [DOI] [PubMed] [Google Scholar]
- 11.Brown J R, Doolittle W F. Proc Natl Acad Sci USA. 1995;92:2441–2445. doi: 10.1073/pnas.92.7.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown J R. In: Thermophiles: The Keys to Molecular Evolution and the Origin of Life? Wiegel J, Adams M H W, editors. London: Taylor & Francis; 1998. pp. 217–230. [Google Scholar]
- 13.Ibba M, Bono J L, Rosa P A, Söll D. Proc Natl Acad Sci USA. 1997;94:14383–14388. doi: 10.1073/pnas.94.26.14383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ibba M, Morgan S, Curnow A W, Pridmore D R, Vothknecht U C, Gardner W, Lin W, Woese C R, Söll D. Science. 1997;278:1119–1122. doi: 10.1126/science.278.5340.1119. [DOI] [PubMed] [Google Scholar]
- 15.Syvanen M. Annu Rev Genet. 1994;28:237–261. doi: 10.1146/annurev.ge.28.120194.001321. [DOI] [PubMed] [Google Scholar]
- 16.Curnow A W, Garcia G A, Kung F L, Koch K. Biochemistry. 1993;32:5239–5246. doi: 10.1021/bi00070a036. [DOI] [PubMed] [Google Scholar]
- 17.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1990. [Google Scholar]
- 18.Chen J, Brevet A, Lapadat-Tapolsky M, Blanquet S, Plateau P. J Bacteriol. 1994;176:2699–2705. doi: 10.1128/jb.176.9.2699-2705.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wich G, Jarsch M, Böck A. Mol Gen Genet. 1984;196:146–151. doi: 10.1007/BF00334107. [DOI] [PubMed] [Google Scholar]
- 20.Bult C J, White O, Olsen G J, Zhou L, Fleischmann R D, Sutton G G, Blake J A, FitzGerald L M, Clayton R A, Gocayne J D, et al. Science. 1996;273:1058–1072. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- 21.Kwak J H, Kim M Y. Anal Biochem. 1995;228:178–180. doi: 10.1006/abio.1995.1335. [DOI] [PubMed] [Google Scholar]
- 22.Sako Y, Nomura N, Uchida A, Ishida Y, Morii H, Koga Y, Hoaki T, Maruyama T. Int J Syst Bacteriol. 1996;46:1070–1077. doi: 10.1099/00207713-46-4-1070. [DOI] [PubMed] [Google Scholar]
- 23.Wells T N, Knill-Jones J W, Gray T E, Fersht A R. Biochemistry. 1991;30:5151–5156. doi: 10.1021/bi00235a006. [DOI] [PubMed] [Google Scholar]
- 24.Agris P F, Söll D, Seno T. Biochemistry. 1973;12:4331–4337. doi: 10.1021/bi00746a005. [DOI] [PubMed] [Google Scholar]
- 25.Ogilvie A. Anal Biochem. 1981;115:302–307. doi: 10.1016/0003-2697(81)90009-9. [DOI] [PubMed] [Google Scholar]
- 26.Brevet A, Chen J, Leveque F, Plateau P, Blanquet S. Proc Natl Acad Sci USA. 1989;86:8275–8279. doi: 10.1073/pnas.86.21.8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsen G J, Woese C R, Overbeek R. J Bacteriol. 1994;176:1–6. doi: 10.1128/jb.176.1.1-6.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barns S M, Delwiche C F, Palmer J D, Pace N R. Proc Natl Acad Sci USA. 1996;93:9188–9193. doi: 10.1073/pnas.93.17.9188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ribas de Pouplana L, Turner R J, Steer B A, Schimmel P. Proc Natl Acad Sci USA. 1998;95:11295–11300. doi: 10.1073/pnas.95.19.11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schimmel P R, Söll D. Annu Rev Biochem. 1979;48:601–648. doi: 10.1146/annurev.bi.48.070179.003125. [DOI] [PubMed] [Google Scholar]
- 31.Ching W M, Alzner-DeWeerd B, Stadtman T C. Proc Natl Acad Sci USA. 1985;82:347–350. doi: 10.1073/pnas.82.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ibba, M., Hong, K.-W., Sherman, J. M., Sever, S. & Söll, D. Proc. Nat. Acad. Sci. USA 93, 6953–6958. [DOI] [PMC free article] [PubMed]
- 33.Tamura K, Himeno H, Hasegawa T, Shimizu M. Nucleic Acids Res. 1992;20:2335–2339. doi: 10.1093/nar/20.9.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiba K, Stello T, Motegi H, Noda T, Musier-Forsyth K, Schimmel P. J Biol Chem. 1997;272:22809–22816. doi: 10.1074/jbc.272.36.22809. [DOI] [PubMed] [Google Scholar]
- 35.Pace N R. Science. 1997;276:734–740. doi: 10.1126/science.276.5313.734. [DOI] [PubMed] [Google Scholar]
- 36.Deckert G, Warren P V, Gaasterland T, Young W G, Lenox A L, Graham D E, Overbeek R, Snead M A, Keller M, Aujay M, et al. Nature (London) 1998;392:353–358. doi: 10.1038/32831. [DOI] [PubMed] [Google Scholar]
- 37.Woese C. Proc Natl Acad Sci USA. 1998;95:6854–6859. doi: 10.1073/pnas.95.12.6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siatecka M, Rozek M, Barciszewski J, Mirande M. Eur J Biochem. 1998;256:80–87. doi: 10.1046/j.1432-1327.1998.2560080.x. [DOI] [PubMed] [Google Scholar]
- 39.Doolittle W F, Logsdon J M. Curr Biol. 1998;8:R209–R211. doi: 10.1016/s0960-9822(98)70127-7. [DOI] [PubMed] [Google Scholar]
- 40.Leveque F, Plateau P, Dessen P, Blanquet S. Nucleic Acids Res. 1990;25:305–312. doi: 10.1093/nar/18.2.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brevet A, Chen J, Leveque F, Blanquet S, Plateau P. J Biol Chem. 1995;270:14439–14444. doi: 10.1074/jbc.270.24.14439. [DOI] [PubMed] [Google Scholar]
- 42.Saks M E, Sampson J R, Abelson J. Science. 1998;279:1665–1670. doi: 10.1126/science.279.5357.1665. [DOI] [PubMed] [Google Scholar]
- 43.Gabriel K, Schneider J, McClain W H. Science. 1996;271:195–197. doi: 10.1126/science.271.5246.195. [DOI] [PubMed] [Google Scholar]
- 44.Beuning P J, Yang F, Schimmel P, Musier-Forsyth K. Proc Natl Acad Sci USA. 1997;94:10150–10154. doi: 10.1073/pnas.94.19.10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sissler M, Eriani G, Martin F, Giege R, Florentz C. Nucleic Acids Res. 1997;25:4899–4906. doi: 10.1093/nar/25.24.4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weiner A M, Maizels N. Proc Natl Acad Sci USA. 1987;84:7383–7387. doi: 10.1073/pnas.84.21.7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woese C. The Genetic Code. New York: Harper & Row; 1967. [Google Scholar]
- 48.Thompson J D, Gibson T J, Plewniak F, Jeanmougin F, Higgins D G. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strimmer K, von Haeseler A. Mol Biol Evol. 1996;13:964–969. [Google Scholar]