

Abstract
Megabase DNA molecules become trapped in agarose gels during electrophoresis if the electric field exceeds a few volts per cm. Fluorescence microscopy reveals that these molecules invariably arrest in U-shaped conformations. The field-vs.-size dependence for trapping indicates that a critical molecular tension is required for trapping. The size of unligated λ-ladders, sheared during gel electrophoresis at a given field, coincides with the size of molecules trapped at that field, suggesting that both processes occur through nick melting near the vertex of the U-shape. Consistently, molecules nicked by exposure to UV radiation trap more readily than unexposed ones. The critical trapping tension at the vertex is estimated to be 15 pN, a force sufficient to melt nicks bent around gel fibers, and, according to our model, trap a molecule. Strategies to reduce molecular tension and avoid trapping are discussed.
Gel electrophoresis is the method of choice for the size fractionation of DNA in analytical biochemistry. Steady-field electrophoresis is commonly used to separate molecules from a few bp to about 20 kbp, while pulsed-field gel electrophoresis (PFGE) methods (1–3) are required to separate molecules beyond this size range, up to 10 megabase pairs (Mbp). In PFGE, the electric field is periodically alternated in two directions and DNA separation depends on the way the molecules reorient through the gel in response to the changing electric field (4, 5). Unfortunately, molecules longer than 1–2 Mbp can become permanently immobilized or trapped after traveling various distances through the gel, leading to band smearing (6). Trapping occurs in both steady-field and pulsed-field experiments if the electric field is higher than some critical value, Ecrit (7). The value of Ecrit falls as the DNA size is increased. Lowering the field below 2 V/cm prevents trapping (6) but at the expense of very long electrophoretic runs (4). Ecrit appears to be higher for PFGE than for steady-field electrophoresis. Trapping in PFGE has been reduced, and sharper bands obtained by interrupting the long electric-field pulses with short high-voltage spikes in the reverse direction (7). These protocols notwithstanding, DNA trapping still remains a practical barrier to high-resolution PFGE above 10 Mbp. Understanding and preventing molecular trapping is essential to overcoming the current size limit of DNA separation and may lead to sharper resolution and reduced running times.
Several mechanisms of DNA trapping have been proposed. Olson (6) suggested that very large molecules pile up against pores in a compressed state. Turmel and coworkers (7) suggest such pileups may occur in agarose cul-de-sacs because of locally high gel concentrations. Alternately, multiple “hernias” or “impaled spirals” (large DNA knots) could be the cause (7). Deutsch (8) has proposed that the molecules may bend around gel fibers with the two arms extending in the direction of the electric field, forming U-shapes. Because of local corrugation in both the DNA and the agarose fibers, a static friction force could develop at the pivot point, pinning the chain and trapping it. Viovy et al. (9) performed trapping experiments in which they observed a population of permanently trapped molecules that, after being exposed to a high field, did not release when the electric field was subsequently lowered below Ecrit. They suggest trapping results from the tightening of a small DNA knot around a gel fiber. Burlatsky and Deutch (10) proposed that a simple form of static friction between DNA and agarose might account for trapping, but Viovy and Duke (11) have pointed out inconsistencies between the friction hypothesis and trapping behavior. To date, the available data have not been sufficient to rule out or support, unambiguously, any of these models.
Here we use both macroscopic (bulk) and microscopic (single-molecule) methods to study the dynamics of very long DNA molecules undergoing steady-field electrophoresis and their behavior leading to field-dependent trapping. A mechanism is proposed that can account for both permanent and reversible DNA trapping in either steady-field electrophoresis or in PFGE.
MATERIALS AND METHODS
For macroscopic trapping experiments, 16 chromosomes from the yeast Saccharomyces cerevisiae were separated on a rotating-gel apparatus by using 120° PFGE. Running conditions were 1% agarose gel (FastLane, FMC) in ½× TBE buffer (45 mM Tris borate/1 mM EDTA, pH 8.3), field strength 5 V/cm for 16 hr and pulse time ramped from 80 to 180 sec. Then the gels were rotated 90° and steady fields of different strengths were applied in the second dimension. To investigate the reversibility of DNA trapping, chromosomes were trapped in the second (analysis) dimension by an electric field of 10 V/cm for 30 min but then the electric field was lowered to 2 V/cm and applied for 7 hr. The temperature was kept at 5°C for all runs.
For imaging experiments, the three largest chromosomes, with lengths of 1.1, 1.6, and 1.9 Mbp, were isolated by PFGE, stained with the fluorescent dye YOYO-1 (12), and placed in an electrophoresis microchamber (13). They were observed by using an epiillumination fluorescence microscope (Axiovert 35M, Zeiss) equipped for real-time imaging experiments (5).
RESULTS AND DISCUSSION
Macroscopic Experiments.
To investigate the relationship between Ecrit and DNA size, the steady-field trapping experiments of Turmel et al. (7) were extended to a wider field-strength range. Yeast chromosomes were first separated in the vertical dimension by PFGE and then a steady field was applied horizontally so the effect of various field strengths could be investigated in this second dimension. Fig. 1a shows that almost all chromosomes migrate without trapping at 2 V/cm. Only the largest (1.9 Mbp) traps partially in the second dimension, as seen by the smear behind its band. With increasing field strength, trapping occurs in smaller chromosomes. This figure illustrates several points about DNA trapping. (i) An inverse relationship exists between the minimum DNA size trapped and the field strength. (ii) Trapping appears as a horizontal smear, indicating that the probability of a molecule becoming trapped increases with its time of exposure to the field. (iii) DNA traps at lower field strengths in steady-field electrophoresis than in PFGE because no band smearing is seen in the PFGE dimension at 5 V/cm, while some trapping is already observed at 2 V/cm in steady-field conditions (Fig. 1a). (iv) As seen in Fig. 1b, molecules can be trapped either reversibly or permanently, i.e., some molecules that were trapped at 10 V/cm regain mobility when the field is lowered to 2 V/cm, while others do not. Fig. 1c depicts the relationship between the DNA size and the critical electric field strength, Ecrit, which is sufficient to cause trapping. These data are best fit by a straight line with a slope of −1, indicating that Ecrit is inversely proportional to the first power of the DNA size. For comparison, the data of Turmel et al. (7), obtained under similar conditions, are also plotted. Those authors proposed a logarithmic relationship for Ecrit, which placed a lower limit of 600 kbp on the trapping size for DNA (7), but their data are reasonably consistent with a simpler inverse relationship. We find that using a sufficiently high field (25 V/cm) traps even the smallest yeast chromosome (225 kbp). Viovy et al. (9) observed a square-law dependence for Ecrit but, unlike the other studies, the trapping field was pulsed. Such pulsing affects long and short chromosomes differently and may have altered the apparent slope of their trapping data in Fig. 1c, as explained below.
Figure 1.

(a) Sixteen yeast chromosomes, ranging from 1.9 Mbp (Top) to 225 kbp (Bottom) resolve vertically into 12 bands using 120° PFGE. Steady fields applied in the horizontal dimension: E = 2 V/cm for 7 hr, E = 6 V/cm for 70 min, E = 8 V/cm for 40 min, E = 10 V/cm for 30 min. (b) (Left) Chromosomal separation. (Center) Trapping at 10 V/cm for 30 min. (Right) Reversibly trapped molecules regain mobility at 2 V/cm and are seen in the intermediate column. (c) Log-log plot of critical field strength vs. size during agarose gel electrophoresis where Ecrit is the field strength sufficient to cause 50% trapping of a band over a 1-cm path. Data from the present study (•) and from Turmel et al. (7) (■) were obtained in steady fields. Data of Viovy et al. (9) (▵) were obtained in PFGE. Their largest size point is also shown (×) corrected for field-averaging effect. λ shear data (○) refer to the largest surviving concatemers of λ (see Fig. 4). Dashed line indicates slope of −1.
Microscope Observations.
To find the molecular mechanisms responsible for trapping, individual DNA chromosomes were visualized under a fluorescence microscope (13) as they migrated through the agarose and became trapped. Imaging megabase-sized DNA molecules requires several modifications to the protocols used for the observation of shorter DNAs (14–16). Gels ≈1 mm thick had to be used to fully contain the larger molecules without distorting their shapes. Because of the molecules’ great length (up to 1 mm), it was necessary to translate the gel past the objective lens while varying the focus as the images were recorded on video tape. Well-focused images of the molecule were then recovered from the tape and pieced together to form a mosaic.
Imaging revealed (13) that megabase molecules migrate through a gel by extending and collapsing in “caterpillar cycles,” as previously described for smaller molecules (4, 17–19). Initially, a molecule is forced into the gel as multiple loops or “hernias,” thus displaying a branched conformation. Farther in the cycle, long branches grow longer by siphoning chain out of the short ones, which eventually disappear. Finally the true ends of the molecule unfold into a surprisingly long U-shape. At only 5 V/cm field, the tip-vertex-tip distance for the U exceeds 50% of its stained contour length, even allowing 40% lengthening because of dye intercalation (20). The molecule then slips off this U-shape with the longer arm pulling the chain. After turning the corner at the vertex, the molecule collapses rapidly before starting a new cycle.
In low-strength fields, e.g., 2 V/cm, the molecules migrate by producing (and slipping off) U-shapes of various symmetries. If the field strength is raised to 10 V/cm, however, the cycle arrests at the U-shape stage, and the molecules become trapped in the gel. At such increased voltage, nearly all molecules above a given size arrest in this conformation, remaining motionless as long as the voltage is not changed. When the field strength is lowered to 2–3 V/cm, the arms of all molecules shorten slightly and then, in some cases, the molecules slip off the U-shape and continue to migrate in caterpillar cycles. One such example is shown in Fig. 2A, where, at 10 V/cm, the chromosome is highly aligned; its visible path is ≈86% of its stained contour length. When the electric field is reduced to 2.5 V/cm, both molecular ends retract so the visible path length is reduced to ≈80% of the contour length (B). Some sliding is already occurring because only one end appears bright, not both. After ≈1 min (C and D), the molecule moves around the vertex into the longer arm. Finally after about 2 min it collapses into a compact shape (E–G). Such behavior is consistent with “chain-pinning” at the base of the U-shape, as suggested before (8).
Figure 2.

(A) Molecule (1.6 Mbp) is trapped reversibly in a high field of 10 V/cm . (B–G) After the electric field is reduced to 2.5 V/cm, the molecule is released and regains motion.
However, some molecules remain permanently trapped in U-shapes even after the field is decreased below 2 V/cm, a value that normally permits migration. For these molecules, if the field is reduced to zero, the arms of the U-shapes shrink back toward the pivot point, indicating that the point of attachment always occurs near the vertex. These two opposite behaviors are likely to correspond, respectively, to the reversibly and permanently trapped molecules characterized in macroscopic experiments (Fig. 1b). The fact that both reversibly and permanently trapped molecules adopt indistinguishable U-shapes suggests that molecules may always trap first reversibly before becoming permanently trapped. If the field was lowered soon after the molecules were initially trapped, only a few exhibited permanent trapping, whereas if the field was maintained for 5–10 min on molecules immobilized in U-shapes, then most of them become permanently trapped. Microscope observations appear to rule out the cul-de-sac, multiple-hernia, or (large) impaled spiral as mechanisms for trapping. The resolution of the microscope is not sufficient, however, to detect the existence of a small tight knot (9) near the vertex of the U.
To localize the points of DNA/gel attachment, the arms of trapped molecules were successively dissected with a focused laser beam. The highly photosensitive YOYO-stained DNA typically breaks within 2–3 sec of exposure to the 488-nm light from an argon ion laser. Two types of behavior were then observed. The first is illustrated in Fig. 3, where a molecule is initially trapped in the gel in a slightly asymmetric U-shape (A). When the laser spot is directed to a point near its lower end (arrow), the molecule breaks and the fragment moves downfield (B), but the majority of the molecule remains trapped. A larger fragment is targeted (C) and liberated (D), but the rest of the molecule remains trapped. When a final piece is removed (E–F), the molecule immediately begins to slip off the U-shape (G–I). Here, either the force imbalance between the arms is sufficient to shear the molecule past its point of attachment or the force pinning the molecule at its vertex is sufficiently reduced to release the molecule. A different type of behavior occurs when successive cutting of the shorter arm does not release the molecule until the cut is made very close to the vertex. This cut leads to the release of the long arm of the molecule, but leaves a visible piece of DNA attached to the gel located between the last cutting point and the short-end arm of the molecule. Presumably this piece contains the permanent point of attachment. It is always located near the vertex, usually within the upper 10% of the U-shape, but it is seldom located precisely at the visible sharp vertex.
Figure 3.

(A) A chromosomal fragment (≈870 kbp) is trapped at 10 V/cm, its lower arm is successively dissected with an argon–ion laser beam (200 mW, 488 nm), and the excised DNA fragments migrate away from the field of view (B–D). Eventually, a point is reached where the molecule starts slipping off the U-shape (F–I).
Molecular Mechanism of DNA Trapping.
Because DNA is uniformly charged, the tension in the molecule builds toward the turning point (vertex) and it is possible that this molecular tension somehow causes local binding to the gel (8–11). Many features of Fig. 1c are explained if some critical tension, Tcrit, is sufficient to induce this interaction between the gel fibers and DNA. Perhaps Tcrit is that tension that would tighten a DNA knot around a gel fiber and make it permanent, as previously suggested (9). Alternatively, the tension developed near the vertex of the U could alter the local DNA structure, leading to an increased interaction between the DNA and the gel fibers. To evaluate these possibilities, it was necessary to measure the tension experienced by the DNA molecule at the vertex of the U.
We first estimated the forces that can be generated inside an agarose gel during steady-field electrophoresis by using the fact that unligated λ-DNA concatemers break apart during gel electrophoresis when exposed to high enough electric fields (21). Unligated λ-DNA concatemers of different sizes (λ-ladders) were loaded into an agarose gel and exposed to different steady fields. The treated DNA samples were then moved to a pulsed-field gel apparatus and analyzed (Fig. 4). Depending on the strength of the initial field, concatemers longer than a given size were selectively removed because forces generated inside the gel broke the 12-bp cos sites holding the monomers together. As the field was increased, the maximum surviving concatemer size decreased. When these maximum sizes are plotted against the applied field strength in Fig. 1c, the results are found to fall on top of the trapping curve for steady-field electrophoresis. Because the force required to break a cos site is nearly equal to the force required to trap a molecule (Tcrit), the molecular mechanism leading to trapping may be the same as that involved in breaking the cos sites. Accordingly, it was never possible to trap λ-concatemers inside gels unless they had first been ligated (data not shown).
Figure 4.

Unligated multimers of λ-phage DNA (48.5 kbp) were transferred by gentle electrophoresis (2 V/cm) from the ladder gel (NEB, Beverly, MA) into a 1% FastLane gel (FMC) in ½× TBE. The DNA band thus formed was excised in a gel plug and placed in a different electrophoresis chamber. Each plug was then subjected to a specific field for 2 min in the forward direction, relaxed in zero field for 2 min, and exposed to that field in the reverse direction for a final 2 min. Seven such plugs, exposed to different fields (values shown in figure in V/cm) were then recast into a 1% gel, and the size of the surviving multimers was analyzed by low-voltage (3 V/cm) PFGE.
The force required to break a cos site directly was next determined by pulling an unligated λ-dimer by its ends with force-measuring laser tweezers (22). About 30 such molecules were pulled with increasing (≈5 pN/sec) force until they broke. The breaking force was 45 ± 3 pN in 0.5 M NaCl buffer and 35 ± 5 pN in electrophoresis buffer (½× TBE). Could such forces develop at the turning point of a U during steady-field electrophoresis? A molecule that assumes a symmetrical U-shape should experience a tension at the vertex
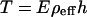 |
1 |
where E is the field strength, h is the projection of the arm length along the direction of the electric field (23), and ρeff is the electrophoretic charge density of the DNA molecule inside the gel. This quantity is the effective charge per unit length remaining after neutralization by counterion condensation and hydrodynamic coupling to nearby counterions moving in solution. Smith and Bendich (24) observed the field-induced extension of anchored DNA molecules in free solution and obtained a value for ρeff of only ≈0.05 e−/phosphate. Long et al. (25) pointed out that in free solution the anchor tension reflects only a hydrodynamic drag between the molecule and its external surroundings and, because such drag scales nonlinearly with molecular length, ρ loses its meaning as an intrinsic property of DNA. Consistent with this interpretation, two things change when DNA is tethered inside a gel. First, nearby gel fibers reduce the velocity of the free counterions and, because the drag exerted by these counterions on the DNA then decreases, the anchor force increases, thus increasing ρeff over its free-solution value. Second, different sections of the DNA molecule become hydrodynamically independent, so ρeff becomes well defined independent of molecular length, but its value depends on the properties of the gel.
To measure directly the tension experienced by DNA molecules tethered inside agarose gel, a dimer of λ-DNA (97 kbp) was attached by one of its ends to a plastic bead and this bead was held in force-measuring laser tweezers (22). The force on the molecule was then measured while an electric field was applied. Either the molecule was tethered in open solution or else it was allowed to penetrate a nearby wall of agarose. In open solution, these molecules gave a tether force corresponding to ρeff = 0.04 e−/phosphate but inside 1.2% FastLane agarose, the tether force increased 4-fold to give ρeff = 0.16 e−/phosphate. Assuming h in Eq. 1 to be slightly shorter than half the contour length, the critical tension developed near the vertex of a U in agarose gel is ≈15 pN. But this tension is less than half of the 35 pN value required to shear individual λ-concatemers by direct pulling. Therefore, cos site breakage in a gel cannot take place by simple tension. Instead, a different mechanism must account for the fact that the shearing data and the trapping data coincide (Fig. 1c).
A molecule’s path through a gel is tortuous and deviates sharply at many locations, most of which are too small to be seen under the microscope. A tension of 15 pN at such a location would be sufficient to bend a molecule around a gel fiber with a radius of curvature <3 nm. The locally stressed DNA could then buckle (26, 27), melt, and break a cos-site when it slides past this location. Significantly, the same mechanism would lead to trapping if the molecules were nicked only in one chain and would explain the coincident behavior of the trapping and the cos sites breaking data. According to this model, when an isolated nick in the molecule slides past a sharp deviation in the gel path, the DNA melts as it turns around a gel fiber under the bending moment developed by the external tension about this point. Because any single-stranded region becomes longer than B-form dsDNA under 15 pN tension (22), the arms of the U-shape will relax by moving (drooping) downfield, thus arresting the molecule’s sliding motion and trapping it at that location.
According to this model, molecules harboring a larger number of nicks should trap more readily. To test this prediction, molecules of DNA were nicked by exposure to UV radiation for increasing periods of time. Fig. 5 shows that molecules exposed for 2 and 10 min to UV light trapped more efficiently than unexposed ones. Moreover, the minimum size of molecules trapped decreases with irradiation time, suggesting that multiple nicks acting simultaneously reduce a molecule’s chance to escape.
Figure 5.

Yeast chromosomes were separated vertically and then exposed for 0, 2, and 10 min to UV light on a transilluminator. Next, a steady field of 10 V/cm was applied in the horizontal direction. The effect of radiation was 3-fold. (i) Medium-sized chromosomes trap faster (farther left) after 2- and 10-min exposure (long thick arrows); (ii) Small chromosomes become trappable after irradiation (long thin arrows); (iii) UV radiation causes some double-stranded breakage (short thin arrows).
To summarize the model, the U shape provides the tension near its vertex, the molecule’s tortuous path through the gel provides the fulcra over which the molecules bend, and the nicks provide a localized potential well. This model explains a number of observations: (i) When the field is reduced below Ecrit, the single-stranded region retracts because of its entropic elasticity. At ≈8 pN tension, ssDNA has the same length as dsDNA (22) and therefore an effective potential well no longer forms as the nick slides over the bend. (ii) Cutting one of the arms of a molecule reduces the tension (depth of the potential well) and increases the shear force on the molecule (pushing it toward one edge of the well). Both effects cause a reversibly trapped molecule to escape the trap (Fig. 3). (iii) The melted regions on both sides of the nick might extend far from the nick and, in time, form their own secondary structure, enclosing gel fibers and thus trapping the molecule “permanently.” Alternatively, imbalance in the U-shape’s arms could shear the molecule past the gel fiber. The displaced strand might then reanneal around the invading gel fiber. (iv) Field-reversal spikes (7) reduce trapping by lifting a nick off a local bend in the gel path. The molecule then slides a relatively long distance before the nick reaches another sharp bend. (v) The field required to trap molecules in PFGE is usually larger than in steady-field electrophoresis because U-shapes must be formed within the duration of a PFGE pulse before molecules can trap. If the field pulses are shorter than the molecular reorientation time, the molecules experience an average field that is smaller than the instantaneous field. Thus Ecrit appears to be increased, as observed in the data of Viovy et al. (9) shown in Fig. 1c. In this study, the largest molecule (≈2 Mbp) should have a reorientation time of ≈200 sec (using 1.5% agarose and E = 6.8 V/cm), but the field reorientation time was only 90 sec. Because the field direction switched through an angle of 120°, the time-averaged field had half the magnitude of the instantaneous field. If the 2-Mbp point in Fig. 1c is lowered from the instantaneous field of 6.8 V/cm to the average field of 3.4 V/cm, it rejoins the other data with a slope of −1 (× in Fig. 1c). (vi) PFGE protocols such as field inversion gel electrophoresis, which rely on U-shaped formation to fractionate DNA, produce more trapping than 120o PFGE in which alternative open forms, called chevron shapes, are usually formed (4, 5, 28).
CONCLUSIONS
The observations presented here suggest the following strategies to reduce trapping and speed up separation of megabase molecules: (i) Avoid nicked DNA molecules by using fresh samples. (ii) Shape the field pulse. Switching the field direction in PFGE causes the collapse of existing U-shapes and begins extending some of these molecules into U-shapes in the opposite direction. Because it takes time to form a U-shape, there should be some grace period just after a field reversal during which the magnitude of the new field can be made arbitrarily large without trapping the molecule. By optimally shaping the field pulses, the molecular reorientation time can be cut in half (compared with constant-height pulses), reducing proportionally the separation time and still avoiding trapping. Assume that the pulse has been reversed for a time t and that the arms extend (from zero length) at the rate μE, where μ is the mobility of the DNA (4, 29). The force builds in time according to F = μρeff E2 t until the molecular contour length, L, is reached, and then F < ρeff EL/2. Shaping the field pulse to decay as E(t) = (Tcrit/2 μρefft)1/2 keeps the tension below Tcrit and avoids trapping. (iii) Use lower-concentration gels that have larger pores. The hydrodynamic coupling between the DNA molecule and its mobile counterions is thus maximized and ρeff is reduced. The tension at the vertex of a U-shape is lowered (Eq. 1), permitting higher electric fields and increasing the velocity of the separation.
Combining strategies ii and iii described above, we have been able to speed up the separation of yeast chromosomes. By using traditional PFGE protocols, the chromosomes of S. cerevisiae normally require a day to separate while those of Schizosaccharomyces pombe require 3 days to a week. By reducing the gel concentration to 0.3% and by shaping the pulses to avoid forces greater than 15 pN during the U-shape conformation, the chromosomes of S. cerevisiae were resolved in 4 hr and those of S. pombe in 20 hr (Fig. 6). A low gel concentration protocol had been developed empirically by Gunderson and Chu (30), obtaining similar results.
Figure 6.

Rotating-gel apparatus separations performed in 0.3% high-strength agarose gels (FMC Gold) cast inside supporting frames of 1% FastLane agarose. (a) S. cerevisiae chromosomes (0.25–2 Mbp) (SC) and λ concatemers (λ) separated in 4 hr by using an electric field of 7.5 V/cm and pulse times ramped from 20 to 45 sec. The two largest chromosomes (1.6 and 2 Mbp) comigrate in the top band. (b) Separation of S. cerevisiae (SC) and S. pombe (SP) chromosomes. The three S. pombe chromosomes (3.5, 4.7, and 5.7 Mbp) are indicated by arrows. To launch the chromosomes from the wells, a field-inversion electrophoresis protocol (30 sec forward, 10 sec backward) was applied for 2 hr, with a field of 1.2 V/cm. Then, 100 periodic rotations of 110° with shaped-field pulses were applied for 18 hr. Pulses started at 10 V/cm (after each rotation), but decayed as E(t) = (Tcrit/2μρefft)1/2, assuming Tcrit = 15 pN, ρeff = 0.084 e−/phosphate, and μ = 6 × 10−9 m2/V·sec.
Human chromosomal DNA ranges in size from 50 to 250 Mbp, i.e., more than five times the size of the largest DNA molecules commonly separated today. As the present study illustrates, the size limit for PFGE separations resides in the mechanical strength of the DNA molecule itself. Protocols that stabilize DNA structure and reduce their mechanical stress will be essential as the separation of larger molecules is attempted.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM-32543) and the National Science Foundation (MBC 9118482 and DBI 9732140). The authors thank Fernanda Bustamante for help with the experiments in Fig. 1c and Yujia Cui for testing cos-site strength.
ABBREVIATIONS
- PFGE
pulsed-field gel electrophoresis
- Mbp
megabase pairs
References
- 1.Cantor C R, Smith C L, Mathew M K. Annu Rev Biophys Biophys Chem. 1988;17:287–304. doi: 10.1146/annurev.bb.17.060188.001443. [DOI] [PubMed] [Google Scholar]
- 2.Birren B W, Lai E, Clark S M, Hood L, Simon M L. Nucleic Acids Res. 1988;16:7563–7582. doi: 10.1093/nar/16.15.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gardiner K. Anal Chem. 1991;63:658–665. doi: 10.1021/ac00007a003. [DOI] [PubMed] [Google Scholar]
- 4.Bustamante C, Gurrieri S, Smith S B. Trends Biotechnol. 1993;11:23–30. doi: 10.1016/0167-7799(93)90071-G. [DOI] [PubMed] [Google Scholar]
- 5.Gurrieri S, Smith S B, Wells K S, Johnson I D, Bustamante C. Nucleic Acid Res. 1996;24:4759–4767. doi: 10.1093/nar/24.23.4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Olson M V. In: Genetic Engineering. Setlow J K, editor. Vol. 11. New York: Plenum; 1989. pp. 183–237. [Google Scholar]
- 7.Turmel C, Brassard E, Slater G W, Noolandi J. Nucleic Acids Res. 1990;18:569–575. doi: 10.1093/nar/18.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deutsch J M. In: Current Communications in Cell and Molecular Biology. Electrophoresis of Large DNA Molecules: Theory and Applications. Lai E, Birren B W, editors. Vol. 1. Plainview, NY: Cold Spring Harbor Lab. Press; 1990. pp. 81–99. [Google Scholar]
- 9.Viovy J V, Miomandre F, Miquel M C, Caron F, Sor F. Electrophoresis. 1992;13:1–6. doi: 10.1002/elps.1150130102. [DOI] [PubMed] [Google Scholar]
- 10.Burlatsky S, Deutch J. Science. 1993;260:1782–1784. doi: 10.1126/science.260.5115.1782. [DOI] [PubMed] [Google Scholar]
- 11.Viovy J-L, Duke T. Science. 1994;264:112–113. doi: 10.1126/science.264.5155.112. [DOI] [PubMed] [Google Scholar]
- 12.Carlsson C, Larsson A, Jonsson M. Electrophoresis. 1996;17:642–651. doi: 10.1002/elps.1150170404. [DOI] [PubMed] [Google Scholar]
- 13.Gurrieri S, Bustamante C. Biochem J. 1997;326:131–138. doi: 10.1042/bj3260131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith S B, Aldridge P K, Callis J B. Science. 1989;243:203–206. doi: 10.1126/science.2911733. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz D C, Koval M. Nature (London) 1989;338:520–522. doi: 10.1038/338520a0. [DOI] [PubMed] [Google Scholar]
- 16.Gurrieri S, Rizzarelli E, Beach D, Bustamante C. Biochemistry. 1990;29:3396–3401. doi: 10.1021/bi00465a036. [DOI] [PubMed] [Google Scholar]
- 17.Gurrieri S. Ph.D. thesis. University of Oregon; 1994. [Google Scholar]
- 18.Deutsch J M. Science. 1988;240:922–924. doi: 10.1126/science.3363374. [DOI] [PubMed] [Google Scholar]
- 19.Deutsch J M. J Chem Phys. 1989;90:7436–7441. [Google Scholar]
- 20.Larsson A, Akerman B. Macromolecules. 1995;28:4441–4454. [Google Scholar]
- 21.Mathew M K, Smith C, L, Cantor C R. Biochemistry. 1988;27:9210–9216. doi: 10.1021/bi00426a020. [DOI] [PubMed] [Google Scholar]
- 22.Smith S B, Cui Y, Bustamante C. Science. 1996;271:795–798. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 23.Lumpkin O J, Dejardin P, Zimm B H. Biopolymers. 1985;24:1573–1593. doi: 10.1002/bip.360240812. [DOI] [PubMed] [Google Scholar]
- 24.Smith S B, Bendich A J. Biopolymers. 1990;29:1167–1173. doi: 10.1002/bip.360290807. [DOI] [PubMed] [Google Scholar]
- 25.Long D, Viovy J-L, Ajdari A. Biopolymers. 1996;39:755–759. [Google Scholar]
- 26.Crick F H C, Klug A. Nature (London) 1975;255:530–533. doi: 10.1038/255530a0. [DOI] [PubMed] [Google Scholar]
- 27.Sussman J L, Trifonov E N. Proc Nat Acad Sci USA. 1978;75:103–107. doi: 10.1073/pnas.75.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Southern E M, Anand R, Brown W R A, Fletcher D S. Nucleic Acids Res. 1987;15:5925–5943. doi: 10.1093/nar/15.15.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oana H, Masubutchi Y, Matsumoto M, Doi M, Matsuzawa Y, Yoshikawa K. Macromolecules. 1994;27:6061–6067. [Google Scholar]
- 30.Gunderson K, Chu G. Mol Cell Biol. 1991;11:3348–3354. doi: 10.1128/mcb.11.6.3348. [DOI] [PMC free article] [PubMed] [Google Scholar]