

Abstract
Fusion proteins of many viruses, including HIV-1 envelope protein (Env), fold into six-helix bundle structures. Fusion between individual Env-expressing cells and target cells was studied by fluorescence microscopy, and a temperature jump technique, to determine whether folding of Env into a bundle is complete by the time fusion pores have formed. Lowering temperature to 4°C immediately after a pore opened halted pore growth, which quickly resumed when temperature was raised again. HIV gp41-derived peptides that inhibit bundle formation (C34 or N36) caused the cold-arrested pore to quickly and irreversibly close, demonstrating that bundle formation is not complete by the time a pore has formed. In contrast, lowering the temperature to an intermediate value also halted pore growth, but the pore was not closed by the bundle-inhibiting peptides, and it enlarged when temperature was again elevated. This latter result shows that bundle formation is definitely required for the fusion process, but surprisingly, some (if not all) bundle formation occurs after a pore has formed. It is concluded that an essential function of the bundle is to stabilize the pore against collapse and ensure its growth.
INTRODUCTION
The process of membrane fusion is central to a diverse range of physiological events (Hernandez et al., 1996). The energy required to induce membrane merger is provided by the conformational changes of the fusion proteins. The initial fusion pore is a small water-filled passageway that connects two formerly separate aqueous compartments. The process of fusion, however, is not completed until the pore enlarges sufficiently to accommodate the contents that must transfer between aqueous compartments for biological function to proceed. To elucidate the mechanism of membrane fusion, the correspondence between conformational changes of the fusion proteins and the lipid rearrangements of pore formation must be identified.
For intracellular fusion in eukaryotic cells, SNARES are the favored candidate proteins (Jahn and Sudhof, 1999), although their precise roles remain controversial (Coorssen et al., 1998; Mayer, 2001). In contrast, the proteins that mediate fusion between viral envelopes and cell membranes have been unambiguously identified for most known viruses. The fusion proteins of many viral families have been crystallographically shown to form a trimer of hairpins, a structure referred to as a six-helix bundle (6HB; Skehel and Wiley, 2000; Eckert and Kim, 2001). A six-helix bundle consists of three N-terminal heptad repeat segments that form a trimeric coiled coil and three C-terminal helical segments that bind, in antiparallel orientation, to hydrophobic grooves along the central coiled coil. The 6HB is an extremely stable structure, melting only at temperatures that are well above the physiologically pertinent one of 37°C (e.g., Lu et al., 1995). (A helical bundle motif may also be important for eukaryotic fusion: the SNARE complex exhibits a thermally stable helical bundle [Poirier et al., 1998; Sutton et al., 1998].)
The fact that 6HBs are found in many viral fusion proteins clearly demonstrates that this structure must be critical in the viral fusion process. The role of the 6HB in fusion has been most extensively studied for the HIV envelope (Env) glycoprotein. Structural (Ji et al., 1999) and mutagenesis (Dubay et al., 1992; Cao et al., 1993; Weng et al., 2000; Lu et al., 2001) studies of the Env fusion subunit, gp41, have provided strong evidence that the 6HB directly participates in fusion. Also, synthetic peptides with the amino acid sequences of the N- and C-terminal heptad repeats of gp41 have been shown to inhibit Env-mediated membrane fusion and viral infection (Jiang et al., 1993; Wild et al., 1994; Munoz-Barroso et al., 1998) by binding to complementary cognate sites on gp41, preventing 6HB formation (Lu et al., 1995; Weissenhorn et al., 1997; Chan and Kim, 1998; Eckert and Kim, 2001). A C-peptide, T20, is currently in phase III of clinical trials as an antiviral therapy (Kilby et al., 1998). The N- and C-peptides are powerful experimental tools for determining the mechanism by which Env refolding promotes membrane fusion.
In general, the fusion peptides (nonpolar stretches of amino acid residues) reside near the N-terminal heptad repeats and the membrane spanning segments reside near the C-terminal repeats. Once a protein has folded into its 6HB structure, the antiparallel arrangement of N- and C-terminal segments should bring the fusion peptides and membrane spanning segments into close proximity (neither of these domains is part of the crystallized fragments). Because fusion peptides are thought to insert into target membranes (Durrer et al., 1996) and the membrane-spanning segments are located in the viral envelope, the two membranes should come into close proximity as Env refolds from its initial structure into the bundle configuration (Weissenhorn et al., 1997; Chan and Kim, 1998; Eckert and Kim, 2001). Because the bundle is a critical structure, whether formation of the bundle is also responsible for events subsequent to membrane approach is central for understanding the fusion mechanism. If the temporal and functional relation between the formation of the bundle, the formation of the pore, and enlargement of the pore were to be identified, such understanding would be greatly advanced (Melikyan et al., 2000b; Russell et al., 2001).
The present study addresses whether Env has folded into a 6HB by the time the pore has formed. We took advantage of the temperature-dependence of pore growth and used peptides that block bundle formation to answer this central question. We found that by the time the pore forms, folding of Env into bundles has not been completed: either none of the Env participating in pore formation have yet fully folded into bundles or some have completed their folding, whereas others have not. Further, completion of folding into bundles after pore formation stabilizes the initial pore against collapse. We propose that binding of the inhibitory peptides to a prebundle configuration of Env not only prevents bundle formation, but also causes these Envs to revert to an even earlier, more upstream prebundle configuration.
MATERIALS AND METHODS
Reagents
Reagents (including the appropriate reference and original source) obtained from the NIH Research and Reference Reagent Program, Division of AIDS, NIAID, NIH were as follows: HeLaT4+ cells (Maddon et al., 1986) provided by Dr. Richard Axel; 3T3.T4.CXCR4 cells (Deng et al., 1997) provided by Dr. Dan R. Littman; CXCR4 mAb 12G5 (Endres et al., 1996) provided by Dr. James Hoxie; and HIV-1 C34 peptide (Chan et al., 1998) provided by Dr. Peter Kim. The TF228.1.16 cell line stably expressing HIV-1 Env glycoprotein (BH10 strain) was a kind gift from Dr. Z. Jonak (Smith Kline Beecham, Philadelphia, PA). We were also generously provided with the gp41-derived peptides C34 and N36 by Dr. Min Lu (Cornell Medical School, New York). We purchased the peptide T22, a CXCR4 antagonist, from Bachem Bioscience (King of Prussia, PA); Q4120, an mAb against CD4, and bovine serum albumin (BSA) from Sigma Chemical Co. (St. Louis, MO); lauroyl-lysophosphatidylcholine (LPC) from Avanti Polar Lipids (Alabaster, AL); and the fluorescent dyes calcein AM and CMAC (7-amino-4-chloromethylcoumarin) from Molecular Probes (Eugene, OR).
Cell Maintenance, Transfection, and Labeling
TF228.1.16 cells were cultivated in RPMI-1640 medium (GIBCO BRL, Gaithersburg, MD) supplemented with 10% Cosmic Calf serum (Hyclone Laboratories, Logan, UT). HeLaT4+ cells that stably express CD4 were grown in DMEM supplemented with 10% Cosmic Calf serum with 0.5 mg/ml G418, and 3T3.T4.CXCR4 cells that stably express CD4 and CXCR4 were grown in the same medium, but without G418. CXCR4 and/or CD4 were transiently expressed with pCDNA3 plasmids (kindly provided by Dr. Robert Doms, University of Pennsylvania) in HeLa cells (ATCC, Rockville, MD) grown in DMEM supplemented with 10% fetal calf serum. The HeLaMonster reagent (Panvera, Madison, WI) was used for transfection according to the manufacturer's instructions. Cells were used for experiments ∼42 h post transfection. The HIV-1 Env-expressing (effector) cells were loaded with 0.4 μM calcein AM per 106 cells and, when required, the CD4+/CXCR4+ cells (target) were loaded with 20 μM CMAC per 106 cells, as described (Melikyan et al., 2000b).
Flow Cytometry
Relative expression levels of CD4 on cell surfaces were determined by incubating HeLaT4+ cells, 3T3.T4.CXCR4 cells or transfected HeLa cells with saturating concentrations of the antibody Q4120. Similarly, relative expression levels of CXCR4 were determined by using the antibody 12G5. In both cases, these procedures were followed by binding goat anti-mouse FITC-labeled IgG (Southern Biotech, Birmingham, AL), as previously described (Melikyan et al., 1999). Cells were analyzed on an ORTHO Cytoron Absolute flow cytometer (Ortho Diagnostic Systems, Raritan, NJ).
Fusion Experiments
The fraction of TF228 and HeLaT4+ (or other target) cells that fuse at 37°C (usually 50–60%) was quantified as the number of cell pairs labeled by both calcein and CMAC, normalized by the total number of effector-target (E/T) cell pairs. In the present study, however, we relied primarily on monitoring, in real-time, calcein transfer between the individual effector and target cell pairs using fluorescence videomicroscopy (Melikyan et al., 2000a). To synchronize the fusion event better than occurs by maintaining 37°C, E/T cells were preincubated at 23°C for 3 h. This procedure has been shown to kinetically advance the HIV Env-mediated fusion process, so that fusion is rapidly induced when temperature is raised to 37°C (Melikyan et al., 2000a). All experiments were recorded on S-VHS format videotape, digitizing images with a frame grabber (Matrox Meteor-2, 8-bit resolution), and analyzing dye spread with custom-written software (Markosyan et al., 2000). Dye transfer was quantified by drawing regions of interest encompassing the target and effector cells and measuring the average fluorescence intensity of that region over the course of time (Markosyan et al., 2000).
To initiate and then quickly arrest a small fusion pore, we used a temperature-jump protocol described in detail elsewhere (Melikyan et al., 2000a, 2000b). Briefly, the experimental chamber, with bottom made of heat-absorbing glass, was mounted in a fluorescence microscope (Axiovert 100A; Carl Zeiss, Thornwood, NY). The solution bathing the cells was maintained at 4°C by mounting the chamber within a Peltier-based temperature-controlled holder (20/20 Technology, Wilmington, NC). Selected E/T cells were irradiated with an infrared laser diode to locally increase temperature to 37°C. The laser was turned off immediately after a detectable amount of calcein moved into the target cell, allowing the temperature to drop back to that of the surrounding bath, 4°C, within ∼10 s through passive heat exchange.
Monitoring the Dynamics of Fusion Pores by Fluorescent Dye Redistribution
The rate of aqueous dye transfer between cells increases with the diameter of the pore lumen. The rate of accumulation of calcein into the target cell (dCt/dt) and the rate of its depletion from the effector cell (−dCe/dt) at any time, t, are proportional to the permeability of the pore at that time, P(t), according to
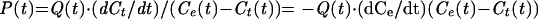 |
1 |
where (Ce(t) − Ct(t)) is the difference in dye concentration across the pore at the time t. The function Q(t) corrects for the diffusion coefficient of the dye as a function of temperature; we used Q(t) because the temperature changes were not instantaneous upon turning the laser on and off. We assumed the diffusion coefficient increases ∼1.3 for every increase of 10°C (i.e., Q10 ∼ 1.3), yielding Q(t) = 1 + 0.03(T0 − T∞)[1− exp(−bt)]. T0 − T∞ is the steady-state difference in temperature before and after the jump and the time constant, b, for temperature changes was obtained by fitting, with a single exponential, the measured time course of the temperature changes (unpublished data). For the fluorescence intensity to be a reliable measure of dye concentration, it is necessary that the fluorescence of the dye not be quenched and that the video camera (Stanford Photonics model from Solamere Technology, Salt Lake City, UT) output vary linearly with incident intensity. We validated the two requirements. For the first, we treated effector cells with saponin to release aqueous dye. If the dye was quenched, there would have been a transient increase in fluorescence as the dye diluted. But there was not. The linearity of the camera output with incident intensity was confirmed by placing neutral density filters in the excitation pathway of the microscope. The average fluorescence intensities of effector (Fe(t)) and target (Ft(t)) cells are therefore proportional to Ce(t) and Ct(t), respectively.
We often improved the sensitivity of dye transfer measurements by increasing the gain of the video camera to the point that the fluorescence of the effector cells saturated the camera's output (maximum 256 brightness units). As a consequence, after calcein had fully redistributed between cells, the fluorescence intensities were within the range of 150–200 brightness units. The pore permeability was calculated from the fluorescence increments of a target cell according to
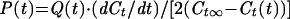 |
2 |
where Ct∞ is the mean fluorescence intensity of the target cell after complete dye redistribution. Eq. (2) assumes that the cytoplasmic dye does not leak into the extracellular solution and that it completely equilibrates between the cells at infinite time, so that Ct∞ = ½Ce(0). Equilibration was often not perfect: a small immobile fraction of calcein (<20%) was often retained in the effector cell, as established by lysing individual calcein-loaded cells with saponin (unpublished data). The occurrence of an immobile fraction of calcein would introduce a systematic error when using Eq. (2) to estimate pore permeability when it was large. Because we are primarily concerned with the evolution of a pore while it is small, such errors were inconsequential. We verified that the pore permeability calculated by Eq. (2) was in fact close to that obtained by Eq. (1) when gain settings of the camera were sufficiently low that they did not saturate the camera output (unpublished data).
RESULTS
Dynamics of Fusion Pore Size Can Be Monitored from the Redistribution of Cytoplasmic Aqueous Dyes
HIV Env-expressing effector (designated “E”) cells loaded with calcein were bound to target (“T”) HeLaT4+ cells (Figure 1A). The E/T cells were preincubated at 23°C for 3 h. Cells so treated proceed toward fusion but will not fuse at this temperature, stopping at what is referred to as a temperature-arrested stage (TAS) (Melikyan et al., 2000b). When temperature is raised to 37°C at this stage, fusion will occur within a few minutes, a much quicker rate than for cells that are bound and allowed to fuse at a constant 37°C. TAS is a stable state: it does not revert to a less advanced state of fusion when temperature is lowered to 4°C. After establishing TAS, the E/T cell pairs were placed in an experimental chamber maintained at 4°C. Fusion was triggered by quickly and locally raising temperature to 37°C by illuminating cells with an IR laser diode.
Figure 1.

Monitoring fusion pore dynamics through fluorescent dye redistribution between effector and target cells. Top panel: (A) Effector (TF228.1.16) cells labeled with calcein and unlabeled target (HeLaT4+) cells were coincubated for 3 h at 23°C. These cells were then placed in an experimental chamber maintained at 4°C. The image of A is a superposition of fluorescence and phase contrast after placement. Fusion was then triggered by quickly raising the temperature to 37°C and monitored by the transfer of calcein from the effector cell to the target cell (marked by arrowheads). (B–D) The onset of dye movement is shown in C (arrow) and after full redistribution in D (the dotted circles mark the region of interest encompassing the effector and the target cell). Bottom panel: Changes in mean fluorescence intensity of the target (lower thick solid line) and the effector (upper thick solid line) cell with time after raising temperature to 37°C. The temperature profile is shown by dotted line. The thin solid line is the sum of the fluorescence intensities of the effector and target cells. Once the fusion pore formed (marked by vertical dotted arrows), it took ∼1 min for the calcein to fully redistribute. The pore permeability profile in arbitrary units (open circles) was calculated from fluorescence intensity changes using Eq. (1) (see MATERIALS AND METHODS).
On fusion, calcein was observed to move from the effector to the target cell (Figure 1, C and D). The average fluorescence intensities within the target and effector cells as a function of time were quantified (Figure 1, bottom panel, thick solid lines) and this allowed us to estimate, by a straightforward method, pore permeability throughout the time course of pore evolution. The dye continued to redistribute until the same intensities were reached in the two cells (image D and equality of fluorescence in bottom panel), showing that a fusion pore was maintained. Leakage of calcein into the external medium was usually small, as shown by the constancy of the sum of fluorescence intensities of the effector and target cells (thin line). The total pore permeability continuously increased over the ∼1-min time period it took for the dye to fully redistribute (Figure 1, bottom panel, ○). This pattern of permeability increase is in agreement with prior electrophysiological measurements and demonstrates quick enlargement of fusion pores induced by HIV Env (Melikyan et al., 2000b). Although our method of determining the evolution of fusion pore size based on changes in fluorescence intensities over time is not as direct as electrical measures of pore size, it can be used for the wide-ranging manipulations of this study whereas, as a practical matter, electrical measures cannot.
Pore Growth Can Be Arrested by Lowering Temperature and Restored by Raising Temperature
Immediately after the onset of calcein redistribution at 37°C, we shut off the IR laser and the temperature of the E/T cells dropped back to the temperature of the surrounding bath (4°C) in <10 s. The rate of calcein accumulation in the target cell (Figure 2A, fluorescence intensity shown by the noisy solid curve) decreased upon dropping temperature to 4°C. The pore permeability (⋄), calculated from the fluorescence trace (using Eq. (2)), ceased to increase shortly after lowering temperature to 4°C and thereafter slowly decreased over the course of several minutes. The low temperature thus not only prevents further growth of the pore, it causes pore shrinkage. On raising the temperature back to 37°C, the rate of calcein transfer rapidly increased because the pore immediately enlarged (nearly vertical line). Thus by maintaining 4°C, small pores can be sustained for several minutes, allowing them to be studied. But if temperature was maintained at 4°C for more than ∼15 min, the pores irreversibly closed, as determined by the absence of additional dye spread upon raising temperature to 37°C (unpublished data).
Figure 2.

Arresting the fusion pore by low temperature. Immediately after a fusion pore formed between TF228.1.16 and HeLaT4+ cells, pore growth was arrested by quickly reducing the temperature from 37 to 4°C (downward arrows in A and B). The fluorescence intensities of the target cell over time are shown by solid lines. (A and B) Upward arrows denote raising temperature back to 37°C (A) or to 23°C (B). The pore permeability (⋄) was calculated from the fluorescence traces using Eq. (2). After elevating the temperature from 4°C, the pore rapidly enlarged (nearly vertical lines in A and B). (C) The waiting time distribution between the fast increase of temperature at TAS and formation of a fusion pore, plotted as the fraction of cells that fused (●) was significantly longer than the distribution for time between exposing the cold-arrested pores to 37°C and their rapid enlargement, plotted as the fraction of pores that enlarged (○).
It was important to establish that the same pore was followed throughout an experiment as temperature was varied. To rule out the possibility that a new pore formed upon raising temperature, we performed two types of experiments. In the first, we raised the temperature to 23°C instead of 37°C (Figure 2B). This protocol resulted in an immediate augmentation of dye spread (Figure 2B, noisy solid data curve): quantitatively the pore quickly enlarged (nearly vertical increase in pore permeability, ⋄). Because fusion pores do not form at 23°C for the E/T cells used here (Melikyan et al., 2000b), the enhanced calcein movement at 23°C must be due to widening of the pore whose size was arrested by 4°C (the “cold-arrested pore”). In fact, the rate of fluorescence increase was so enhanced at 23°C, compared with the initial rate at 37°C, that the pore must have progressed in some way during its time at 4°C, even though pore permeability did not increase at this low temperature. In the second set of experiments, we measured the times between raising the temperature of E/T pairs at TAS to 37°C and the observation of dye spread. The kinetics of pore formation after raising temperature to 37°C at TAS (Figure 2C, ●) was significantly slower than the kinetics of augmentation of dye spread upon raising temperature of the cold-arrested pore (○). We do not have a measure for the actual number of pores that form between cells, but we can describe the situation as if it were one pore connecting the cells. Although we cannot determine how many pores existed before temperature was lowered, we can unambiguously determine that they have all closed once there is a cessation of dye transfer. Although it would obviously be informative to know how many pores were present at every point in an experiment, we can determine whether a manipulation will reliably cause the elimination of all pores.
Peptides that Inhibit Formation of a Six-helix Bundle Can Cause Quick and Irreversible Closure of a Cold-arrested Pore
During the normal course of fusion, its various states are so transitory that they cannot be characterized. Characterization becomes possible, however, by arresting states. Peptides that inhibit 6HB formation can be added to the cold-arrested fusion pore to determine whether bundle formation has been completed at this point. We anticipated that if bundles had not formed, subsequent bundle formation would be central to pore enlargement and therefore that the peptides would inhibit pore growth when temperature is elevated.
After creating the cold-arrested pores, we added the peptides C34 or N36 and allowed them to bind to their cognate sites on gp41 (C34 to the N-terminal coiled coil and N36 to the C-terminal heptad repeat) for ∼5 min before raising the temperature to 37°C. Strikingly, both C34 (Figure 3A) and N36 (Figure 3B) quickly stopped calcein transfer through the pores, showing that these pores either closed entirely or shrank to sizes too small to accommodate calcein. It is much more likely that the pores closed, and did so irreversibly, because raising the temperature to 37°C after incubation with the peptides did not induce any additional dye transfer (Figure 3, A and B). Even when C34 was allowed to bind for only 3 min and unbound peptide was removed by washing, raising temperature to 37°C failed to promote pore reopening in 3 of 4 experiments (unpublished data). Thus the slow spontaneous closure of the cold-arrested pores (Figure 2A) was drastically accelerated by the presence of C34 and N36. These experiments show that formation of 6HBs is not completed by the time the pore forms. Clearly, some (perhaps even all) of the gp41 that participate in pore evolution have not yet folded into a bundle by the time a pore forms and these copies of gp41 are somehow critical for maintaining a pore in an open state. We refer to pores that close in the presence of the inhibitory peptides as “labile.” The pores defined as labile by this criterion are also the ones that shrink and close within 15 min at 4°C.
Figure 3.

Cold-arrested fusion pores were closed by peptides that block 6HB formation but not by other inhibitors of fusion. The time at which the temperature was dropped to 4°C to block pore growth is marked by downward arrows in each panel. The fluorescence intensities of target cells (HeLaT4+, solid lines) were used to calculate pore permeability from Eq. (2) (⋄). Once pore growth was blocked at 4°C, inhibitory agents were added (indicated by ▵), allowed to bind for ∼5 min, and the temperature was then raised back to 37°C (upward arrows). The inhibitors were (A) 47 nM C34 peptide, (B) 4 μM N36 peptide, (C) 0.4 μg/ml T22 peptide, and (D) 0.2 mg/ml LPC.
To determine whether interactions between chemokine receptors and Env play a role in pore evolution, we added a high concentration of the peptide T22 (which is a CXCR4 antagonist that greatly reduces pore formation if added at TAS; Melikyan et al., 2000b) to the cold-arrested pore; pore behavior was not affected (Figure 3C). Thus, whereas formation of a pore from TAS still requires gp120-chemokine receptor interactions, once a pore has formed these interactions are no longer relevant for either maintenance or growth of pores. The absence of an effect by T22 implies that pore stabilization and enlargement do not require additional Env proteins to be activated by binding to coreceptors. This absence of an effect when adding a peptide that does not target gp41 also serves as a control; it shows that the inhibitory actions of C34 and N36 are specific: they cause pore closure because they prevented formation of a 6HB.
Not only was pore dynamics independent of chemokine receptor activity, but it was independent of lipid composition as well: LPC, as a lipid component of membrane bilayers, inhibits Env-induced cell–cell fusion when added before or at TAS (Melikyan et al., 2000b). However, when LPC was added to cells connected by cold-arrested pores, neither pore stability at 4°C nor subsequent pore growth at 37°C was noticeably affected (Figure 3D). This result suggest that fusion proteins, rather than lipids, maintain the pore in an open state.
In general, individual fusion pores grow in a similar manner, but that growth is quantitatively variable. To characterize the response of a pore to a treatment, we calculated the average pore permeability profile (Figure 4B) from multiple experiments that track calcein transfer over time (Figure 4A). In the control experiments, calcein continued to accumulate within target cells (Figure 4A, ▪) for at least 4–5 min at 4°C, showing that these fusion pores remained open. But dye transfer was fully blocked soon after adding either C34 (○) or N36 (▵). The averaged pore permeability calculated from the individual fluorescence profiles clearly revealed a relatively stable (although slowly declining) pore size in the absence of inhibitory peptides (Figure 4B, ▪). But in the presence of C34 (circles) or N36 (triangles), the average pore closed rapidly and irreversibly, within about 1 min.
Figure 4.

Average rate of pore closure after adding inhibitors of 6HB formation. The fluorescence (A) and pore permeability (B) after the addition (indicated by downward arrow) at 4°C of the inhibitory peptides C34 (47 nM, ○, n = 5) or N36 (4 μM, ▵, n = 7) or, as a control, BSA (▪, n = 8), as averaged from multiple experiments. In the control experiments, we changed the external solution by the same procedure used to add peptides, to ensure that the peptides themselves were causing the inhibitory effect, rather than a simple shear stress on the E/T cell pairs. (A) Fluorescence traces from individual experiments were aligned at the time of peptide addition, and the average fluorescence from multiple experiments over the next 200 s was calculated. Bars, SEs of mean. (B) Pore permeabilities, calculated from the averaged fluorescence intensities (traces in A). The addition of C34 or N36, but not BSA, caused pore closure.
At High Surface Density of Coreceptors, Pores Are Resistant to Blockers of 6HB Formation
Assuming 6HB formation is irreversible (Chen et al., 1995; Kliger and Shai, 2000), it is clear that at least some gp41 trimers that participate in maintaining the cold-arrested pore are not yet in 6HBs at this stage because the addition, at this point, of peptides that prevent bundle formation can cause pore closure. The number of gp41 trimers that can undergo the conformational changes necessary for bundle formation should depend on the density of receptors and coreceptors in the target membrane. The standard target cells HeLaT4+ that were used in the above experiments express a high level of the receptor CD4, but a low level of the coreceptor CXCR4 (Maddon et al., 1986). We therefore also used target cells that express high levels of both CD4 and CXCR4 to allow more extensive activation of Env. For these experiments we either transiently expressed CD4 and CXCR4 in HeLa cells (the parental cells used to produce HeLaT4+) or we used a stable 3T3 cell line, 3T3.T4.CXCR4 (Deng et al., 1997). By expressing CD4 and CXCR4 in HeLa cells (which we denote HeLa/CD4/X4) we were able to keep aspects of the target membrane (such as membrane mechanics) constant yet vary receptor and coreceptor density. The relative expression levels of CD4 and coreceptor were assessed by FACS analysis for both HeLa/CD4/X4 and HeLaT4+ cells (Table 1). As expected, both the mock-transfected HeLa and HeLaT4+ cells expressed comparable low levels of CXCR4. The HeLa/CD4/X4 cells, in contrast, expressed much higher levels of coreceptor and the levels of CD4 were made comparable to those of HeLaT4+ cells.
Table 1.
Relative levels of CD4 and CXCR4 expression on cell surfaces, as determined by flow cytometry
| Protein | Cells
|
||
|---|---|---|---|
| HeLaT4+ constitutive expressiona (% transfected HeLa) | HeLa/CD4/CXCR4 transient expressiona (% transfected HeLa) | 3T3.T4.CXCR4 constitutive expressiona (% transfected HeLa) | |
| CD4 | 130 ± 21 | 100 ± 22 | ND |
| CXCR4 | 0b | 100 ± 26 | 113 ± 8 |
| Labile pore | Robust pore | Robust pore | |
After subtraction of the background signal that was measured in the parental cell line (columns I and III) or in the mock-transfected cells (column II).
CXCR4 expression did not exceed that from the parental HeLa cell lines.
For the HeLa/CD4/X4 cells, the enlargement of fusion pores was reversibly arrested by 4°C, using the same protocols as those followed for the HeLaT4+ cells. Although the pores with HeLaT4+ cells rapidly closed upon addition of C34, here the cold-arrested pores did not close even in the presence of high concentrations of C34 (up to 900 nM), and they readily enlarged when the temperature was raised from 4 to 37°C (Figure 5, A and B, ○). In contrast, for HeLa cells transfected only with CD4 (HeLa/CD4 cells), pores closed in the presence of C34 at 4°C and remained close when the temperature was elevated to 37°C (Figure 5, A and B, ●). This behavior, the same as that observed for HeLaT4+ cells as target, shows that for low density of CXCR4 (and high density of CD4), the pores can be closed by C34, but those that form for a high level of CXCR4 in the target cell cannot be so closed. We refer to pores that remain open in the presence of the 6HB peptides as “robust.” The calculated average pore permeability at 4°C quantitatively demonstrates that cells expressing a high level of CXCR4 exhibit relatively stable pore sizes at 4°C, independent of whether C34 peptide was present (Figure 5C, ○) or not present (▵). But for target cells expressing a low level of coreceptor, all pores quickly closed at 4°C in the presence of C34 peptide. This was demonstrated by the fact that the average total permeability decreased to zero (●).
Figure 5.

Cold-arrested pores for target cells expressing a high level of CXCR4 were robust, whereas those expressing a low level of CXCR4 were labile. HeLa cells were transfected with either CD4- and CXCR4-bearing plasmids (HeLa/CD4/X4, high CXCR4) or a CD4-bearing plasmid alone (HeLa/CD4, low CXCR4). Immediately after arresting pore growth by lowering temperature to 4°C, 150 nM of C34 peptide was added to the extracellular solution. (A) Fluorescence intensity changes of transfected HeLa cells. (B) Pore permeability profiles calculated from the traces in A. (C) Average permeability as a function of time at 4°C for HeLa/CD4/X4 cells with (○, n = 6) and without (▵, n = 5) C34 peptide, and for HeLa/CD4 cells in the presence of C34 peptide (●, n = 5). Average pore permeability was calculated as described in the legend to Figure 4.
We tested the generality of our finding that coreceptor density controlled pore properties by carrying out experiments similar to those described for a very different target cell, 3T3.T4.CXCR4. As expected for target cells that express a high density of receptors and coreceptors (Table 1), the cold-arrested pores remained open in the presence of C34 and enlarged when the temperature was raised to 37°C (Figure 6A, ○). The calculated average permeability of the arrested pore shows that the addition of C34 did not cause pore closure (Figure 6B, open circles for C34, filled circles for no peptide). Thus, regardless of the specific cell line, a high density of receptor and coreceptor in the target cells ensures that the pore that forms is resistant to peptides that inhibit formation of the 6HB.
Figure 6.

The cold-arrested pore is robust for 3T3. T4. CXCR4 cells as targets. (A) C34 (150 nM, ○) or a blank solution (●) was added at 4°C to cold-arrested fusion pores formed between TF228.1.16 and 3T3.T4.CXCR4 cells. (B) Average pore permeability profiles at 4°C in the presence (○) and in absence (●) of C34 peptide.
Bundles Do Not Form before Membrane Merger at High as well as Low Chemokine Receptor Densities
We previously showed that for HeLaT4+ cells (which have low CXCR4 densities), bundles do not form before pore opening (Melikyan et al., 2000b). Because the inhibitory peptides had no effect on the robust pores that formed for target cells expressing a high density of coreceptor, it was possible that in this case bundles had formed before membrane merger. We tested this for 3T3.T4.CXCR4 cells using the same strategy we used previously: We created TAS and added LPC to the E/T cell pairs. The LPC, as a lipid component within the membranes (Chernomordik et al., 1995), blocked fusion upon subsequent exposure to 37°C. After the temperature was lowered to 23°C, the LPC in solution and within the membrane was removed by washing, and fusion still did not occur (Figure 7, second bar). The intermediate of fusion created by this procedure is referred to as a lipid-arrested stage (LAS; Melikyan et al., 2000b). If at this point, temperature is again raised to 37°C, the full extent of fusion was observed (last bar), demonstrating that the effect of LPC was reversible. If before raising temperature at LAS, C34 (third bar) or N36 (fourth bar) was added, fusion was suppressed, showing that bundles had not formed at the point of LAS and that, as in the case of low chemokine receptor density, bundle formation requires membrane merger. We have thus now shown that for high, as well as low, chemokine receptor density, bundle formation requires membrane merger.
Figure 7.

Peptides that prevent the formation of six-helix bundles still inhibit fusion when added at an LPC-arrested stage for high CXCR4-expressing cells. TF228.1.16 cells were coincubated with 3T3.T4.CXCR4 cells for 3 h at 23°C followed by incubation at 37°C. The full extent of fusion is indicated (first bar). A lipid-arrested stage (LAS) of fusion was created by adding 0.3 mg/ml LPC to cells for 3 min at 23°C, raising temperature to 37°C for 15 min and then removing LPC by repeated washings with a BSA-containing solution at 23°C. The extent of fusion was negligible (second bar). The addition of 150 nM C34 (third bar) or 4 μM N36 (fourth bar) at LAS abolished any additional fusion when temperature was raised to 37°C, showing that 6HBs have not yet formed. When peptides were not added after creating LAS, raising temperature to 37°C led to full extent of fusion (fifth bar). Bars, SEs of mean (n = 4).
Labile Pores Are Converted to Robust Pores when Arrested at an Intermediate Temperature
For pores that form at 37°C with a low density of CXCR4 in the target membrane, not all gp41 participating in pore maintenance has folded into 6HBs, as judged by the ability of C34 and N36 to close pores at 4°C. We have shown that for high chemokine receptor densities, the fusion pore reaches a stage in which it is unaffected by peptides that prevent bundle formation. But we had not yet shown that this stage is reached for low densities of chemokine receptors. We therefore sought, for low chemokine receptor density, a temperature that prevented pore enlargement yet allowed the labile pore to convert to a robust pore. We used HeLaT4+ cells and lowered temperature from 37 to 15°C (instead of 4°C), once a pore formed. These pores did not close after addition of C34. This held true in records of individual experiments (Figure 8A) and in averaging from many experiments (Figure 8B). The labile pore therefore evolves into a robust pore if temperature is not too low. As expected for a robust pore, the pores arrested at 15°C enlarged when temperature was increased to 37°C in the presence of C34 (Figure 8A). In 10 of 14 experiments in which a high concentration (150 nM) of C34 was added at 15°C, the pores fully enlarged. Clearly, for low densities of chemokine receptors, at the time a pore initially forms, not all gp41 trimers engaged in the process have folded into 6HBs; at least some trimers must fold into bundles subsequent to pore formation. High densities of chemokine receptors in the target cell should better facilitate synchronous formation of 6HBs and, in fact, caused a pore to quickly become insensitive to C34. These results suggest that the window of time during which the pore evolves from labile to robust is highly dependent upon temperature and chemokine receptor density. The sensitivity of pore evolution to temperature and to peptides immediately suggests that the fate of the initial pore is basically under the control of fusion proteins rather than membrane lipids. Our results strongly indicate that the conversion of a labile pore to a robust pore is still not spontaneous. Rather, fusion proteins must sustain the pore, and free energy released by protein conformational changes subsequent to pore formation is utilized to stabilize the pore in an open state, allowing it to further enlarge.
Figure 8.

Pores arrested at 15°C are robust for target cells expressing low levels of CXCR4. (A) Fluorescence trace of calcein accumulation within the target cells (solid line). Temperature was lowered from 37 to 15°C (long downward arrow) and 150 nM C34 peptide was then added, as indicated. The pore permeability (⋄) was calculated from the curve of fluorescence intensity using Eq. (2). The pore fully enlarged (nearly a vertical line) immediately after temperature was raised back to 37°C (upward arrow). (B) The average pore permeability (●) calculated from the average fluorescence profile (○) as a function of time at 4°C in the presence of C34. HeLaT4+ cells were used as targets. Bars, SEs of mean (n = 5).
DISCUSSION
It has long been realized that the 6HB must be critical to fusion because ectodomains of many viral fusion proteins, when in solution, fold into bundles, and because peptides that block bundle formation have been shown to prevent fusion (Skehel and Wiley, 2000; Eckert and Kim, 2001). Experiments in which peptides have been used to prevent fusion have only shown that bundles have not yet formed at the point of peptide addition (Munoz-Barroso et al., 1998; Melikyan et al., 2000b; Gallo et al., 2001; Russell et al., 2001; Golding et al., 2002). They have not explicitly provided an experimental demonstration that bundles do form during the fusion process. We have now provided that demonstration by showing the conversion of a fusion pore from one that can be closed by peptides (a labile pore) to one that cannot be closed (a robust pore). Furthermore, in the present study, we have made the surprising discovery that bundle formation plays a role subsequent to pore formation. Both the time of formation (post pore opening) and the role of the bundle (to maintain the pore in an open state) were unexpected findings. It is so widely believed that bundle formation is a requisite for pore formation that it is almost assumed that this has been experimentally verified. But, in fact, it has not. Based on our own acceptance that bundles are required for fusion pore opening, when we demonstrated that 6HBs do not form before membrane merger, we had concluded that the pore and the bundle form simultaneously (Melikyan et al., 2000b). It remains possible that some bundles participate in pore formation, whereas others act subsequently, but we now have hard data that bundle formation stabilizes the open pore. If the common expectation that bundles are necessary for pore formation is not correct, then the configurations of Env that induce the fusion pore remain to be identified. It would also mean that peptides, such as C34, that block formation of 6HBs, inhibit fusion by affecting conformational changes upstream of the bundle.
It is known that the early pore of intracellular fusion is a labile structure that can still close, and that its growth is controlled by protein (Jahn and Sudhof, 1999). Because there are structural similarities between the SNARE complex and viral fusion proteins (e.g., they both contain helical bundles), an understanding of six-helix bundle folding and labile-to-robust pore conversion could provide a general conceptual framework for aspects of intracellular fusion.
Env Folds into Six-helix Bundles More Readily after Pore Formation
When fusion peptides and membrane-spanning segments of Env are inserted in opposite membranes, the ability to form a bundle will be much more constrained than bundle formation of isolated ectodomains in solution. Bundle formation by membrane-anchored gp41 is made even more difficult by the threefold symmetry of Env: The three C-terminal heptad repeats must somehow separate from each other and move so that each fits into one of the three grooves of the N-terminal trimeric coiled coil in an antiparallel orientation. The relatively small number of amino acid residues (∼20) between the repeats and the membrane-spanning domains should limit the possible ways the repeat segments can move. If the three C-terminal heptad repeats bind into the grooves sequentially, the movement of each repeat into a groove would affect the binding of the subsequent ones. One would expect that because fusion peptides and membrane-spanning domains must come into proximity for bundles to form, Env should be able to fold into a bundle only at the site of a pore. The membrane continuity created by a pore would facilitate the movement of membrane-spanning domains that is needed for them to fold in a threefold symmetric manner around the central coiled-coil.
It is thought that multiple copies of Env act in concert to form the initial pore. One could imagine that additional Envs are recruited into the region where the pore has formed as a means of ensuring pore stabilization. There is, however, no experimental evidence to support this and some evidence to suggest that no recruitment takes place: the permeabilities (and hence diameters) of arrested labile pores slowly decreased with time. If additional Envs had been recruited into the labile pore, one would expect the pore to enlarge or at least to maintain itself. But some changes must have occurred during arrest—possibly altered interactions between Env trimers, formation or merger of cholesterol/sphingolipid rafts, or other phase separation at 4°C, since the pore enlarged much more rapidly after raising temperature (to 23 or 37°C) than when temperature was maintained at 37°C. A mechanistic explanation that we presently favor for the more rapid enlargement after arrest is that lipid leaves the pore during the arrest. This would account for smaller pore lumens and lead to greater density of gp41 at the site of the pore, without recruiting additional protein. A higher density of gp41 would facilitate any cooperative actions between copies of gp41 that are part of pore enlargement.
We have previously shown that if membrane merger is prevented by incorporating LPC into membranes, addition of the peptides C34 or N36 inhibits fusion when the LPC is removed. This indicates that formation of a 6HB requires membrane merger (Melikyan et al., 2000b). If it were assumed that formation of a pore requires Env to fold into bundles, then it would follow that pore formation and bundle formation must be occurring simultaneously. But in the present study we have found that inhibitory peptides can cause the pore to close. The fact that these peptides have any effect at all at this stage signifies that after a pore has opened, at least some of the relevant Envs remain in a prebundle state. It also means that some of these prebundle configurations must, in some way, be helping to maintain the pore in an open state, and because the pore closes quickly after adding peptide, peptide binding must be altering the prebundle configurations.
Binding of Inhibitory Peptides Reduces Late Prebundle Conformations of Env
There are undoubtedly many conformations before the six-helix bundle in which the heptad repeats of Env are exposed. We separate states upstream of the bundle into two classes—“early” and “late” prebundle configurations (Figure 9). We define an early prebundle configuration as activated Env in which none of the C-segment heptad repeats are bound into a groove of the coiled coil, and a late prebundle configuration as a structure in which one or two of the three C-segment heptad repeats have bound. Once the bundle has formed, it does not, as a practical matter, convert back to a prebundle configuration (Chen et al., 1995; Kliger and Shai, 2000; Markosyan et al., 2002). Because early and late prebundle states are not expected to be as stable as the final six-helix bundle configuration (in which all three C-segment heptad repeats are bound within the grooves), we assume that transitions between early and late prebundles are reversible. That is, the bound C-segments can dissociate from the coiled coil before completion of the threefold symmetric 6HB. The inhibitory peptides can block bundle formation and inhibit fusion by binding to the heptad repeats of either early or late bundles.
Figure 9.

A proposed mechanism for the conformational changes of gp41 in pore formation and stabilization. Left panels: Conversion of Env in early prebundles (top) at a fusion site into late prebundles (middle) creates a labile pore, and subsequent folding into the final six-helix bundle configuration establishes the robust pore (bottom). The gp120 is depicted as large circles; N-terminal helices (purple rectangles) of gp41 are adjacent to the fusion peptides (arrows) and C-terminal helices (red rectangles) are adjacent to the membrane-spanning domain (this domain and cytoplasmic regions are solid red lines). Early prebundles are shown as an extended conformation with N- and C-terminal helices well separated from each other; late prebundles are depicted with one C-terminal helix bound into a groove of the triple-stranded coiled-coil. Arresting a labile pore by 4°C leads to reduction in pore size and eventual closure (transition from middle to top). The addition of 6HB blockers (C34, red rectangle) to the labile pore results in rapid pore closure (dashed arrow). Late prebundles stabilize the pore, but only when the six-helix bundle is formed is the pore permanently stabilized against closure. Top right: The proposed energy profile of gp41 during the fusion reaction at 37°C (solid line) and at 4°C (dashed line). In the reaction scheme, N stands for the native conformation, EPB and LPB are early and late prebundle conformations, respectively. Because the formation of the 6HB should be constrained by limitations in protein reorientation, its activation barrier would be controlled by entropy whose energetic consequences are more sensitive to temperature than is enthalpy. The transition from late prebundles to bundles is therefore expected to be more temperature-sensitive than the other transitions.
How could the inhibitory peptides alter the prebundle configurations to cause pore closure? Binding of C34 into a groove of the coiled coil would prevent binding of the C-terminal segments of gp41 into that groove; binding of N36 (which naturally oligomerizes into its own coiled coils) to gp41 C-terminal segments would similarly prevent these segments from fitting into the grooves of the gp41 coiled coil. Binding of peptides to early prebundles would deter progression to late prebundles. In contrast, binding of peptides to late prebundles would not hinder the dissociation of any of the previously bound C-segments of gp41. This means that the transition from early to late prebundles would be reduced, but transition from late to early prebundles would be unaffected. Peptide binding would thereby cause a buildup of early prebundle configurations at the expense of the late prebundles. Because the inhibitory peptides can cause pore closure, we propose, as a central conjecture, that Env in a late prebundle configuration stabilizes the pore against closure, but an early prebundle does not provide this support. Because a late prebundle can reverse into an early prebundle, the pore is not permanently open—that is, robust—until folding into a 6HB is complete. If lowering temperature to 4°C reduced the folding of Env into a bundle to a greater extent than it reduced transitions between early and late prebundles, more late prebundles would convert back to early prebundles. According to our central conjecture that it is the late prebundle (and bundle) that stabilizes the pore, we would expect the pore to eventually close at 4°C.
The Role of Bundle Formation
The simplest explanation of all data would then be that transitions into late prebundle states are responsible for pore formation, and the actual bundles do not occur until after a pore forms. In this scenario, when Env does fold into a bundle, it converts a labile pore into a robust pore. In an alternative model, some trimers of Env fold into bundles simultaneously with the formation of the initial pore. The resulting membrane continuity allows additional trimers to fold into bundles, which stabilizes the pore. In this latter model, not all Env is in the same configuration when the pore initially forms. In both models, once the pore is robust, all relevant copies of Env have folded into bundles. In contrast to our conclusion that the bundle does not form before the pore, it has been argued that 6HBs form before pore formation and are responsible for fusion on the evidence that antibodies raised against 6HBs can inhibit fusion (Golding et al., 2002). But these antibodies recognize Env that has been activated by soluble CD4 alone (de Rosny et al., 2001). Therefore the antigenic sites recognized by the antibodies may be exposed by prebundle configurations, and if so, the inhibition of fusion by the antibodies would be expected according to either of our models described above.
The Initial Pore Is Labile
In actual infection, the cells that are targets for HIV-1 have a low density of chemokine receptors compared with transfected cells (Lee et al., 1999; Tokunaga et al., 2001), and thus experiments using cells with low densities of coreceptor as target might best model the physiological situation. At low densities, the addition of C34 or N36 induced rapid closure of the pore arrested at 4°C. But at 15°C, the pore quickly (<30 s) evolved into a robust one and the addition of the peptides was of no consequence. We conclude that at 15°C, Env folds into six-helix bundles at the site of a pore before peptides can be bound to Env. Before pore formation, however, bundles do not form at 15°C, as evidenced by the ability of the peptides to inhibit fusion when added at TAS (Melikyan et al., 2000b). We have thus been able to experimentally verify the expectation that bundles form more readily after membrane merger than before. The higher the density of chemokine receptors, the greater the number of Env trimers that become activated. At high chemokine receptor densities, pores were robust by the time (5–10 s) temperature could be lowered from 37 to 4°C. The greater percentage of Env that was activated would account for this effect.
We suggest that the initial pore is always labile, no matter what the temperature or chemokine receptor density. Biologically, this would mean that when the pore first forms at 37°C, not all (perhaps none) of the gp41 have yet folded into bundles at the site. A higher peptide concentration is required to close labile pores (e.g., ∼50 nM of C34) (unpublished data) than is needed to prevent their formation (e.g., ∼15 nM of C34) (Melikyan et al., 2000b). This provides more experimental validation for the concept that bundle formation is made easier by the membrane continuity provided by the fusion pore. The greater amount of peptide required to close pores than to prevent their opening indicates that multiple copies of Env act in a more concerted manner to create a pore than to ensure that it remains open. Conditions that facilitate folding of Env into a late prebundle will obviously increase the likelihood for bundle formation and creation of a robust pore. Thus initial pores readily become robust at high temperature or at high chemokine receptor densities. Labile pores remain small. Once a fusion pore becomes robust, it can enlarge if temperature is not too low.
Two central questions in the field of viral membrane fusion are: At what point in the fusion process do 6HBs form? and What are the roles of bundles? In this article we have addressed and answered some core aspects of these questions. By quickly lowering temperature after pore formation, we were able to identify a point of bundle formation as occurring between the time the pore forms and the time it becomes robust. The membrane continuity created by a fusion pore facilitates the formation of 6HBs. In our model, binding of inhibitory peptides to prebundle structures prevents the late prebundles from folding into bundles and instead causes them to revert back to early prebundles (Figure 9). The copies of Env that are in late prebundle configurations and in 6HBs serve as buttresses that support the walls of the pore. But reversion of late prebundles removes buttresses and the pore collapses. Only the bundle configuration is stable and so the pore is not robust until all the relevant Envs fold into bundles. In viral infection, late prebundles convert to bundles, an irreversible step that permanently supports the wall of the pore so that it does not close. Once the pore is robust, it is committed to enlarge to sizes that permit passage of the viral nucleocapsid.
ACKNOWLEDGMENTS
We thank Sofya Brener for excellent technical assistance. We appreciate the critical reading of this manuscript and helpful discussions with Dr. Leonid Chernomordik. We thank the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health (NIH) for providing HeLaT4+ and 3T3.T4.CXCR4 cells, C34 peptide, and 12G5 MAb; Dr. Min Lu for C34 and N36; and Dr. Robert Doms for CD4- and CXCR4-expressing plasmids. This work was supported by National Institutes of Health grants GM-27367 and GM-54787.
Abbreviations used:
- 6HB
six-helix bundle
- C34
a 34 residue-long C-terminal peptide from the HIV-1 gp41 ectodomain
- E/T
effector and target cells in contact
- LAS
lipid-arrested stage of fusion
- LPC
lyso-phosphatidylcholine
- Env
HIV envelope protein
- N36
a 36-residue-long N-terminal peptide from the HIV-1 gp41 ectodomain
- TAS
temperature-arrested stage of fusion
Footnotes
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E02–09–0573. Article and publication date are at www.molbiolcell.org/cgi/doi/10.1091/mbc.E02–09–0573.
REFERENCES
- Cao J, Bergeron L, Helseth E, Thali M, Repke H, Sodroski J. Effects of amino acid changes in the extracellular domain of the human immunodeficiency virus type 1 gp41 envelope glycoprotein. J Virol. 1993;67:2747–2755. doi: 10.1128/jvi.67.5.2747-2755.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci USA. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC, Kim PS. HIV entry and its inhibition. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- Chen CH, Matthews TJ, McDanal CB, Bolognesi DP, Greenberg ML. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: implication for viral fusion. J Virol. 1995;69:3771–3777. doi: 10.1128/jvi.69.6.3771-3777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L, Kozlov MM, Zimmerberg J. Lipids in biological membrane fusion. J Membr Biol. 1995;146:1–14. doi: 10.1007/BF00232676. [DOI] [PubMed] [Google Scholar]
- Coorssen JR, Blank PS, Tahara M, Zimmerberg J. Biochemical and functional studies of cortical vesicle fusion: the SNARE complex and Ca2+ sensitivity. J Cell Biol. 1998;143:1845–1857. doi: 10.1083/jcb.143.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rosny E, Vassell R, Wingfield PT, Wild CT, Weiss CD. Peptides corresponding to the heptad repeat motifs in the transmembrane protein (gp41) of human immunodeficiency virus type 1 elicit antibodies to receptor-activated conformations of the envelope glycoprotein. J Virol. 2001;75:8859–8863. doi: 10.1128/JVI.75.18.8859-8863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HK, Unutmaz D, KewalRamani VN, Littman DR. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature. 1997;388:296–300. doi: 10.1038/40894. [DOI] [PubMed] [Google Scholar]
- Dubay JW, Roberts SJ, Brody B, Hunter E. Mutations in the leucine zipper of the human immunodeficiency virus type 1 transmembrane glycoprotein affect fusion and infectivity. J Virol. 1992;66:4748–4756. doi: 10.1128/jvi.66.8.4748-4756.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrer P, Galli C, Hoenke S, Corti C, Gluck R, Vorherr T, Brunner J. H+-induced membrane insertion of influenza virus hemagglutinin involves the HA2 amino-terminal fusion peptide but not the coiled coil region. J Biol Chem. 1996;271:13417–13421. doi: 10.1074/jbc.271.23.13417. [DOI] [PubMed] [Google Scholar]
- Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- Endres MJ, et al. CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell. 1996;87:745–756. doi: 10.1016/s0092-8674(00)81393-8. [DOI] [PubMed] [Google Scholar]
- Gallo SA, Puri A, Blumenthal R. HIV-1 gp41 six-helix bundle formation occurs rapidly after the engagement of gp120 by CXCR4 in the HIV-1 Env-mediated fusion process. Biochemistry. 2001;40:12231–12236. doi: 10.1021/bi0155596. [DOI] [PubMed] [Google Scholar]
- Golding H, et al. Dissection of human immunodeficiency virus type 1 entry with neutralizing antibodies to gp41 fusion intermediates. J Virol. 2002;76:6780–6790. doi: 10.1128/JVI.76.13.6780-6790.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez LD, Hoffman LR, Wolfsberg TG, White JM. Virus-cell and cell-cell fusion. Annu Rev Cell Dev Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- Jahn R, Sudhof TC. Membrane fusion and exocytosis. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- Ji H, Shu W, Burling FT, Jiang S, Lu M. Inhibition of human immunodeficiency virus type 1 infectivity by the gp41 core: role of a conserved hydrophobic cavity in membrane fusion. J Virol. 1999;73:8578–8586. doi: 10.1128/jvi.73.10.8578-8586.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Lin K, Strick N, Neurath AR. HIV-1 inhibition by a peptide. Nature. 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- Kilby JM, et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- Kliger Y, Shai Y. Inhibition of HIV-1 entry before gp41 folds into its fusion-active conformation. J Mol Biol. 2000;295:163–168. doi: 10.1006/jmbi.1999.3368. [DOI] [PubMed] [Google Scholar]
- Lee B, Sharron M, Montaner LJ, Weissman D, Doms RW. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci USA. 1999;96:5215–5220. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Blacklow SC, Kim PS. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- Lu M, Stoller MO, Wang S, Liu J, Fagan MB, Nunberg JH. Structural and functional analysis of interhelical interactions in the human immunodeficiency virus type 1 gp41 envelope glycoprotein by alanine-scanning mutagenesis. J Virol. 2001;75:11146–11156. doi: 10.1128/JVI.75.22.11146-11156.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddon PJ, Dalgleish AG, McDougal JS, Clapham PR, Weiss RA, Axel R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell. 1986;47:333–348. doi: 10.1016/0092-8674(86)90590-8. [DOI] [PubMed] [Google Scholar]
- Markosyan RM, Cohen FS, Melikyan GB. The lipid-anchored ectodomain of influenza virus hemagglutinin (GPI-HA) is capable of inducing nonenlarging fusion pores. Mol Biol Cell. 2000;11:1143–1152. doi: 10.1091/mbc.11.4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markosyan RM, Ma X, Lu M, Cohen FS, Melikyan GB. The mechanism of inhibition of HIV-1 Env-mediated cell-cell fusion by recombinant cores of gp41 ectodomain. Virology. 2002;302:174–184. doi: 10.1006/viro.2002.1593. [DOI] [PubMed] [Google Scholar]
- Mayer A. What drives membrane fusion in eukaryotes? Trends Biochem Sci. 2001;26:717–723. doi: 10.1016/s0968-0004(01)01984-3. [DOI] [PubMed] [Google Scholar]
- Melikyan GB, Lin S, Roth MG, Cohen FS. Amino acid sequence requirements of the transmembrane and cytoplasmic domains of influenza virus hemagglutinin for viable membrane fusion. Mol Biol Cell. 1999;10:1821–1836. doi: 10.1091/mbc.10.6.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Markosyan RM, Brener SA, Rozenberg Y, Cohen FS. Role of the cytoplasmic tail of ecotropic moloney murine leukemia virus Env protein in fusion pore formation. J Virol. 2000a;74:447–455. doi: 10.1128/jvi.74.1.447-455.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol. 2000b;151:413–424. doi: 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Barroso I, Durell S, Sakaguchi K, Appella E, Blumenthal R. Dilation of the human immunodeficiency virus-1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41. J Cell Biol. 1998;140:315–323. doi: 10.1083/jcb.140.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat Struct Biol. 1998;5:765–769. doi: 10.1038/1799. [DOI] [PubMed] [Google Scholar]
- Russell CJ, Jardetzky TS, Lamb RA. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 2001;20:4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Tokunaga K, Greenberg ML, Morse MA, Cumming RI, Lyerly HK, Cullen BR. Molecular basis for cell tropism of CXCR4-dependent human immunodeficiency virus type 1 isolates. J Virol. 2001;75:6776–6785. doi: 10.1128/JVI.75.15.6776-6785.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- Weng Y, Yang Z, Weiss CD. Structure-function studies of the self-assembly domain of the human immunodeficiency virus type 1 transmembrane protein gp41. J Virol. 2000;74:5368–5372. doi: 10.1128/jvi.74.11.5368-5372.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc Natl Acad Sci USA. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]