

Abstract
We report here the direct measurement of intra-tRNA distances during annealing of the tRNA primer to the HIV RNA genome. This key step in the initiation of retroviral reverse transcription involves hybridization of one strand of the acceptor arm of a specific lysine tRNA to the primer binding site on the RNA genome. Although the mechanism of tRNA unwinding and annealing is not known, previous studies have shown that HIV nucleocapsid protein (NC) greatly accelerates primer/template binary complex formation in vitro. An open question is whether NC alone unwinds the primer or whether unwinding by NC requires the RNA genome. We monitored the annealing process in solution by using fluorescence resonance energy transfer (FRET). Distance measurements demonstrate unequivocally that the tRNA acceptor stem is not substantially unwound by NC in the absence of the RNA genome, that is, unwinding is not separable from hybridization. Moreover, FRET measurements show that both heat- and NC-mediated annealing result in an ≈40-Å increase in the separation of the two ends of the tRNA acceptor arm on binding to the template. This large increase in separation of the two ends suggests a complete displacement of the nonhybridized strand of the acceptor stem in the initiation complex.
An early step in the initiation of retroviral reverse transcription is binary complex formation between the RNA genome and a host-cell tRNA that is used as a primer by reverse transcriptase. The annealing process requires hybridization of the 3′ strand of the tRNA acceptor arm to the complementary primer binding site (PBS) sequence on the RNA genome. In HIV-1, human tRNALys-3 is selectively packaged for use as the replication primer (1). After the primer/template complex is formed, reverse transcriptase extends the 3′ hydroxyl at the terminus of the tRNA to produce an initial cDNA product.
Both the RNA genome and the tRNA primer undergo extensive conformational changes on binary complex formation. Previous studies have shown that recombinant HIV-1 nucleocapsid protein (NC) acts as an RNA “chaperone” to facilitate RNA rearrangements (recently reviewed in ref. 2) and, in particular, gel electrophoresis studies have demonstrated that NC promotes tRNA annealing to the RNA genome (3, 4). However, little is known about the annealing mechanism or about the tertiary structure of the tRNA/NC and tRNA/NC/RNA genome complexes.
Based on near-UV circular dichroism studies, NC was proposed to denature or unwind free tRNAs in the absence of Mg2+, whereas the extent of unwinding was significantly reduced when Mg2+-renatured tRNA was used (5). However, the spectral changes observed in circular dichroism studies of Mg2+-free tRNA have been interpreted by other researchers to be caused by changes in base stacking interactions rather than helix unwinding (6). NMR studies performed in the presence of Mg2+ also are inconsistent with significant unfolding of tRNA helices by NC (7). The Pb2+-ribozyme self-cleaving activity of tRNAPhe, however, was completely inhibited by one molar equivalent of NC, suggesting that even in the presence of Mg2+, NC may alter the tertiary conformation of the tRNA (7).
In this work, we use fluorescence resonance energy transfer (FRET) to directly probe the tRNA acceptor stem structural rearrangements that occur on primer annealing to the PBS. In addition, we use this technique to resolve the question of whether NC unwinding of the tRNA acceptor stem occurs in the absence of the RNA genome or whether NC requires the template as a cofactor. By using a recombinant C-terminal histidine-tagged form of NCp15 (8), we measure intra-tRNA distance changes between the 3′ and 5′ ends of the acceptor arm by using FRET. We established that NCp15 does not induce substantial acceptor stem helix unwinding or strand separation in the absence of template. We also determined for the first time the distance between the 3′ and 5′ ends of the tRNA in both heat- and NCp15-annealed primer/template binary complexes.
MATERIALS AND METHODS
Materials.
Cystamine, sodium cyanoborohydride, sodium periodate, and sodium hypophosphite were purchased from Sigma. Tetramethylrhodamine-5-iodoacetamide (TMR-IA), 5-iodoacetamidofluorescein (IAF), and tris-(2-carboxyethyl)phosphine hydrochloride (TCEP) were purchased from Molecular Probes. DNA plasmids used for preparing tRNALys-3 and the 394-nt HIV-1 HXB2 RNA genome have been described (8). Plasmid pNCH6, which encodes the gene for HIV-1 NC with poly(histidine) at the C terminus, was a gift from S. Le Grice (Case Western Reserve University, Cleveland).
Protein Purification.
NCp15 was prepared from BL21(DE3) cells carrying plasmids pNCH6 and pREP4 (Qiagen). Cells (3 liters) were grown in Luria–Bertani (LB) media containing 100 μg /ml ampicillin and 25 μg /ml kanamycin at 37°C to an OD600 of 0.7. Isopropyl-β-d-thiogalactopyranoside was added to 1 mM, and cells were incubated at 37°C for 4 hr. The following steps were performed at 4°C using freshly degassed buffers. Cells were resuspended in 90 ml of buffer A (50 mM sodium phosphate/5 mM Tris⋅HCl, pH 8.0/10 mM 2-mercaptoethanol/10 μM ZnCl2) containing 20 mM imidazole. Phenylmethylsulfonyl fluoride (0.2 mM) and lysozyme (1 mg /ml) were added. After stirring for 15 min, NaCl (0.2 M) and Triton X-100 (1% vol/vol) were added. The mixture was stirred for 1 hr, followed by addition of NaCl to 1.3 M. After 30 min of stirring, 45 ml of buffer A/20 mM imidazole/0.5 M NaCl/18% (wt/vol) polyethylene glycol (PEG 8000) were added. After centrifugation at 25,800 × g for 45 min, the supernatant was added to 270 ml of buffer A/0.5 M NaCl and loaded onto a pre-equilibrated, 1-ml Ni2+–nitrilotriacetic acid column (Qiagen, Chatsworth, CA). The column was washed with 45 ml of buffer A/0.5 M NaCl/20 mM imidazole, 20 ml of buffer A/100 mM NaCl/15% glycerol/0.1% Triton X-100/6 M guanidine hydrochloride, and 25 ml of the same buffer without guanidine hydrochloride. NCp15 was eluted with 25 ml of buffer B (50 mM sodium phosphate, pH 7.6/100 mM NaCl/15% glycerol/0.1% Triton X-100/10 μM ZnCl2/10 mM 2-mercaptoethanol) containing 100 mM imidazole, followed by 25 ml of buffer B with 200 mM imidazole. DTT (5 mM) and ZnCl2 (75 μM) were added. The protein was next chromatographed on a 27-ml CM-Sepharose Fast Flow column (Pharmacia) pre-equilibrated with buffer C (50 mM sodium phosphate, pH 7.6/100 mM NaCl/10% glycerol/75 μM ZnCl2/5 mM DTT). NCp15 was eluted by a 100-ml linear gradient of NaCl from 100 mM to 1.5 M in buffer C. Peak fractions eluted at 650 mM NaCl. NCp15 was concentrated to 1 mg /ml by using Centricon 10 (Amicon) concentrators, followed by a 4-fold dilution with storage buffer (50 mM sodium phosphate, pH 7.6/5 mM DTT/75 μM ZnCl2) and the addition of glycerol to 15% (vol/vol). The protein was stored at −80°C in 10-μl aliquots. This purification routinely yields NCp15 (2–3 mg) that was judged to be >95% pure by using SDS/PAGE. T7 RNA polymerase was purified as described (9).
RNA Preparation.
Human tRNALys-3 was transcribed as described (8), except the GTP concentration was reduced to 1 mM and 5 mM GMP was included in the reaction. The tRNA was refolded by incubation at 85°C and 60°C for 2 min each in 25 mM Tris⋅HCl (pH 7.5), followed by addition of MgCl2 to 10 mM. The sample was then placed on ice. The 5′ 394-nt HXB2 RNA template was prepared as described (8). The RNA template was refolded in 25 mM Tris·HCl (pH 7.5) and 100 mM NaCl by incubation at 85°C (2.5 min) and 50°C (8 min), followed by addition of MgCl2 to 8 mM. The RNA was then incubated at 37°C for 10 min and placed on ice.
Covalent Attachment of Fluorescent Probes to tRNA.
The 5′ monophosphorylated tRNALys-3 was purified on an 8% denaturing PAGE gel, and the 5′ end was reacted with cystamine as described (10). After LiCl/ethanol precipitation, excess cystamine was removed by using a NAP-10 column (Pharmacia). The 5′-cystaminylated tRNA (97 μM) and TCEP (3.25 mM) were incubated in 400 mM Hepes/2 mM EDTA (pH 7.6) at room temperature for 15 min, followed by addition of an equal volume of 9.7 mM TMR-IA in dimethyl sulfoxide. After a 3-hr incubation in the dark, RNA was recovered by using NaCl/ethanol precipitation, redissolved in water, extracted twice with an equal volume of phenol, and precipitated. The 5′-TMR-labeled tRNA (TMR–tRNA) was purified by using HPLC with a Rainin Microsorb MV C-8 reversed phase column (300 Å). The elution profile was developed at 0.8 ml/min as described (11). The 3′ end of TMR–tRNA (100 μM) was next oxidized by incubation in 20 mM NaIO4 at room temperature in the dark for 20 min. Excess NaIO4 was destroyed as described (12). RNA was recovered by NaCl/ethanol precipitation. The oxidized RNA (86 μM) was reductively aminated with cystamine (340 μM) and NaCNBH3 (6 mM) in 100 mM sodium phosphate (pH 6.3) at room temperature overnight (12, 13). Excess reagents were removed by using a NAP-10 column. The 3′-cystaminylated tRNA was reacted with IAF by using conditions similar to those used in the attachment of TMR to the 5′ end. The TMR-fluorescein doubly labeled tRNA (TMR–tRNA–FL) was separated from TMR–tRNA by using HPLC as described in the legend to Fig. 1. Singly 3′ fluorescein-labeled tRNA (tRNA–FL) was prepared similarly.
Figure 1.

HPLC trace showing separation of TMR–tRNA–FL from TMR–tRNA. TMR and FL are represented by a circle and square, respectively. Following a 10-min wash with buffer A, the elution profile was developed by increasing the percentage of buffer B as follows: 0 → 50% over 10 min, 50 → 55% over 30 min, and 55 → 100% over 5 min (Inset) Denaturing PAGE gel (8%) analysis of fluorescently labeled tRNALys-3. One microgram each of tRNA–FL (lane I), TMR–tRNA (lane II), and TMR–tRNA-FL (lane III) was denatured by heating at 85°C in 66% formamide for 2 min, followed by cooling on ice water for 2 min. RNA samples were visualized by ethidium bromide staining.
Heat-Mediated Annealing Assay.
A modification of the method of Arts et al. (14) was used to promote tRNA/RNA genome annealing by heat. Briefly, in a 100-μl volume, dye-labeled tRNA (0.32 μM) and template (0.49 μM) were heat-annealed in the presence of 0.105 M Tris⋅HCl (pH 8.3) and 0.21 M KCl by successive incubation at 85, 50, and 37°C for 2.5, 8, and 10 min, respectively. Diethyl pyrocarbonate-treated water (400 μL) was added to the samples before recording the emission spectrum. To determine the extent of annealing, samples were concentrated by using ethanol precipitation, analyzed by gel-retardation assays as described (8), and visualized by observing the fluorescence of the dyes by using a Bio-Rad Gel Doc 1000 video gel documentation system (302 nm excitation).
NCp15-Mediated Annealing Assay.
NCp15 annealing conditions were as described (8, 15) with the following modifications. Dye-labeled tRNA (6.5 nM) and template (15.8 nM) were incubated at 22°C for 5 min before an initial fluorescence measurement. NCp15 was then added to an RNA nucleotide to NCp15 ratio of 8:1 unless noted otherwise. After 10 min at 22°C, another fluorescence spectrum was taken. The extent of binary complex formed was determined by quantification of a gel assay performed under identical conditions by using 32P-labeled tRNALys-3.§
Fluorescence Measurements.
Steady-state fluorescence spectra (corrected) were recorded by exciting the samples at 468 nm at 22°C under magic-angle conditions. Time-resolved fluorescence data were determined by time-correlated single-photon counting (TCSPC) at 22°C with 468-nm vertically polarized excitation pulses (≈100-fs duration) at a repetition rate of 9.5 MHz. Emission was collected at 90° through a 520-nm interference filter and detected with a Hamamatsu Photonics (Bridgewater, NJ) R3809U-01 microchannel plate photomultiplier tube. The time-resolved fluorescence anisotropy, r(t), was calculated by the usual method (16) from time-resolved emission data collected alternatively with the analyzing polarizer in a vertical (parallel) or horizontal (perpendicular) orientation. All other time-resolved data were recorded with the polarizer set at the magic angle.
FRET Distance Calculations.
The following equation was used to calculate the FRET efficiency (ℰ) from steady-state data:
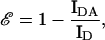 |
1a |
where IDA and ID are the peak fluorescence intensity of FL in TMR–tRNA–FL and tRNA–FL, respectively. The efficiency of energy transfer is related to the distance between the two probe molecules (R) according to Förster theory (17):
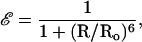 |
1b |
where Ro is the Förster radius (see below). To calculate R from TCSPC data, a nonlinear least square fitting procedure was used to fit the decay data to a sum of exponential decays convolved with the instrument response (≈100 ps Full Width Half Maximum). From the best-fit parameters, R was calculated according to Eq. 2c. Under conditions where efficient FRET was observed, the FRET dynamics could be analyzed in detail. The data were fit to a Gaussian distribution [g(R)] of fast decays (Eq. 2a), where 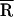 and σ are the mean and SD of the distribution, respectively (Eq. 2b), and τD is the fluorescence lifetime of the donor in the absence of the acceptor (Eq. 2c). The data were corrected for a minor slow-decay component (see Table 1 footnote). Using a scaling factor 𝒜,
and σ are the mean and SD of the distribution, respectively (Eq. 2b), and τD is the fluorescence lifetime of the donor in the absence of the acceptor (Eq. 2c). The data were corrected for a minor slow-decay component (see Table 1 footnote). Using a scaling factor 𝒜, 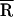 , and σ as the adjustable parameters, a trial decay [IFl(t)] was calculated and convolved with the instrument response. The resultant decay was compared with the corrected raw data, and the best fit was obtained by using a downhill simplex searching algorithm. The integral in Eq. 2a was evaluated by using the Gaussian quadratures method (18). The mean donor-acceptor distance (
, and σ as the adjustable parameters, a trial decay [IFl(t)] was calculated and convolved with the instrument response. The resultant decay was compared with the corrected raw data, and the best fit was obtained by using a downhill simplex searching algorithm. The integral in Eq. 2a was evaluated by using the Gaussian quadratures method (18). The mean donor-acceptor distance (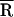 ) was obtained from the best fit.
) was obtained from the best fit.
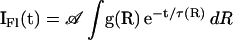 |
2a |
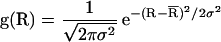 |
2b |
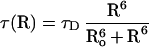 |
2c |
Table 1.
Time-resolved fluorescence data and analysis
| Sample | τdecay,* ns |
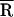 (Å)† (Å)†
|
σ (Å)† |
|---|---|---|---|
| No heat or NCp15 | |||
| tRNA-FL | 4.3 | — | — |
| TMR–tRNA–FL | 0.3 (85)‡ | 32 | 6 |
| tRNA–FL/T | 4.2 | — | — |
| TMR–tRNA–FL/T | 0.4 (74)‡ | 34 | 7 |
| Heat-annealed | |||
| tRNA–FL/T | 4.3 (76)§ | — | — |
| TMR–tRNA–FL/T | 3.9 (70)§ | 73 | — |
| NCp15-annealed | |||
| tRNA–FL | 4.3 (68)¶ | — | — |
| TMR–tRNA–FL | 0.4 (77)‡ | 35 | 5 |
| tRNA–FL/T | 4.3 (68)§ | — | — |
| TMR–tRNA–FL/T | 3.9 (52)§ | 73 | — |
Values are averages of at least five determinations. Numbers in parentheses represent typical amplitudes (percent total signal) with SD ≤ 8%.
Based on a Gaussian distribution [g(R)] analysis, where 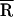 is the mean donor–acceptor distance and σ is the SD of g(R). SD ≤ 3 Å for
is the mean donor–acceptor distance and σ is the SD of g(R). SD ≤ 3 Å for 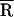 and ≤2 Å for σ.
and ≤2 Å for σ.
Mean donor lifetimes (SD ≤ 36%) from the Gaussian distribution analysis. A minor component (τD ∼ 4.3 ns), which may be the result of FL’s inability to undergo efficient FRET because of misfolded tRNA, was subtracted from the raw data before fitting was performed.
Based on a bi-exponential fit (SD ≤ 5%). A second component (τ ∼ 1.2 ns) can be resolved from these data.
Based on a single measurement.
Determination of the Förster Radius (Ro).
The Förster radius was determined to be 50 Å, assuming a value of 2/3 for the geometrical factor κ2 (see below) and by using 1.33 for the refractive index of water at 22°C. The quantum yield of the donor in the absence of acceptor was determined to be 0.29 by using fluorescein at pH 13 as a standard [quantum yield = 0.92 ± 0.03 (19)]. The spectral overlap integral was determined to be 2.76 × 1015 M−1⋅cm−1·nm4 by numerical integration of the emission spectrum of tRNA–FL and the absorption spectrum of TMR–tRNA.
RESULTS
Labeling of tRNALys-3 with Fluorescent Reporter Groups.
Fig. 2 illustrates schematically changes that may occur to the tertiary structure of human tRNALys-3 on NCp15 binding (Fig. 2, Lower Left) and annealing (Fig. 2, Lower Right) to the HIV-1 genome. In this work, we first established a labeling procedure for attachment of fluorescent dyes to the 3′ and 5′ ends of the tRNA for use as reporter groups to monitor conformational changes in the acceptor stem domain via FRET. The overall yield of 5′ tRNA modification with TMR was about 80%, as judged from HPLC profiles (data not shown). The subsequent steps afforded approximately 30% conversion of TMR–tRNA to TMR–tRNA–FL (Fig. 1). The doubly labeled species migrates as a doublet on the HPLC, most likely because of RNA conformational heterogeneity on this nondenaturing HPLC column. This was confirmed by using denaturing gel electrophoresis, where we observe that the TMR–tRNA–FL, as well as the singly labeled tRNAs, migrates as single sharp bands (Fig. 1 Inset). A UV–visible spectrum of the purified TMR–tRNA–FL shows well separated absorbance peaks for RNA (260 nm), FL (494 nm), and TMR (560 nm). Extinction coefficients were calculated to be approximately 70,000 M−1⋅cm−1 for both dyes after conjugation.
Figure 2.

Transfer RNA unwinding and annealing monitored by FRET. Schematic illustration of tRNA structural changes probed in this work. R1 and R2 represent the distance between the probes on NCp15 addition in the absence (Lower Left) and presence (Lower Right) of template. TMR and FL are represented by a circle and square, respectively.
Characterization of Singly and Doubly Dye-Labeled tRNALys-3.
To evaluate the rotational freedom of the tRNA-attached dye molecules that are shown in Fig. 3, steady-state and time-resolved anisotropy measurements, r(t), were carried out. The FL donor emission in free TMR–tRNA–FL and TMR–tRNA–FL heat-annealed to the template has an initial anisotropy, r(0), of ≈0.2, which decayed to ≤0.03 in ≈0.4 ns. Because of the unavailability of a suitable direct excitation source for TMR, its emission anisotropy was determined only by steady-state methods, yielding a value of 0.15 ± 0.02. Based on the small anisotropy values of both FL and TMR, we assumed a value of 2/3 for κ2 in the evaluation of Ro (20).
Figure 3.

Structure of doubly labeled human tRNALys-3 used in the study. TMR and FL are shown attached to the 5′ and 3′ ends of the tRNA, respectively. Nucleotide sequence G59–A76 is complementary to the PBS.
Fig. 4A shows the emission spectrum of tRNA–FL (green) excited at 468 nm. The spectrum displays a single emission band centered at 520 nm. The fluorescence decay of tRNA–FL is shown in Fig. 4B (green). The decay is very well described by a single exponential with a lifetime of 4.3 ns (Table 1). The steady-state measurements of TMR–tRNA–FL show very efficient energy transfer from FL to TMR on 468 nm excitation. For example, the emission intensity of FL at 520 nm in TMR–tRNA–FL (Fig. 4A, red) is substantially quenched when compared with that of FL in tRNA–FL (Fig. 4A, green). A concomitant increase in the TMR fluorescence intensity at 580 nm in TMR–tRNA–FL also is observed. Under these conditions, TMR emission caused by direct excitation of TMR at 468 nm was found to be negligible (data not shown). Therefore, the 580-nm emission is entirely the result of energy transfer from the FL donor to the TMR acceptor. Based on the steady-state measurements, the FRET efficiency in free TMR-tRNA–FL was calculated to be 93 ± 2%, corresponding to an average distance of 33 ± 1 Å between the two probes.
Figure 4.

(A) Steady-state fluorescence spectra of 80 nM tRNA–FL (green), 80 nM TMR–tRNA–FL (red), 80 nM TMR–tRNA–FL with 760 nM NCp15 (black), and 64 nM TMR-tRNA-FL heat-annealed to 98 nM RNA template (blue). All spectra were normalized by integrating the area between 490 and 630 nm. (B) Scaled fluorescence decays and best fits (see Materials and Methods) of 80 nM tRNA–FL (green), 80 nM TMR–tRNA–FL (red), 80 nM TMR–tRNA–FL with 760 nM NCp15 (black), 64 nM TMR–tRNA–FL heat-annealed to 98 nM template (blue), and 6.5 nM TMR–tRNA–FL NCp15-annealed to 9.8 nM template (orange). Inset shows fluorescence decay of 80 nM TMR–tRNA–FL in the absence (red) and presence (black) of 760 nM NCp15 before scaling.
The TCSPC data for TMR–tRNA–FL were moderately well fit to a bi-exponential decay (Fig. 4B, red). This suggests that the emission may originate from two populations of FL in TMR–tRNA–FL, a fast decay with a lifetime of ≈0.3 ns that accounts for ≈85% of the emission, and a slower 4.3-ns decay that resembles the lifetime of FL in tRNA–FL (Table 1). The fast decay is likely a result of rapid FRET in TMR–tRNA–FL and is consistent with the decrease in the 520-nm emission observed in the steady-state experiments (Fig. 4A).
FRET measurements of donor–acceptor distances in biomolecules are better described by a model that uses a Gaussian distribution of distances rather than only a single distance (21). The former approach (see Materials and Methods) accounts for the range of donor–acceptor distances that presumably result from the conformational distribution of the linkers and the single-stranded CCA 3′ end of the tRNA. For example, for free TMR–tRNA–FL (no NCp15 or template), the fitting procedures reveal a significant spread of donor–acceptor separations (σ = 6 Å). From the mean of the distribution (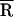 ), an average donor–acceptor distance of 32 Å was determined for free TMR–tRNA–FL (Table 1). This distance agrees well with the value obtained from steady-state measurements.
), an average donor–acceptor distance of 32 Å was determined for free TMR–tRNA–FL (Table 1). This distance agrees well with the value obtained from steady-state measurements.
Heat-Annealing of tRNALys-3 to the HIV-1 RNA Template.
To assess the effects of the conjugated fluorescent probes on tRNA annealing to the RNA template, we performed heat-annealing gel-retardation assays (8, 15) using singly and doubly labeled tRNAs under identical experimental conditions to those used in FRET measurements. As shown in Fig. 5, the presence of the fluorophores does not affect the annealing process.
Figure 5.

Gel showing heat-annealing of fluorescently labeled tRNAs and RNA template. The identity of each primer/template combination is given above the lanes. The absence (−) and presence (+) of RNA template also is indicated.
We next used FRET to monitor tRNALys-3 acceptor stem structural changes on binary complex formation. Heat-annealing TMR-tRNA–FL to the template (T) resulted in a dramatic increase in steady-state FL emission intensity, with a concomitant decrease in TMR emission at 580 nm (Fig. 4A, blue) relative to free TMR–tRNA–FL (red). These data show that there is much less efficient FRET from donor to acceptor in the binary complex and thus, a larger donor–acceptor separation. Based on the steady-state FL emission intensity of tRNA–FL/T and TMR–tRNA–FL/T complexes, we calculated a 19.9% FRET efficiency in the heat-annealed complex, corresponding to an average donor–acceptor separation of 63 Å.
Time-resolved measurements on the donor emission also show dramatic evidence of a substantial decrease in FRET on heat-annealing. The heat-annealed TMR–tRNA–FL/T complex (Fig. 4B, blue) exhibits a relatively slow decay (Table 1) that is qualitatively similar to that observed for free tRNA—FL (Fig. 4B, green), tRNA–FL/T in the absence of heat, and heat-annealed tRNA–FL/T (Table 1). Strikingly, the TCSPC data reveal that the 0.3-ns decay observed in the analysis of TMR–tRNA–FL alone (Fig. 4B, red) completely disappears in the heat-annealed complex (Table 1). Taken together, these data indicate that a dramatic conformational change occurs on heat-annealing, leading to a donor–acceptor separation that precludes efficient energy transfer. Based on the 3.9-ns decay, the FRET efficiency is estimated to be 9.6%, leading to a donor–acceptor separation of 73 Å (Table 1). This value is somewhat larger than the distance determined from the steady-state analysis (63 Å); however, both reveal that a major structural change occurs in the acceptor stem of the tRNA on heat-annealing to the template.
Effect of NCp15 on Mg2+-Renatured tRNALys-3 in the Absence of Template.
We next addressed the question of whether NCp15 alone induces substantial unwinding of the acceptor stem in the absence of the template. Addition of NCp15 to TMR–tRNA–FL did not affect the relative fluorescence intensities of FL (520 nm) and TMR (580 nm) in the steady-state measurements (Fig. 4A, compare red and black curves). This result suggests that NCp15 has a minimal effect on the structure of the acceptor stem of tRNALys-3 in the absence of template. A comparison of time-resolved measurements of TMR–tRNA–FL in the presence of NCp15 (Fig. 4B, black) to that of heat-annealed TMR–tRNA–FL/T (Fig. 4B, blue) clearly shows that NCp15 alone is not capable of inducing dramatic acceptor stem unwinding and the corresponding decrease in FRET. However, a comparison of the scaled time-resolved data of TMR–tRNA–FL in the presence (Fig. 4B, black) and absence (Fig. 4B, red) of NCp15 reveals some obvious differences that apparently reflect less dramatic NCp15-dependent changes in the acceptor stem conformation.
The Inset in Fig. 4B compares the unscaled fluorescence decays of TMR–tRNA–FL in the absence and presence of NCp15 at early times. Interestingly, the initial emission intensity increases almost 2-fold when NCp15 is present (Fig. 4B, Inset, black). The lower initial intensity observed in the absence of NCp15 suggests the presence of some fast dynamics not resolvable by the ≈100-ps instrument response of the TCSPC apparatus. We hypothesize that the formation of the TMR–tRNA–FL/NCp15 complex slows down the initial FRET dynamics, making it less susceptible to distortion by the instrument response, resulting in a higher apparent initial intensity. This interpretation suggests that NCp15 shifts the donor–acceptor separation distribution toward slightly larger values, but the change is much smaller than with heat annealing. In fact, an analysis of the data using the Gaussian distribution model yields values of 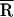 and σ that are identical within the experimental error in the absence (
and σ that are identical within the experimental error in the absence (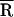 ≈ 32 Å) and presence (
≈ 32 Å) and presence (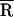 ≈ 35 Å) of NCp15 (Table 1). Moreover, no major change in
≈ 35 Å) of NCp15 (Table 1). Moreover, no major change in 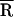 and σ was observed when additional NCp15 was added to TMR–tRNA–FL up to an RNA nucleotides to NCp15 ratio of 3:1 (data not shown).
and σ was observed when additional NCp15 was added to TMR–tRNA–FL up to an RNA nucleotides to NCp15 ratio of 3:1 (data not shown).
NCp15-Annealing of tRNALys-3 to the HIV-1 RNA Template.
Before FRET determinations in the presence of both NCp15 and template, we used a conventional gel-retardation assay and 32P-labeled tRNALys-3 to optimize NCp15-annealing activity at relatively low RNA concentrations to avoid sample aggregation. The time-resolved FRET measurements, therefore, were performed by using conditions where ≥85% annealing is routinely observed as monitored by the radioactive gel assay.
The time-resolved FRET data for the NCp15- and heat-annealed TMR–tRNA–FL/T complexes are very similar (Fig. 4B, compare orange and blue curves). The fast components observed for free TMR–tRNA–FL (≈0.3 ns) and TMR–tRNA–FL/T (no heat or NCp15; ≈0.4 ns), disappear on NCp15-mediated annealing to the template (Table 1). Thus, in the absence of heat, global unwinding of the tRNA acceptor stem requires the simultaneous presence of both NCp15 and template.
A FRET analysis of the major (3.9-ns) kinetic component of the fluorescence decay of the NCp15-annealed complex leads to an estimate of 73 Å for the donor–acceptor separation. Time-resolved data for the heat- and NCp15-annealed complexes exhibit an additional (≈1-ns) decay component (Table 1) that is assigned tentatively to quenching of the FL emission by the template. It is unlikely that this component is the result of FRET because it also is observed when no TMR acceptor is present, i.e., heat- and NCp15-annealed tRNA–FL/T. In particular, the quenching of the FL emission may be because of one or more guanosine residues in or near the PBS in a subset of annealed complexes that have an appropriate conformation for quenching through electron transfer (22). The differences in the observed amplitudes of the fast component suggest that there may be small structural differences between the NCp15- and heat-annealed binary complexes. Alternatively, the variations may be caused by the presence of NCp15 and Mg2+ ions that are closely associated with the NCp15-annealed complex but absent in the heat-annealed complex.
DISCUSSION
We established a synthetic strategy for covalent attachment of fluorescence energy donor and acceptor probes to the 3′ and 5′ ends of tRNALys-3. The probes do not significantly affect the capability of the tRNA to hybridize to the template. Based on the known three-dimensional structure of yeast tRNAPhe (23, 24), the distance between the ribose sugar of 3′ A76 and the phosphate of 5′ G1 is 23 Å. After taking the linkers into account, we estimate that the maximum distance between the center of mass of TMR and FL in TMR–tRNA–FL is ≈48 Å.¶ Thus, the average distance between the probes in TMR–tRNA–FL determined by using both steady-state and time-resolved FRET measurements (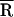 ≈ 32 Å) is consistent with the predicted range.
≈ 32 Å) is consistent with the predicted range.
Our studies show that only minor NCp15-induced structural changes occur in the acceptor stem of tRNALys-3 in the absence of template. A very similar donor–acceptor distance is measured for tRNA in the presence of NCp15 (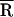 ≈ 35 Å) as for tRNA alone (
≈ 35 Å) as for tRNA alone (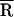 ≈ 32 Å). The only noticeable change in the fluorescence spectra when NCp15 is added to free TMR–tRNA–FL is a change of the overall emission intensity. The minor changes observed in both steady-state and time-resolved fluorescence measurements on NCp15 addition to free tRNA are entirely consistent with local base unstacking rather than global acceptor stem unwinding. The effect of NC on tRNA structure in the absence of template was previously investigated by CD (5, 6), NMR spectroscopy, and Pb2+ cleavage assays (7). In these studies, evidence both for and against NC-induced tRNA structural changes was reported. Although most of the previously reported data also are inconsistent with global tRNA acceptor stem unwinding by NC, many of these earlier experiments were carried out with tRNAs other than tRNALys-3 and under conditions (low temperature or ≥10 mM Mg2+) where we observe little NCp15-mediated tRNALys-3 annealing to the RNA genome in vitro (B.C. and K.M.-F., unpublished data). We carried out FRET measurements by using conditions where in vitro NCp15-annealing activity (in the presence of template) is high (≥85%). Nevertheless, NCp15 binding to tRNALys-3 does not induce substantial acceptor stem unwinding or strand separation in the absence of the RNA genome.
≈ 32 Å). The only noticeable change in the fluorescence spectra when NCp15 is added to free TMR–tRNA–FL is a change of the overall emission intensity. The minor changes observed in both steady-state and time-resolved fluorescence measurements on NCp15 addition to free tRNA are entirely consistent with local base unstacking rather than global acceptor stem unwinding. The effect of NC on tRNA structure in the absence of template was previously investigated by CD (5, 6), NMR spectroscopy, and Pb2+ cleavage assays (7). In these studies, evidence both for and against NC-induced tRNA structural changes was reported. Although most of the previously reported data also are inconsistent with global tRNA acceptor stem unwinding by NC, many of these earlier experiments were carried out with tRNAs other than tRNALys-3 and under conditions (low temperature or ≥10 mM Mg2+) where we observe little NCp15-mediated tRNALys-3 annealing to the RNA genome in vitro (B.C. and K.M.-F., unpublished data). We carried out FRET measurements by using conditions where in vitro NCp15-annealing activity (in the presence of template) is high (≥85%). Nevertheless, NCp15 binding to tRNALys-3 does not induce substantial acceptor stem unwinding or strand separation in the absence of the RNA genome.
In contrast to the experiments performed in the absence of template, large changes in the average donor-acceptor distance are measured after the tRNA is NCp15-annealed to the RNA genome. The 41-Å increase in probe separation is indicative of complete displacement of the 5′ strand of the acceptor arm on 3′-strand hybridization to the RNA genome.
A secondary structure model of the heat-annealed tRNA/HIV RNA genome initiation complex has been proposed based on chemical and enzymatic structure probing studies (25). This model predicts that there are extensive structural rearrangements of both the tRNA primer and the RNA genome on binary complex formation. In particular, it has been proposed that in the binary complex, the 5′ strand of the tRNA acceptor arm forms a new intramolecular duplex with complementary nucleotides located at positions 47–54 in the TΨC stem (Fig. 3). If such an intramolecular duplex forms, the distance between the 5′ and 3′ ends of the tRNA could increase by ≈40 Å. This distance change assumes an A-form helix geometry extending 22 bases from U54, which is paired to the 5′ G1 in the proposed binary complex, to the 3′ A76. Thus, the intra-tRNA distances we measured for both heat- and NCp15-annealed complexes are consistent with predictions based on the secondary structure model. Although the distances we measured for both complexes are essentially the same, additional experiments are needed to establish structural differences, if any, in other regions of the heat- and NCp15-annealed complexes. Because mature HIV-1 NC is derived from the gag polyprotein precursor and tRNALys-3 is placed onto the PBS in protease-negative virus, gag and/or gag-pol polyprotein are likely to be responsible for tRNA annealing in vivo (2, 26). Future in vitro studies using the gag polyprotein are thus also of interest.
In summary, we used FRET to directly monitor intra-tRNA distance changes induced by NCp15 in the formation of the initiation complex for HIV-1 reverse transcription. Our data indicate that NCp15 binding alone does not induce strand separation and are consistent with only minor base unstacking in the tRNA acceptor stem. Our analysis demonstrates that substantial unwinding of the acceptor arm occurs only in the presence of both the RNA genome and NCp15. Therefore, tRNA unwinding is not separable from hybridization.
Acknowledgments
We thank Drs. Stuart Le Grice, Jean-Luc Darlix, and F. William Studier for kindly providing plasmids and strains used in this study, Ms. Maria Nagan for assistance with computer modeling, and Dr. Paul Schimmel for providing helpful comments on the manuscript. This work was supported by Grant NP-953 from the American Cancer Society (to K.M.-F.), Grant CHE-9628523 from the National Science Foundation (to P.F.B.), and an Interdisciplinary Research Grant from the University of Minnesota. K.W. was a postdoctoral fellow of the National Institutes of Health.
ABBREVIATIONS
- FL
fluorescein
- FRET
fluorescence resonance energy transfer
- NC
nucleocapsid protein
- PBS
primer binding site
- T
template
- TCSPC
time-correlated single photon counting
- TMR
tetramethylrhodamine
- TMR–tRNA
5′ TMR-labeled tRNA
- TMR–tRNA–FL
5′ TMR-labeled and 3′ FL-labeled tRNA
- tRNA–FL
3′ FL-labeled tRNA
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Direct quantitation of the extent of NCp15 annealing using the dye-labeled tRNA was technically difficult because of the low RNA concentrations maintained in the FRET measurements. However, the similar fluorescence dynamics of heat- and NCp15-annealed complexes demonstrate that the dyes do not interfere with the capability of NCp15 to anneal the tRNA to the template.
The structure of human tRNALys-3 with the attached fluorophores was modeled based on the known structure of yeast tRNAPhe (23, 24) by using insightII (Biosym Technologies, San Diego). Energy minimization of the fluorophores and linkers was carried out with discover by using the Consistent Valence Force Field (27).
References
- 1.Kleiman L, Caudry S, Boulerice F, Wainberg M A, Parniak M A. Biochem Biophys Res Commun. 1991;174:1272–1280. doi: 10.1016/0006-291x(91)91559-u. [DOI] [PubMed] [Google Scholar]
- 2.Rein A, Henderson L E, Levin J G. Trends Biochem Sci. 1998;23:297–301. doi: 10.1016/s0968-0004(98)01256-0. [DOI] [PubMed] [Google Scholar]
- 3.Barat C, Lullien V, Schatz O, Keith G, Nugeyre M, Grüninger-Leitch F, Barré-Sinoussi F, Le Grice S F J, Darlix J-L. EMBO J. 1989;8:3279–3285. doi: 10.1002/j.1460-2075.1989.tb08488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Rocquigny H, Gabus C, Vincent A, Fournié-Zaluski M-C, Roques B, Darlix J-L. Proc Natl Acad Sci USA. 1992;89:6472–6476. doi: 10.1073/pnas.89.14.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan R, Giedroc D P. J Biol Chem. 1992;267:6689–6695. [PubMed] [Google Scholar]
- 6.Gregoire C J, Gautheret D, Loret E P. J Biol Chem. 1997;272:25143–25148. doi: 10.1074/jbc.272.40.25143. [DOI] [PubMed] [Google Scholar]
- 7.Khan R, Chang H-O, Kaluarachchi K, Giedroc D P. Nucleic Acids Res. 1996;24:3568–3575. doi: 10.1093/nar/24.18.3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan B, Musier-Forsyth K. Proc Natl Acad Sci USA. 1997;94:13530–13535. doi: 10.1073/pnas.94.25.13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grodberg J D, Dunn J J. J Bacteriol. 1988;170:1245–1253. doi: 10.1128/jb.170.3.1245-1253.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh S S, Kao P M, McCue A W, Chappelle H L. Bioorg Chem. 1990;1:71–76. doi: 10.1021/bc00001a009. [DOI] [PubMed] [Google Scholar]
- 11.Odom O W, Picking W D, Hardesty B. Biochemistry. 1990;29:10734–10744. doi: 10.1021/bi00500a004. [DOI] [PubMed] [Google Scholar]
- 12.Proudnikov D, Mizabekov A. Nucleic Acids Res. 1996;24:4535–4542. doi: 10.1093/nar/24.22.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borch R F, Bernstein M D, Durst H D. J Am Chem Soc. 1971;93:2897–2904. [Google Scholar]
- 14.Arts E J, Li X, Gu Z, Kleiman L, Parniak M A, Wainberg M A. J Biol Chem. 1994;269:14672–14680. [PubMed] [Google Scholar]
- 15.Lapadat-Tapolsky M, Pernelle C, Borie C, Darlix J-L. Nucleic Acids Res. 1995;23:2434–2441. doi: 10.1093/nar/23.13.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birch D J S, Imhof R E. In: Topics in Fluorescence Spectroscopy. Lakowicz J R, editor. Vol. 1. New York: Plenum; 1991. pp. 64–71. [Google Scholar]
- 17.Förster T. Ann Phys. 1948;2:55–75. [Google Scholar]
- 18.Press W H, Teukolsky S A, Vetterling W T, Flannery B P. Numerical Recipes in C. New York: Cambridge Univ. Press; 1992. [Google Scholar]
- 19.Shen J, Snook R D. Chem Phys Lett. 1989;155:583–586. [Google Scholar]
- 20.Haas E, Katchalski-Katzir E, Steinberg I Z. Biochemistry. 1978;17:5064–5070. doi: 10.1021/bi00616a032. [DOI] [PubMed] [Google Scholar]
- 21.Hochstrasser R A, Chen S-M, Millar D P. Biophys Chem. 1992;45:133. doi: 10.1016/0301-4622(92)87005-4. [DOI] [PubMed] [Google Scholar]
- 22.Tuite E, Kelly J M, Beddard G S, Reid G S. Chem Phys Lett. 1994;226:517–524. [Google Scholar]
- 23.Suddath F L, Quigly G J, McPherson A, Sneden D, Kim J J, Kim S H, Rich A. Nature (London) 1974;248:20–26. doi: 10.1038/248020a0. [DOI] [PubMed] [Google Scholar]
- 24.Robertus J D, Ladner J E, Finch J T, Rhodes D, Brown R S, Clark B F C, Klug A. Nature (London) 1974;250:546–551. doi: 10.1038/250546a0. [DOI] [PubMed] [Google Scholar]
- 25.Isel C, Ehresmann C, Keith G, Ehresmann B, Marquet R. J Mol Biol. 1995;247:236–250. doi: 10.1006/jmbi.1994.0136. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Khorchid A, Gabor J, Wang J, Li X, Darlix J-L, Wainberg M A, Kleiman L. J Virol. 1998;72:3907–3915. doi: 10.1128/jvi.72.5.3907-3915.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maple J R, Hwang M-J, Stockfisch T P, Dinur U, Waldman M, Ewig C S, Hagler A T. J Comput Chem. 1994;15:162–182. [Google Scholar]